

Abstract
Alzheimer's disease is associated with an age-related accumulation of Abeta and inflammation. The inflammatory mediator, TNFα activates a signaling cascade involving NFκB translocation to the nucleus and a beneficial or detrimental transcriptional response, depending on the age of the neurons and the type of stress applied. Relative to treatment with Abeta42 alone, previously we found that TNFα plus Abeta42, applied to old rat neurons (24 month) is toxic, while the same treatment of middle-age neurons (10 month) is protective. In contrast to improved survival of middle-age rat cortical neurons, neurons from old rats are killed by TNFα plus Abeta42 despite greater p50 nuclear translocation. In middle-age neurons, blocking TNFR1 does not affect NFκB translocation, whereas blocking TNFR2 results in an increase in NFκB translocation. For old neurons, blocking either receptor, does not change NFκB translocation, but improves cell survival. To account for these effects on cell viability in response to TNF + Abeta, measures of the Bcl-2/Bax ratio positively correlate with survival. In the setting of old neurons, these results suggest that overactivated nuclear translocation of NFκB and lower Bcl-2 levels promote death that is reduced by inhibition of either TNFR1 or R2.
Keywords: TNFR1, TNFR2, blocking antibodies, cortical neurons
Introduction
The inflammatory response in the brain of Alzheimer's disease (AD) patients may exacerbate neurodegeneration (Akiyama et al., 2000). Increases in inflammatory cytokines in the brain with age (Davies et al., 2003; Knoblach et al., 1999; Gemma et al., 2002), likely contribute to neurodegeneration. Elevated beta-amyloid (Abeta) may be another factor contributing to inflammation. Abeta forms soluble oligomers and insoluble fibrillar aggregates that deposit in the brain forming senile plaques that are associated with neurodegeneration. Abeta both directly and indirectly activates microglia. The deposition of Abeta activates reactive microglia and other inflammatory responses (Sheng et al., 1997). Using the scavenger receptor, Abeta fibrils attach to microglia, which in turn, release TNFα and produce reactive oxygen species (ROS) (El Khoury et al., 1998; Johnstone et al., 1999). Microglia are chronically activated due to the presence of Abeta, therefore chronically releasing TNFα. TNFα is neurotoxic and with other factors elicits neuronal cell death (Combs et al., 2001; Kang et al., 2001). In comparing, cerebrospinal fluid (CSF) levels of TNFα, TNFβ, IL-1β, IL-1 receptor antagonist (IL1ra), and IL-6 in AD patients and healthy age-matched controls, TNFα was the only cytokine found to be elevated in AD patients (Tarkowski et al., 2003). There was no significant difference between CSF levels of the two groups for TNFβ, IL-1β, IL-6; however, IL-1ra levels were lower in AD patients. We have previously shown age-related increases in TNFα production by rat microglia in response to Abeta (Viel et al., 2001) and age-related toxicity from the combination of TNFα plus Abeta40 or Abeta42 (Patel and Brewer, submitted).
Recently, Abeta has been shown to activate NFκB through the tumor necrosis factor receptor 1 (TNFR1) signaling cascade that results in apoptosis of neurons (Li et al., 2004; Valerio et al., 2006). However, others have shown that NFκB activates anti-apoptotic genes and protects neurons from excitotoxicity and ischemic injury in the mature brain (Schneider et al., 1999; Pennypacker et al., 2001; Bhakar et al., 2002, Mattson, 2005). We hypothesize that age-related down-regulation to the TNFR1 and TNFR2 signaling results in failure of NFκB translocation to the nucleus that fails to provide a neuroprotective response against Abeta42 toxicity by TNFα. Our results show that both middle-age and old neurons translocate NFκB to the nucleus in response to TNFα plus Abeta42 treatment, but NFκB accumulates in old neuron nuclei. This effect is unchanged when TNFR1 is blocked in both middle-age and old neurons. When TNFR2 is blocked, middle-age neurons increase translocation of NFκB, but old neurons do not. These results suggest that promotion of survival or death in the presence of TNF + Abeta depends on the amount of nuclear NFκB and Bcl-2/Bax ratio.
Materials and Methods
Adult Neuronal Cell Culture
Cortical neurons were isolated from 10-12 month old (middle-age) and 24-25 month old (old) male rats (Fisher 344, Harlan, Indianapolis, IN; Brewer, 1997; Brewer and Torricelli, 2007). Isoflurane (Baxter, Deerfield, IL) was used to anaesthetize the animals. Rats were decapitated and their brains removed. The cortices were isolated in Hibernate A (www.BrainBitsLLC.com)/ 2% B27/0.5 mM Glutamax (Invitrogen, Carlsbad, CA) and chopped into 500 μm slices (McIwain tissue chopper, Mickle Laboratory Engineering, Guilford, UK). Next, the tissue was placed in 5 ml Neurobasal A/ 2% B27/0.5 mM Glutamax for 8 minutes in a shaking water bath at 30°C. The tissue was then treated with 2 mg/ml papain (Worthington, Lakewood, NJ) for 30 minutes in a shaking water bath at 30°C. The cortical tissue was triturated 3 times. Using two 4 ml density gradients of Optiprep (Sigma, St. Louis, MO), neurons were separated from other non-neuronal cells by centrifugation at 800 × g for 15 minutes. Fractions containing neurons were collected and mixed with 5 ml of Hibernate A/2% B27/0.5 mM Glutamax and centrifuged at 200 × g for 2 minutes. The supernatant was aspirated and the pellet was resuspended in 5 ml of Hibernate A/2% B27/0.5 mM Glutamax and centrifuged a third time at 200 × g for 2 minutes. The pellet was resuspended in 8 ml of Neurobasal A/2% B27/0.5 mM Glutamax and viable cells counted by trypan blue exclusion. Cells were plated in Neurobasal A medium supplemented with 2% B27/0.5 mM Glutamax, 1 μg/ml Gentamycin (Invitrogen) and 5 ng/ml human fibroblast growth factor 2 (FGF2, Invitrogen). Neurons were plated on glass coverslips (Carolina Biologicals, Burlington, NC), coated with 100 μg/ml poly-d-lysine (Sigma), at a density of 32,000 cells/cm2. The coverslips were either 1 cm2 for imaging or 12 cm2 for gel shift assay. Adult cultures were incubated at 37°C in 5% CO2 and 9% O2 (Thermo-Forma, Waltham, MA) until 8-10 days in vitro. Cultures were immunostained with neuronal and glial markers to determine percentage of neurons (Brewer, 1997). Adult cultures were > 80% neurons (data not shown).
Neuron treatments
Cultures were treated with TNFα (0.01- 1 μg/ml Peprotech, Rocky Hill, NJ) and/or 10 μM fibrillar Abeta (1-42; US Peptides, Rancho Cucamonga, CA). Fibrillar Abeta42 was prepared by dissolving solid peptide in water at 2.5 mM and diluting in Dulbecco's phosphate-buffered saline (dPBS, Invitrogen) to a final concentration of 0.5 mM. Next the Abeta42 was incubated for 3 days at 37°C for aggregation before use (Simmons et al., 1994). Adult cortical cultures at 8-10 days in vitro were placed in fresh medium containing Neurobasal A/B27/Glutamax and TNFα. Untreated controls also underwent 100 % medium changes. The cells were incubated for 24 hours at 37°C in 5% CO2 and 9% O2. After 24 hours, 10 μM Abeta was added to the cultures and were incubated for another 24 hours at 37°C in 5% CO2 and 9% O2. The cultures were now used in viability assays, fixed for immunocytochemistry, and nuclear protein extraction. Middle-age and old neurons were treated with TNFα for 24 hrs followed by 24 hrs of Abeta42 treatment, as before (Patel and Brewer, in press). Experiments using blocking antibodies against TNFR1 (Anti-mouse TNFR1, R&D Systems, Minneapolis, MN) and TNFR2 (Anti-mouse TNFR2, Biolegend, San Diego, CA), cultures were treated with the antibody for 1 hour before TNFα was added. Antibody concentrations were chosen by determining the dose that produced the highest survival of old neurons.
Immunocytochemistry of NFκB, Bcl-2 and Bax
Neurons were treated as described above and then fixed in 4% paraformaldehyde, blocked with 5% normal goat serum (NGS, Invitrogen,) and 0.5% TX-100 (Sigma) in phosphate buffered saline (PBS). Antibodies were diluted in 5% NGS and 0.05% TX-100 in PBS. Primary rabbit antibody against the p50 subunit of NFκB (1:500 dilution; QED Bioscience, San Diego, CA) or mouse anti-Bcl-2 IgG (1:150 dilution, BD Transduction, San Diego, CA) and rabbit anti-mouse Bax antiserum (1:1000 dilution, BD Pharmingen, San Diego, CA) were added to the neurons for overnight incubation at 4°C. Twenty-four hours later, after four rinses with PBS, the secondary antibody, Alexa fluor 568 goat anti-rabbit (1:2000 dilution; Molecular Probes, Eugene, OR) and/or Alexa fluor 488 anti-mouse IgG (1:1000, Molecular Probes) were added for 60 minutes at room temperature. Duplicate coverslips were rinsed two times with PBS and incubated in 1 μg/ml Bisbenzamide (Sigma) for 2 minutes, followed by two rinses with PBS. Coverslips were mounted onto a clean glass microscope slide with Aquamount (Fisher). These mounted slides were imaged using an Olympus epifluorescence microscope under 60× oil objective (N.A. 1.42) with Dapi and Tritc filters and digital Q-imaging camera (Media Cybernetics, Silver Springs, MD). Exposure time and gain were be determined by finding the brightest neurons within treatment and age. Two sets of six consecutive fields per coverslips were imaged at the set exposure time and gain. The images were be analyzed using Image-Pro Plus Software (Media Cybernetics). Density and area threshold ranges were chosen based on the lowest density and area values within treatment and age and kept constant for all neurons imaged. Each individual cell was analyzed for density and area. NFκB translocation to the nucleus was analyzed by first using the bisbenzamide images to determine the outline of the nucleus, and then the outlines were overlaid onto the p50 images. Density and area were determined only within the nuclear outlines. Mean background cellular fluorescence of non-specific binding of secondary antibodies alone was subtracted from all images to account for autofluorescence of neurons. Fluorescence from lipofuscin was either far below that of specific antibody labels and/or was lost by turn-over during regeneration. The amount of cytoplasm imaged was minimal because of the high numerical aperture objective used to selectively focus on the nucleus.
Nuclear extraction preparation
Middle-age and old cortical cultures at 9 days in vitro were treated with 100 ng/ml TNF only, 10 μM Abeta42 only or 100 ng/ml TNF plus 10 μM Abeta42 together. Nuclear extraction was conducted based on Dignam et al. (1983). Cultures were rinsed twice in ice-cold PBS. Cytoplasmic extracts were prepared in buffer A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol (DTT), 0.2 % nonidet P 40 (NP-40), and protease inhibitor cocktail (Sigma)). The cells were scraped off coverslips in Buffer A, vortexed for 15 seconds, and incubated on ice for 10 minutes. Next 10% NP-40 is added to each tube (final NP-40 concentration =1%), vortexed for 5 seconds, incubated on ice for 1 minutes and centrifuged at 16,000 × g for 5 minutes at 4°C. The supernatant was the cytoplasmic fraction and the pellet was the nuclear fraction. The pellet was resuspended in buffer B (20 mM HEPES, pH 7.9, 25% glycerol, 0.42 mM NaCl, 15 mM MgCl2, 0.5 mM DTT, 0.2 mM EDTA and protease inhibitor cocktail), vortexed for 30 seconds at highest setting and rotated at 4°C for 30 minutes. Next the nuclei were centrifuged at 16,000 × g for 15 minutes at 4°C. The supernatant contains nuclear proteins and is used for electrophoretic mobility shift assays. Protein was quantified using the Micro BCA protein assay kit (Pierce).
Annealing oligonucleotides for electrophoretic mobility shift assay
Sense and anti-sense oligos were purchased from Bioneer (Alameda, CA). The sense strand was biotinylated. The following were NFκB sense strand sequence: 5′ ACTTGAGGGGACTTTCCCAGGC 3′ and antisense sequence 5′ GCCTGGGAAAGTCCCCTCAAGT 3′. The oligonucleotides were reconstituted in 10 mM Tris, 1 mM EDTA, and 50 mM NaCl pH 8.0. The complementary strand oligonucleotides were mixed together at 1:1 molar concentrations and annealed in a thermocycler. Each set of oligos were incubated at 95°C for 5 minutes, followed by 70 cycles at which the temperature decreased by 1°C every cycle. At the end of the cycles the oligos were aliquoted and stored at -70°C.
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were used to determine NFκB translocation to the nucleus. Ten micrograms of protein were incubated with 280 fmol of either double stranded biotinylated oligos for NFκB, 10 μg/ml BSA, 2.5% glycerol, 1 μg Poly dI.dC, 0.1% Triton-x for 1 hour at room temperature. All solutions were from Pierce's Lightshift Chemiluminescent EMSA kit (Rockford, IL) and samples were electrophoresed according to manufacturer's instructions. Samples were electrophoresed on 6% nondenaturing gel at 200 volts for 1 hour and transferred to charged nylon membrane for 1.5 hours at 200 mA. The membrane was UV cross-linked for 15 minutes and blocked for 15 minutes, followed by incubation with strepavidin-horse radish peroxidase conjugate in blocking buffer. The membrane was then washed for 20 minutes and then incubated for 5 minutes in substrate equilibration buffer. Next it was incubated for 5 minutes in luminal/enhancer solution and peroxide solution and visualized using a charged-coupled device camera (Hitachi Genetic Systems, MiraiBio Inc., Alameda, CA). Bands were quantified using Image-Pro Plus software. Super shifts were performed to verify p50 subunit using antibody against p50 (data not shown, Santa Cruz Biotechnology, Santa Cruz, CA).
Summary and Statistical Methods
Experiments were conducted on cultured cortical neurons and cortical extracts from 10 month, and 25 month old rats. Each culture consisted of 16,000 to 1.3 million neurons depending on surface area of plating surface. Student's t-test was conducted to determine effect of treatment within each age. Mean and Standard errors are reported.
Results
Our aging neuron model successfully extracts live cortical neurons from middle-age (11 month) or old (24 month) Fisher rats (Brewer, 1997; 1998; Brewer & Torricelli, 2007), near their median lifespan (Solleveld et al., 1984). Similar percentages of neurons survive the dissociation process to regenerate axons and dendrites (Brewer, 1997; 1998; Patel and Brewer 2003). Compared to middle-age neurons, the neurons from old animals take up glucose at the same rate (Patel and Brewer, 2003), have similar passive membrane properties and generation of action potentials (Evans et al., 1998). The resting membrane potentials of old and middle-age neurons are similar (Cady et al., 2001). Therefore, we are not isolating old neurons that are noticeably more fragile or deficient in their ability to regenerate axons and dendrites. Based on previous observations that 100 ng/ml TNFα protected against killing by 10 μM Abeta42 in middle-age, but enhanced toxicity in old neurons, we hypothesized that NFκB translocation from the cytoplasm to the nucleus would be greater for middle-age neurons than old neurons when TNFα, Abeta42, or TNFα plus Abeta42 were added. We examined NFκB translocation to the nucleus by nuclear immunoreactivity of the active subunit p50 (Fig 1). Both middle-age and old neurons show greater p50 nuclear translocation than untreated controls (Fig 1A-H). Digital analysis of these and other images (Fig 1M) shows significantly greater p50 signal in the nucleus for middle-age neurons compared to old neurons. However, when treated with 100 ng/ml TNFα plus 10 μM Abeta42 (Fig 1M), both middle-age and old neurons show a significant increase in p50 immunoreactivity in the nucleus. This increase in p50 immunoreactivity is significantly greater for the old neurons than middle-age neurons (p=0.02). Compared to untreated neurons, nuclear p50 signal increased 20% in middle-age neurons and 60 % in old neurons treated with TNFα plus Abeta42. The results also show that old neurons are capable of NFκB activation by p50 translocation to the nucleus.
Figure 1.
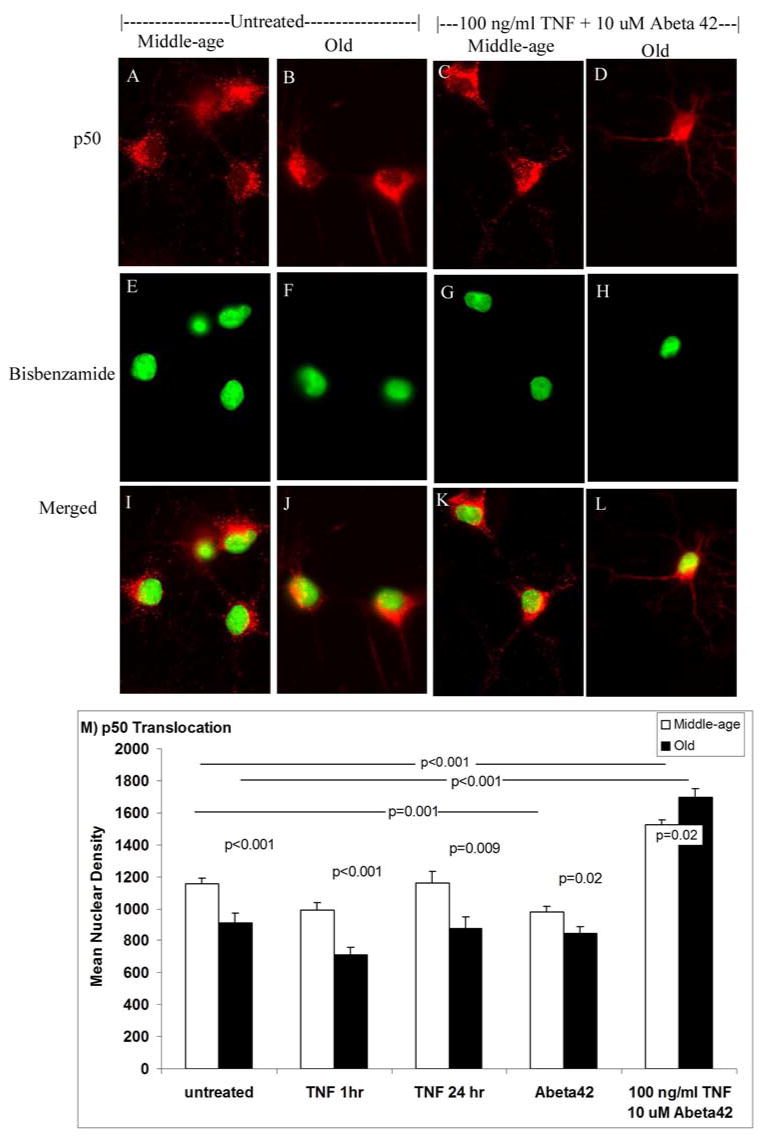
Anti-p50 subunit of NFκB (A-D) and nuclear bisbenzamide (E-H) labeling of untreated middle-age or old neurons compared to neurons treated with the combination of 100 ng/ml TNFα and 10 μM Abeta42. I-L shows the digitally merged image with yellow indicating overlap. Scale = 10 μm. B) Analysis of p50 translocation to the nucleus. Middle-age and old neurons were left untreated or treated with 100 ng/ml TNFα, 10 μM Abeta42, or 100 ng/ml TNFα plus 10 μM Abeta42. Middle-age neurons have higher nuclear density values for untreated, TNFα 1hr, TNFα 24 hr, and Abeta42 treatments compared to old neurons. TNFα plus Abeta42 treated middle-age and old neurons have significantly higher density compared to untreated middle-age (p<0.001) and old neurons (p<0.001). Results are from 3 independent experiments (n=73-150 cells, 3 animals).
We also examined NFκB translocation by binding to the DNA consensus sequence in nuclear extracts. We expected to find results similar to the immunocytochemistry experiment. However, the crude nature of nuclear extracts showed that middle-age neurons have greater binding activity than old neurons when untreated, treated with TNFα or TNFα plus Abeta42 (Fig 2). When middle-age and old neurons were treated with Abeta42 alone they show similar levels of NFκB translocation. We used blocking antibodies to determine if blocking TNFα binding to TNFR1 or TNFR2 would interfere with neuron survival. We hypothesized that blocking TNFR1 would result in greater cell death for TNFα plus Abeta42 treated old neurons, whereas blocking TNFR2 would result in greater cell survival. In Fig. 3A, middle-age neurons show a significant reduction in cell death when treated with blocking antibodies for TNFR1 (p = 0.03) or TNFR2 (p=0.05). Old neurons also show significant reductions in cell death when treated with either TNFR1 (p <0.01) or TNFR2 (p<0.01) blocking antibodies. This suggests that both middle-age and old neurons need both receptors for the activation of cell death signals. When either receptor was blocked, cell death signals were decreased.
Figure 2.
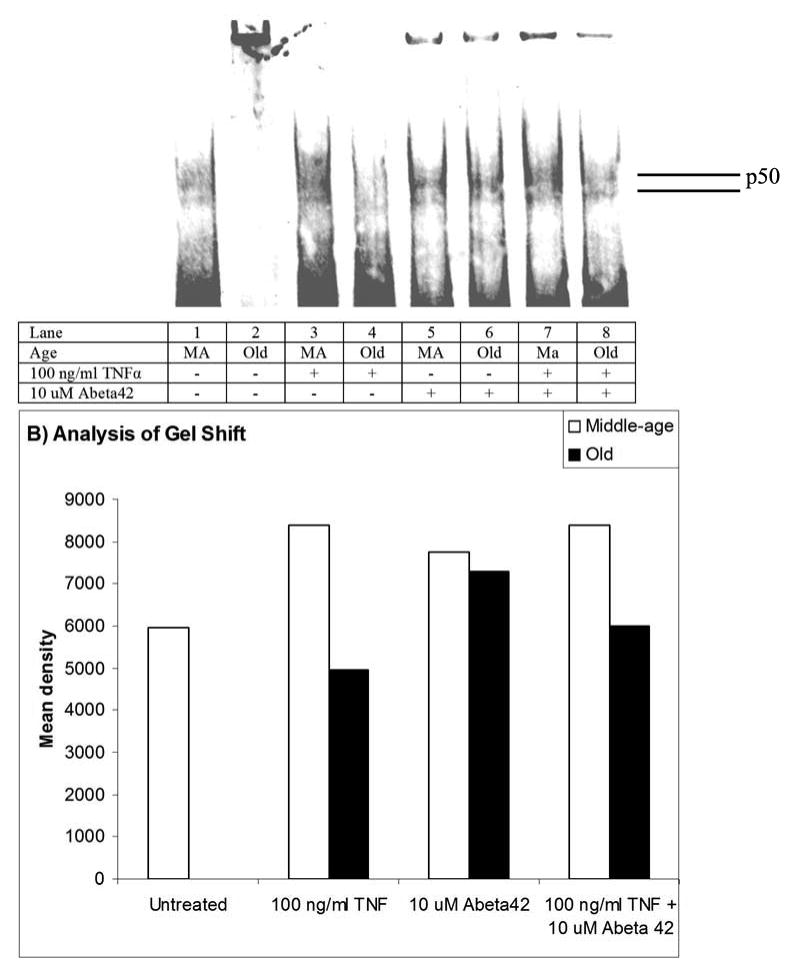
A) Gel-shift of p50 from nuclear extracts of middle-age and old neurons treated as indicated. The band at the top of the page is protein or nucleic acid that was left in the well.
B) Middle-age neurons show significantly greater NFκB translocation than old neurons when treated with TNFα and TNFα plus Abeta42. Middle-age and old neurons show differences in NFκB translocation when treated with Abeta42.
Figure 3.

Middle-age and old neuron viability after treatment with TNFR1 and TNFR2 blocking antibodies. A) Middle-age and old neurons were treated with 100 ng/ml TNF plus 10 μM Abeta42 ± TNFR1 or TNFR2 blocking antibody. When R1 is blocked, both middle-age and old neurons show a decrease in cell death percentage, however, old neurons (p=0.001) show a greater decrease than middle-age neurons (p=0.03). When R2 is blocked again both middle (p=0.006) and old neurons (p=0.05) show a decrease in cell death percentage (n= 24-48, 2-4 animals). B) Immunocytochemisty of p50 translocation to nucleus of middle-age and old neurons. Middle-age and old neurons were treated with 100 ng/ml TNF plus 10 uM Abeta42 either TNFR1 or TNFR2 blocking antibody. When TNFR1 blocking antibody was added; neither middle-age and old neurons decreased nuclear p50 immunoreactivity. When TNFR2 blocking antibody was added; only middle-age neurons increased nuclear p50 immunoreactivity (p<0.001). Old neurons do not show a change in nuclear p50 immunoreactivity (n= 30-75 cells, 2-3 animals). The band at the top of each well is protein plus nucleic acid that did not enter the gel.
We examined NFκB translocation to the nucleus, as in Figs. 1, but in the presence of blocking antibodies. The activation of TNFR has been shown to result in NFκB translocation to the nucleus (Miyamoto et al., 1994; Hsu et al., 1995). We hypothesized that blocking TNFR1 for either age would result in lower nuclear p50 immunoreactivity and blocking TNFR2 would not result in a reduction in p50 immunoreactivity for either age compared to unblocked controls. Fig 3B shows that when TNFR1 blocking antibody acted on middle-age and old neurons, p50 nuclear immunoreactivity did not change for either age. When TNFR2 blocking antibody is used, middle-age neurons increased p50 nuclear immunoreactivity, but old neurons did not show any change in p50 nuclear immunoreactivity.
To explain the greater toxicity of old neurons treated with TNF + Abeta, we reasoned that neuroprotective Bcl-2 would be downregulated in old neurons compared to middle-age neurons and/or proapoptotic Bax immunoreactivity would be greater in old neurons than middle-age neurons. We examined Bcl-2 and Bax immunoreactivity following similar treatments (Fig. 4). In Fig. 4A-B, untreated old neurons show higher Bcl-2 immunoreactivity than untreated middle-age neurons. After treatment with TNF + Abeta, middle-age neurons have similar levels of Bcl-2 as the untreated controls (Fig. 4A, C), however, the old neurons after TNF + Abeta treatment downregulated Bcl-2 levels (Fig 4B, D). Digital analysis of these and other images show age-related and treatment differences in Bcl-2 and Bax (Fig. 4E-H). For middle-age neurons treated with TNF or Abeta, Bcl-2 or Bax levels did not change compared to untreated controls (Fig. 4E). Compared to middle-age neurons treated with TNF, middle-age neurons treated with TNF + Abeta (p=0.05) had lower levels of Bax, while Bcl-2 levels did not change. This lowering of proapoptotic Bax in middle-age neurons treated with TNF + Abeta correlates with the observed neuroprotection for these conditions (Fig. 3A). Compared to middle-age neurons treated with Abeta, the combined treatment of TNF + Abeta elicited lower levels of Bcl-2 (p=0.05). For middle-age neurons, treatment with Abeta alone produced higher Bcl-2 levels and lower Bax levels that were now significantly different (p=0.001). Compared to TNF + Abeta treatment alone, treatment of middle-age neurons with R1 blocking antibody and TNF + Abeta, elevates Bcl-2 (p=0.01) and Bax levels (p<0.001, Fig. 4E)
Figure 4.
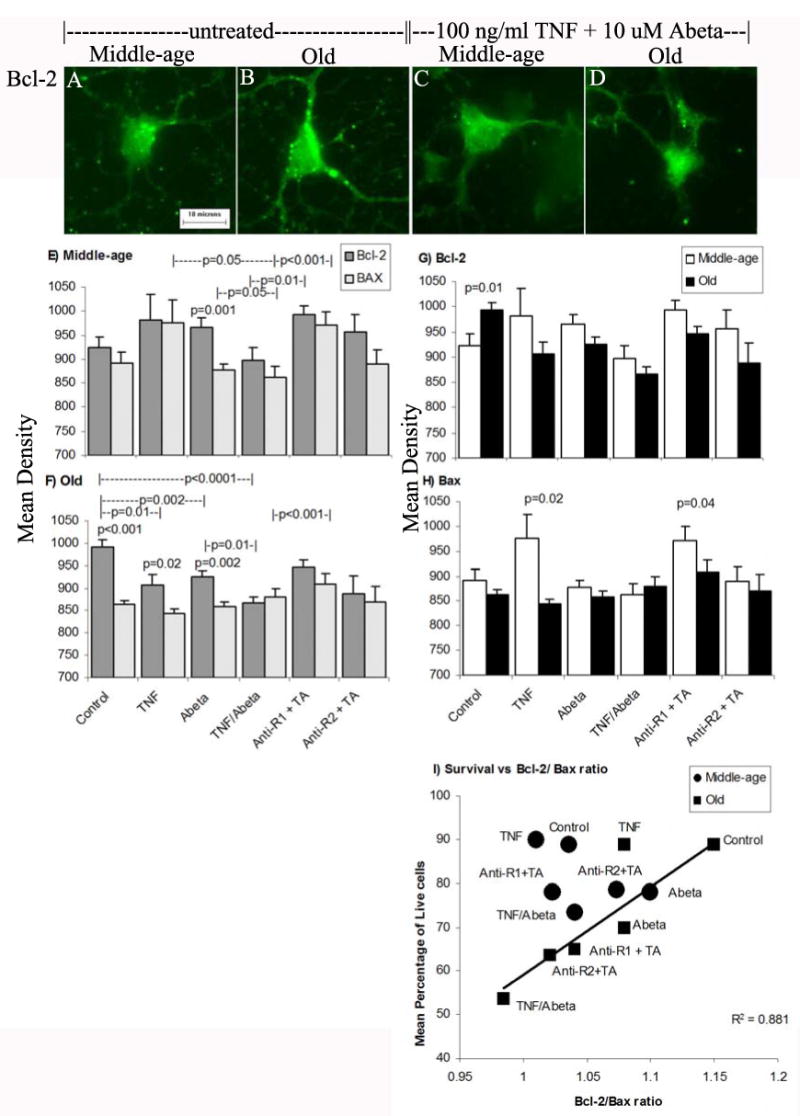
A-D) Bcl-2 immunoreactivity in middle-age and old neuron. Scale = 10 μm E) Bcl-2 and Bax immunoreactivity for middle-age neurons. F) Bcl-2 and Bax immunoreactivity for old neurons. Rearrangement of the same data indicates in G) higher control levels of Bcl-2 immunoreactivity in old neurons compared to middle-age neurons. H) in middle-age neurons Bax immunoreactivity is higher than in old neurons for TNF treatment and TNFR1 blocking antibody with TNF + Abeta (TA). I) Survival versus Bcl-2/Bax ratio of middle-age and old neurons shows a strong correlation for all conditions except middle-age neurons untreated or treated with TNF and/or TNFR1 blocking antibody and old neurons treated with TNF alone.
In contrast to middle-age neurons, untreated old neurons showed significantly less Bax than Bcl-2 (p<0.001, Fig.4F). Compared to untreated old neurons, Bcl-2 immunoreactivity is significantly downregulated following treatment with TNF alone (p=0.01), Abeta alone (p=0.002), TNF + Abeta alone (p<0.0001), TNFR1 blocking antibody with TNF + Abeta (p=0.03) or TNFR2 blocking antibody with TNF + Abeta (p=0.02). Compared to Abeta treatment of old neurons, TNF + Abeta treatment of old neurons significantly lowered Bcl-2 levels (p=0.01). Adding TNFR1 blocking antibody with TNF + Abeta significantly raised Bcl-2 levels compared to only TNF + Abeta treatment, suggesting a role for TNFR1 in lowering Bcl-2 (p<0.001). Bcl-2 levels were significantly higher than Bax levels when old neurons were treated with TNF (p=0.02) or Abeta (p=0.002, Fig. 4F). However, Bax levels were not significantly affected by any treatment of old neurons.
The same data was rearranged for easier age-related comparisons of Bcl-2 (Fig. 4G) and Bax (Fig. 4H). Compared to untreated middle-age neurons, untreated old neurons had significantly higher Bcl-2 immunoreactivity (p=0.01, Fig. 4G). All other treatments reversed this age-related ratio for Bcl-2 and Bax. Compared to untreated middle-age neurons, Bax levels were not different for untreated old neurons. Bax levels in middle-age neurons after TNF treatment were higher than those in old neurons (p=0.02) or after TNFR1 blocking with TNF + Abeta treatment (p=0.04, Fig. 4H). Bax levels between middle-age and old neurons did not change with other treatments.
Since TNF receptor activation of NFκB translocation did not easily account for the effects on viability of old neurons, we examined correlations of viability to Bcl-2/Bax ratios. Fig. 4I shows that survival positively correlated with Bcl-2/Bax ratio for nearly all experimental conditions, except three middle-age conditions and TNF treatment of old neurons. Bax levels did not change in middle-age neurons either treated with Abeta or untreated controls. There was an increase in the Bcl-2/Bax ratio with Abeta treatment. Based on previous work (Patel & Brewer, in press), both TNFR1 and TNFR2 were higher in middle-age than in old neurons treated with TNF plus Abeta. This shows that both prosurvival and proapoptotic proteins are critical components of the TNF signaling pathway along with NFκB translocation.
Discussion
Our data are consistent with the hypothesis that age-related changes to the TNFR1 and TNFR2 signaling results increased NFκB translocation as p50 translocation to the nucleus that provides neuroprotection to middle-age neurons, but the same translocation in old neuron fails to protect against TNFα plus Abeta42 toxicity. Untreated old neurons show significantly higher Bcl-2 levels compared to middle-age neurons and other experimental conditions. The immunoreactive Bcl-2/Bax ratio positively correlates with cell survival for middle-age and old neurons treated with Abeta, TNFα + Abeta or R2 blocking antibody with TNFα + Abeta. Others have shown that Abeta toxicity in neuroblastoma cells occurs by Bcl-2 downregulation and upregulated Bax expression (Clementi et al., 2006). Elevated Bcl-2 in old neurons greatly decreased following treatment with TNFα + Abeta and is only partly restored by blocking TNFR1. The positive correlation between survival and Bcl-2/Bax ratio suggests that levels of those proteins exert stronger control over viability in old neurons than does NFκB translocation to the nucleus. Fig. 5 summarizes the age-related differences between middle-age and old neurons after TNFα + Abeta treatment. Old neurons increase NFκB translocation to the nucleus, but downregulate Bcl-2/Bax ratio, which correlates with greater old neuron death. As discussed below, previous work has shown old neurons fail to upregulate TNFR1 and TNFR2 receptors (Patel and Brewer, submitted), increase ROS (Parihar and Brewer, 2007) and oxidized redox levels (Parihar et al., in revision) in comparison to middle-age neurons.
Figure 5.

TNF receptor mediated signaling in middle-age and old neurons. A) Middle-age neurons treated with 100 ng/ml TNF plus 10 μM Abeta42 show significantly lower cell death compared to old neurons. Old neurons have higher p50 translocation than middle-age neurons, which might promote survival, but old neurons have less Bcl-2 that appears to override or prevent this survival signal.
Experiments for nuclear translocation of NFκB were performed by immunostaining neurons for p50 antibody. Middle-age and old neurons treated with either TNFα or Abeta42 alone failed to translocate NFκB above untreated levels. It is possible that neurons have constitutive NFκB activation (Meffert et al., 2003); therefore we did not see a difference in p50 translocation when the neurons were treated with proven activators of NFκB in other cell types. Old and middle-age neurons both show significant up-regulation of p50 translocation when treated with TNFα plus Abeta42. Old neurons show greater NFκB translocation than middle-age neurons treated with TNFα plus Abeta42. The larger amount of NFκB translocation in old neurons versus middle-age in response to TNFα + Abeta shows the capacity of NFκB is patent in old neurons (Fig 1M), but does not account for the loss of viability in old neurons. Fig. 4F and I show that the same TNFα + Abeta treatment that leaves the Bcl-2/Bax ratio unchanged in middle-age neurons greatly reduces the neuroprotective Bcl-2 in old neurons.
The differences between the immunocytochemistry experiments and gel shift may be due to the differences in sensitivity of either technique. The immunocytochemistry is a more sensitive and spatially more specific method than a gel shift assay. The homogenized average of gel shift assays requires a large population of cells and therefore, may not be sensitive to subtle differences in activation between age and treatment groups. Antibody binding for immunocytochemistry is a stable reaction compared to DNA binding activity of gel shift assays. The NFκB and oligonucleotide complex is sensitive to temperature, pH, protease activity, and salt concentration, therefore may produce variability in signal between experimental conditions.
We conducted viability experiments using TNFR1 and TNFR2 blocking antibodies. Blocking antibodies were added to the adult cultures prior to TNFα and Abeta42 addition. When either TNFR1 or TNFR2 blocking antibodies were added, cell death decreased for both ages. However, old neurons showed a greater decrease in cell death than middle-age neurons, which suggests that old neurons are more dependent on the ratio of surface receptor expression than middle-age neurons. This suggests that both ages need activation of both receptors for apoptosis or that p50 is inactive, possibly because it dimerizes with an inhibitory p65 rather than an activating p50 homodimer or that bcl-2 sites are available for activation in middle-age neurons, but blocked epigenetically in old neurons. The protection seen in old neurons may be better attributed to the Bcl/Bax ratio and appears less dependent on NFκB translocation.
TNFR1 blocking antibody addition to either middle-age and old neurons did not change NFκB translocation (p50 immunoreactivity) for either age. This means that TNFR2 can still be stimulated and that TNFR2 does not mediate NFκB activation in either age neuron. However, when TNFR2 blocking antibodies were added to middle-age neurons, NFκB translocation was upregulated, whereas the old neurons do not show a change in NFκB translocation. This suggests that stimulation of TNFR1 activates NFκB, but only in the middle-age neurons. TNFR1 and its intracellular mediators may activate common pathways that lead to degradation of IκBα, which allows translocation of NFκB from the cytoplasm into the nucleus (Miyamoto et al., 1994; Hsu et al., 1995). In middle-age neurons, NFκB appears to activate prosurvival pathways as demonstrated by Kaltschmidt et al. (1999). But in old neurons, the NFκB that translocates to the nucleus fails to activate these prosurvival pathways.
Based on many other studies, the role of NFκB in survival appears to depend on the age of neurons and the type of prior stress applied to neurons. In embryonic neurons, NFκB translocation stimulated by TNF alpha promotes neuron survival (Nicholas et al., 2001; Fernyhough et al., 2005), possibly by inducing the expression of anti-apopotic proteins such as Bcl-2 and manganese superoxide dismutase (Mattson, 2005). Also, Lu et al. (2006) showed that 10 month old p50 knockout mice had lipofuscin granule accumulation, abnormal capillaries, apoptotic glia cells, and caspase-3 positive neurons. These findings support our middle-age data that in the presence of TNFα and Abeta42, NFκB protects neurons from cell death.
However, in embryonic hippocampal neurons, Abeta40 induced cell death by binding to TNFR1 and activating its signaling pathway which includes NFκB translocation (Li et al., 2004). This mechanism was mediated by apoptotic protease-activating factor 1 (Apaf-1) that induced NFκB nuclear translocation. Also, Tamatani et al. (1999) found that TNFα induces Bcl-2 expression through NFκB activation in embryonic hippocampal neurons. TNFR1 knockouts showed downregulation of Apaf-1 activation and NFκB nuclear translocation. Our studies show that middle-age and to a greater extent old neurons are able to translocate NFκB in response to TNF + Abeta42, but cell death increased only for old neurons. Valerio et al. (2006) showed in postnatal cerebellar granule cells that Abeta mediated death caused NFκB activation, which resulted in cytochrome c release and decreased neuroprotective Bcl-XL. Inhibitors of NFκB prevented neuronal damage, cytochrome c release, and restored Bcl-XL levels. Toliver-Kinsky et al. (1997) found an age-related up-regulation of NFκB in 30 month old rats that resulted in decreased cholinergic activity that could contribute to age-related cognitive deficits.
Failure of NFκB to activate prosurvival pathways in old neurons in response to TNF + Abeta could depend on at least two factors: epigenetic regulation of these genes or redox control of activity. Whether nuclear NFκB promotes transcription of key protective genes depends on the epigenetic state of these genes. Either critical CPG islands could be methylated or start sites blocked by histones in old neurons. According to some studies, the translocation of NFκB in embryonic and young neurons might play an anti-apoptotic role (Guo et al., 1998; Kaltschmidt et al., 1999) or an apoptotic role (Schneider et al., 1999; Li et al., 2004) as in old neurons.
The second possible explanation for failure of NFκB to activate prosurvival pathways involves higher ROS production (Parihar and Brewer 2007) and an oxidized redox state in old neurons (Parihar et al., in press). According to Sulciner et al. (1996) the redox state of NFκB could be a control mechanism that determines the level of NFκB activation or post-translational modifications of genes activated by NFκB (Toledano et al., 1991). The differences in the redox state of old versus middle-age neurons could result in differences in NFκB activation, thus resulting in greater cell death for old neurons compared to middle-age neurons.
Our previous study (Patel and Brewer, in press) showed that old neurons treated with TNFα plus Abeta42 fail to upregulate TNFR1 and TNFR2 like the two-fold increase in middle-age neurons. Old neurons failed to up-regulate TNFR1 density levels above baseline levels. This failure correlates with low survival of old neurons under the same conditions. In old neurons, this combination of R1 and R2 failure to upregulate and a loss of neuroprotective Bcl-2 appears to explain the low survival for old neurons. This supports the hypothesis that either old neurons have an imbalance of signaling from R1 and R2 and/or that nuclear translocation is ineffective in old neurons. The positive correlation between survival and Bcl-2/Bax ratio suggests that ultimately the Bcl-2/Bax ratio is more important for survival than NFκB translocation. These studies demonstrate dramatic differences between middle-age and old neurons when age-related hormonal, inflammatory and vascular complications are controlled.
Further studies are necessary to understand age-related differences in adult neurons versus embryonic neurons and possible epigenetic controls. Aging of the brain creates deficits and/or epigenetic changes that contribute to the development of neurodegenerative disease such as Alzheimer's disease (AD). These age-related changes could be regulated alterations in gene or protein expression that are “programmed aging” or pathologic metabolic responses to generalized senescent deterioration of the brain. Age-related altered regulation could be a response to subtle early damage created by environmental factors (i.e. sedentary lifestyle, head trauma, oxidative stress). Age-related changes in response to inflammatory mediators could further contribute to disease development.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J Neurosci. 2002;22:8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer GJ. Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods. 1997;71:143–155. doi: 10.1016/s0165-0270(96)00136-7. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Age-related toxicity to lactate, glutamate, and beta-amyloid in cultured adult neurons. Neurobiol Aging. 1998;19:561–568. doi: 10.1016/s0197-4580(98)00091-8. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR. Isolation and culture of adult neurons and neurospheres. Nature Protocols. 2007;2:1490–1498. doi: 10.1038/nprot.2007.207. [DOI] [PubMed] [Google Scholar]
- Cady C, Evans MS, Brewer GJ. Age-related differences in NMDA responses in cultured rat hippocampal neurons. Brain Res. 2001;921:1–11. doi: 10.1016/s0006-8993(01)03063-3. [DOI] [PubMed] [Google Scholar]
- Clementi ME, Pezzotti M, Orsini F, Sampaolese B, Mezzogori D, Grassi C, Giardina B, Misiti F. Alzheimer's amyloid beta-peptide (1-42) induces cell death in human neuroblastoma via bax/bcl-2 ratio increase: an intriguing role for methionine 35. Biochem Biophys Res Commun. 2006;342:206–213. doi: 10.1016/j.bbrc.2006.01.137. [DOI] [PubMed] [Google Scholar]
- Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha- dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies HA, Kelly A, Dhanrajan TM, Lynch MA, Rodriguez JJ, Stewart MG. Synaptophysin immunogold labelling of synapses decreases in dentate gyrus of the hippocampus of aged rats. Brain Res. 2003;986:191–195. doi: 10.1016/s0006-8993(03)03251-7. [DOI] [PubMed] [Google Scholar]
- Dignam JD, Lebovitz RM, Roeder RG. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC. Microglia, scavenger receptors, and the pathogenesis of Alzheimer's disease. Neurobiol Aging. 1998;19:S81–S84. doi: 10.1016/s0197-4580(98)00036-0. [DOI] [PubMed] [Google Scholar]
- Evans MS, Collings MA, Brewer GJ. Electrophysiology of embryonic, adult and aged rat hippocampal neurons in serum-free culture. J Neurosci Meth. 1998;79:37–46. doi: 10.1016/s0165-0270(97)00159-3. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Smith DR, Schapansky J, Van Der PR, Gardiner NJ, Tweed CW, Kontos A, Freeman L, Purves-Tyson TD, Glazner GW. Activation of nuclear factor-kappaB via endogenous tumor necrosis factor alpha regulates survival of axotomized adult sensory neurons. J Neurosci. 2005;25:1682–1690. doi: 10.1523/JNEUROSCI.3127-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemma C, Mesches MH, Sepesi B, Choo K, Holmes DB, Bickford PC. Diets enriched in foods with high antioxidant activity reverse age-induced decreases in cerebellar beta-adrenergic function and increases in proinflammatory cytokines. J Neurosci. 2002;22:6114–6120. doi: 10.1523/JNEUROSCI.22-14-06114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Robinson N, Mattson MP. Secreted beta-amyloid precursor protein counteracts the proapoptotic action of mutant presenilin-1 by activation of NF-kappaB and stabilization of calcium homeostasis. J Biol Chem. 1998;273:12341–12351. doi: 10.1074/jbc.273.20.12341. [DOI] [PubMed] [Google Scholar]
- Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Johnstone M, Gearing AJ, Miller KM. A central role for astrocytes in the inflammatory response to beta- amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol. 1999;93:182–193. doi: 10.1016/s0165-5728(98)00226-4. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Uherek M, Wellmann H, Volk B, Kaltschmidt C. Inhibition of NF-kappaB potentiates amyloid beta-mediated neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:9409–9414. doi: 10.1073/pnas.96.16.9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Park EJ, Jou I, Kim JH, Joe EH. Reactive oxygen species mediate A beta(25-35)-induced activation of BV- 2 microglia. Neuroreport. 2001;12:1449–1452. doi: 10.1097/00001756-200105250-00030. [DOI] [PubMed] [Google Scholar]
- Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95:115–125. doi: 10.1016/s0165-5728(98)00273-2. [DOI] [PubMed] [Google Scholar]
- Li R, Yang L, Lindholm K, Konishi Y, Yue X, Hampel H, Zhang D, Shen Y. Tumor necrosis factor death receptor signaling cascade is required for amyloid-beta protein-induced neuron death. J Neurosci. 2004;24:1760–1771. doi: 10.1523/JNEUROSCI.4580-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZY, Yu SP, Wei JF, Wei L. Age-related neural degeneration in nuclear-factor kappaB p50 knockout mice. Neuroscience. 2006;139:965–978. doi: 10.1016/j.neuroscience.2005.12.062. [DOI] [PubMed] [Google Scholar]
- Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res. 2005;30:883–893. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Maki M, Schmitt MJ, Hatanaka M, Verma IM. Tumor necrosis factor alpha-induced phosphorylation of I kappa B alpha is a signal for its degradation but not dissociation from NF-kappa B. Proc Natl Acad Sci U S A. 1994;91:12740–12744. doi: 10.1073/pnas.91.26.12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas RS, Compston A, Brown DR. Inhibition of tumour necrosis factor-alpha (TNFalpha)-induced NF-kappaB p52 converts the metabolic effects of microglial-derived TNFalpha on mouse cerebellar neurones to neurotoxicity. Journal of Neurochemistry. 2001;76:1431–1438. doi: 10.1046/j.1471-4159.2001.00141.x. [DOI] [PubMed] [Google Scholar]
- Parihar MS, Brewer GJ. Simultaneous age-related depolarization of mitochondrial membrane potential and increased mitochondrial reactive oxygen species production correlate with age-related glutamate excitotoxicity in rat hippocampal neurons. J Neurosci Res. 2007;85:1018–1032. doi: 10.1002/jnr.21218. [DOI] [PubMed] [Google Scholar]
- Parihar MS, Kunz E, Brewer GJ. Age-related decreases in NAD(P)H and glutathione cause redox declines before ATP loss during glutamate treatment of hippocampal neurons. J Neurosci Res. 2008 doi: 10.1002/jnr.21679. in press. [DOI] [PubMed] [Google Scholar]
- Patel JR, Brewer GJ. Age-related changes in neuronal glucose uptake in response to glutamate and beta-amyloid. J Neurosci Res. 2003;72:527–536. doi: 10.1002/jnr.10602. [DOI] [PubMed] [Google Scholar]
- Patel JR, Brewer GJ. Age-related changes to TNF receptors affect neuron survival in the presence of beta-amyloid. J Neurosci Res. 2008 doi: 10.1002/jnr.21663. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennypacker KR, Kassed CA, Eidizadeh S, Saporta S, Sanberg PR, Willing AE. NF-kappaB p50 is increased in neurons surviving hippocampal injury. Exp Neurol. 2001;172:307–319. doi: 10.1006/exnr.2001.7817. [DOI] [PubMed] [Google Scholar]
- Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. doi: 10.1038/8432. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer's disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol (Berl) 1997;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- Simmons LK, May PC, Tomaselli KJ, Rydel RE, Fuson KS, Brigham EF, Wright S, Lieberburg I, Becker GW, Brems DN. Secondary structure of amyloid beta peptide correlates with neurotoxic activity in vitro. Mol Pharmacol. 1994;45:373–379. [PubMed] [Google Scholar]
- Solleveld HA, Haseman JK, McConnell EE. Natural history of body weight gain, survival, and neoplasia in the F344 rat. J Natl Cancer Inst. 1984;72:929–940. [PubMed] [Google Scholar]
- Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P, Finkel T. rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol Cell Biol. 1996;16:7115–7121. doi: 10.1128/mcb.16.12.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFκB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Liljeroth AM, Minthon L, Tarkowski A, Wallin A, Blennow K. Cerebral pattern of pro- and anti-inflammatory cytokines in dementias. Brain Res Bull. 2003;61:255–260. doi: 10.1016/s0361-9230(03)00088-1. [DOI] [PubMed] [Google Scholar]
- Toledano MB, Leonard WJ. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc Natl Acad Sci U S A. 1991;88:4328–4332. doi: 10.1073/pnas.88.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toliver-Kinsky T, Papaconstantinou J, Perez-Polo JR. Age-associated alterations in hippocampal and basal forebrain nuclear factor kappa B activity. J Neurosci Res. 1997;48:580–587. doi: 10.1002/(sici)1097-4547(19970615)48:6<580::aid-jnr11>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Valerio A, Boroni F, Benarese M, Sarnico I, Ghisi V, Bresciani LG, Ferrario M, Borsani G, Spano P, Pizzi M. NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur J Neurosci. 2006;23:1711–1720. doi: 10.1111/j.1460-9568.2006.04722.x. [DOI] [PubMed] [Google Scholar]
- Viel JJ, McManus DQ, Smith SS, Brewer GJ. Age- and concentration-dependent neuroprotection and toxicity by TNF in cortical neurons from beta-amyloid. J Neurosci Res. 2001;64:454–465. doi: 10.1002/jnr.1097. [DOI] [PubMed] [Google Scholar]