

Abstract
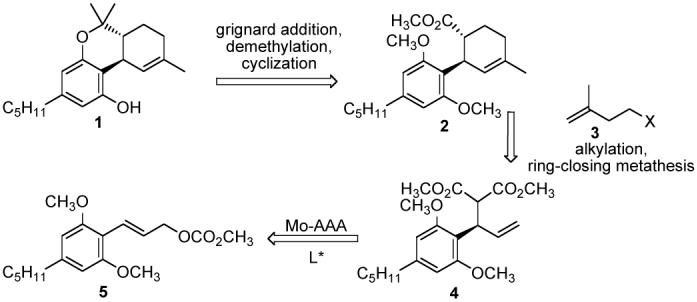
Δ9-THC is synthesized in enantiomericaly pure form, where all of the stereochemistry is derived from the molybdenum catalyzed asymmetric alkylation reaction of the extremely sterically congested bis-ortho substituted cinnamyl carbonate in high regio- and enantioselectivity.
(-)-Δ9-trans-Tetrahydrocannabinol (Δ9-THC) 1, isolated1 from female Cannabis sativa L. in 1964 has been identified as the primary psychomimetic component of marijuana. It is also known to show antiemetic, antiglaucoma, and analgesic properties. Currently it is administered as an antinauseant to patients undergoing chemotherapy. Discovery of the cannabinoid receptors CB1 and CB2 and Δ9-THC analogues2 that bind selectively to them has led to a need for the development of a flexible synthetic route which would yield target compounds easily in high yields and in sterochemically pure form.
THC itself has been prepared numerous times, though most routes are either racemic or derive their chirality from chiral building blocks.3 Only Evan’s4 route targets the natural product enantioselectively from achiral starting materials. The problems associated with the synthesis of THC involve the control of cis-trans stereochemistry at the cyclohexene ring and the position of the double bond i.e. Δ9 vs. Δ8, the latter being thermodynamically more stable.
Our reterosynthetic analysis of the molecule envisioned all of the stereochemistry resulting from a single Mo-catalyzed asymmetric allylic alkylation (AAA) reaction (See abstract). Alkylation of malonate adduct 4 with 3 should furnish the substrate for ring-closing metathesis (RCM). The RCM in itself results in a fixed geometry of double bond thus solving the problem of co-formation of Δ8-THC seen in many syntheses. Decarboxlylation of the malonate after RCM was planned to result in the required trans stereochemistry of 2. Grignard addition to ester 2 followed by demethylation to the free phenol and cyclization yields 1. Varying the aromatic group in 5, the alkylating partner 3, or the Grignard reagents one can potentially prepare different analogues of THC without fundamentally changing the chemistry involved.
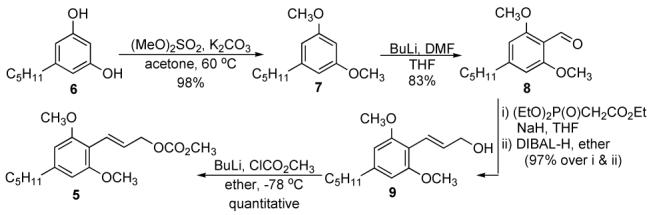
Allyl alcohol 9 was easily prepared in high yield from commercially available olivetol, 6 in 4 steps (Scheme 1). Lithiated dimethyl olvitol was quenched with dry DMF to yield the aldehyde 8 in 83% yield. Wadsworth-Horner-Emmons reaction of 8 with sodium triethyl-phosphonoacetate resulted in the corresponding α,β-unsaturated ethyl ester, which was subjected without further purification to DIBAL-H reduction to yield 97% of 9. Preparation of carbonate 5 proved to be a little tricky because the compound is sensitive to both acid and base, including silica chromatography conditions. A preparation of 5 clean enough to take into the Mo-alkylation reaction, was achieved by titrating alcohol 9 with BuLi at -78 °C in ether and quenching the resulting alkoxide with methyl chloroformate also at -78 °C. Washing the organic layer with ice-cold water followed by solvent removal gave carbonate 5 as a waxy solid, which is stable to storage.
Scheme 1.
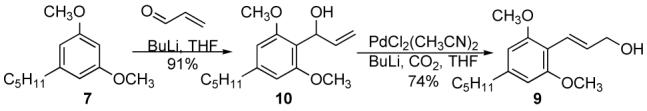
The branched allyl alcohol 10 is prepared by condensation of acrolein with lithiated 7 in one step in 91% yield (Scheme 2). We planned to use the carbonate prepared from 10 in the Mo-AAA reaction to reduce the number of steps and increase the efficiency of the synthesis. Unfortunately it proved impossible to prepare either the carbonate or the acetate of 10, due to their facile decomposition aided by the highly electron rich aromatic ring.
Scheme 2.
We did, however, take advantage of this to isomerize the branched alcohol 10 into the linear alcohol 95. Treatment of the lithium alkoxide of 10 with dry CO2 gas gives the corresponding carbonate which rearranges in the presence of PdCl2(CH3CN)2 to yield the more stable linear carbonate which decomposes to the alcohol 9 on workup (Figure 1). A 74% yield of 9 was observed with 6% recovered 10. To the best of our knowledge this reaction has not been previously reported. This alternate strategy using this novel isomerization should prove to be a more efficient, atom economical approach to cinnamyl alcohols then the more traditional protocol based upon olefination.
Figure 1.
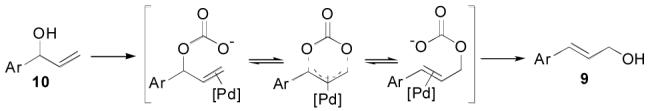
Isomerization of branched 10 to linear alcohol 9.
With the carbonate in hand we began examining the allylic alkylation reaction. Although molybdenum is known for giving the product resulting from attack at the more substituted carbon, we were concerned that the two ortho methoxy groups on the aryl ring would make this reaction sterically unfavorable. Nevertheless, the reaction of carbonate 5 with sodium dimethyl malonate under standard conditions of 10 Mol % [Mo(CO)3C7H8] and 15 mol % chiral ligand S,S-11 proceeded sluggishly but gratifyingly gave only the branched product 4 in 95% yield and 95% enantiomeric excess. The reaction was optimized to reduce catalyst loading and the optimized conditions are shown in Eq. 1.
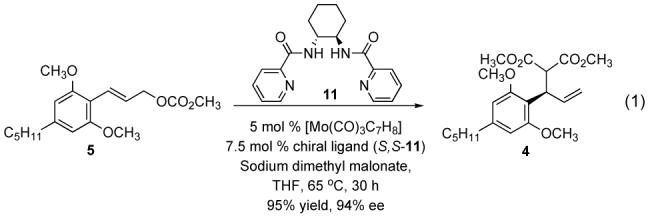 |
Attempts to alkylate the malonate adduct 4 with 3 under numerous conditions met with no success (Scheme 4). The electrophile 3 slowly disappeared under the reaction conditions due to 1,2-elimination, giving rise to isoprene. To examine whether this or the steric demands of making a quaternary center at the congested malonate carbon was the cause for failure, we tried the addition of MeI, but observed only trace amounts of the desired adduct. This led us to believe steric congestion was cause of failure, and hence we prepared the monoester 12 under Krapcho decarboxylation6 conditions in 83% yield, which reduced steric demands. Since the enolates of methyl esters are generally stable only below -35 °C we attempted to carry out the alkylation at -40 °C. Our best result was only a 50% yield of the adduct 13 using triflate 3c (Scheme 3). The competing elimination reaction was still a problem due to the slower rate of alkylation at the reduced temperature.
Scheme 4.
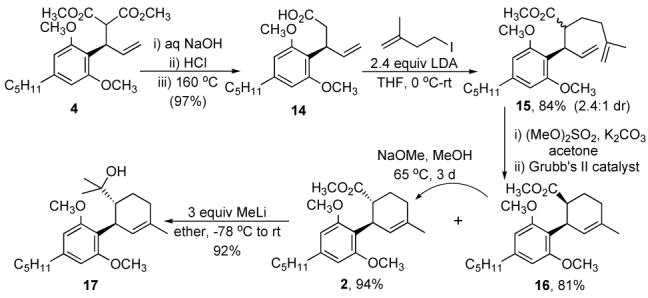
Scheme 3.
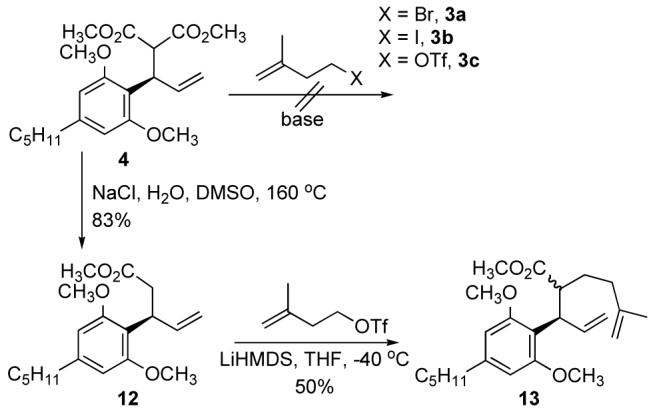
Therefore a nucleophile whose enolate would be stable at room temperature or higher was required. We believed that the dianion of the acid 14 would meet our needs (Scheme 4). This acid is prepared from 4 using classical conditions in 97% yield, as shown in Scheme 5. The dianion of acid 14 cleanly underwent alkylation with iodide 3b to yield 84% of 15 as a 2.4:1 mixture of anti and syn isomers at the newly formed center. The two isomers were separated using column chromatography, converted to their respective methyl esters, and subjected to ring closing metathesis using the second generation Grubb’s ruthenium carbene catalyst,7 providing the anti and syn cyclohexene compounds 2 and 16. The syn compound 16 was easily recycled by equilibrating to anti 2 by treatment with NaOMe in MeOH for 3 days. Addition of MeLi to the cyclized ester 16 at -78 °C resulted in formation of the tertiary alcohol 17 in 92% yield (Scheme 4).
Scheme 5.
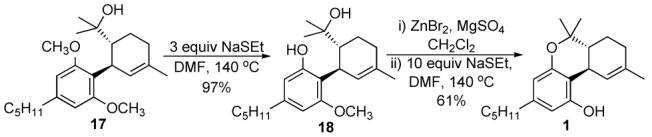
With the end in sight, compound 17 was treated with BBr3 to yield the free phenol by demethylation of both ethers (Scheme 5). We hoped that the free bis phenol would subsequently cyclise under the acidic reaction conditions to give 1. Unfortunately we got a complex mixture of products. The use of other acidic reagents for demethylation resulted in similar fates. Finally we decided to carry out the reaction in a stepwise manner using NaSEt in DMF which is known to stop at mono-deprotection of aryl diethers.8 The reaction proceeded as expected to yield 97% of 18, [α]23D -52 (c 0.79, CHCl3), (lit.9 [α]26D -44 (c 0.20, CHCl3)). Formation of the cyclised ether was achieved by treating 18 with ZnBr2 in the presence of MgSO4. The resulting compound was subjected to NaSEt in DMF without further purification to yield the desired Δ9-THC 1 in 61% yield [α]25D -152 (c 0.46, CHCl3), (lit.6 [α]28D -150 (c 1.0, CHCl3).
In summary, the use of the molybdenum catalyzed asymmetric alkylation reaction developed in this group provided the stereochemical framework to synthesize Δ9-THC in enantiomericaly pure form and 30% overall yield from olivetol dimethyl ether. The regio- and enantio-selectivity of the Mo AAA is undiminished even in this sterically congested example. Furthermore a simple and efficient two step protocol for the synthesis of cinnamyl alcohols has also been developed.
Supplementary Material
Acknowledgment
We thank the General Medical Sciences Institute and the National Institutes of Health (GM 13598) and Merck for their generous support of our programs. We are also indebted to Johnson Matthey for a generous gift of palladium salts. Mass spectra were provided by the Mass Spectrometry Regional Center of the University of California, San Francisco supported by the NIH Division of Research Resources.
References
- (1).Mechoulam R, Gaoni Y. J. Am. Chem. Soc. 1964;86:1646. [Google Scholar]
- (2)a).Mahadevan A, Siegel C, Martin BR, Abood ME, Belestskaya I, Razdan RK. J. Med. Chem. 2000;43:3778. doi: 10.1021/jm0001572. [DOI] [PubMed] [Google Scholar]; b) Drake DJ, Jensen RS, Busch-Petersen J, Kawakami JK, Fernandez-Garcia MC, Fan P, Makriyannis A, Tius MA. J. Med. Chem. 1998;41:3596. doi: 10.1021/jm960677q. [DOI] [PubMed] [Google Scholar]; c) Harrington PE, Stergiades IA, Erickson J, Makriyannis A, Tius MA. J. Org. Chem. 2000;65:6576. doi: 10.1021/jo000716c. [DOI] [PubMed] [Google Scholar]; d) Tius MA, Makriyannis A, Zou XL, Abadji V. Tetrahedron. 1994;50:2671. [Google Scholar]
- (3).Selected examplesMechoulam R, Gaoni Y. J. Am. Chem. Soc. 1965;87:3273. doi: 10.1021/ja01092a065.Mechoulam R, Braun P, Gaoni Y. J. Am. Chem. Soc. 1967;89:4552. doi: 10.1021/ja00993a072.J. Am. Chem. Soc. 1972;94:6159. doi: 10.1021/ja00772a038.Fahrenholtz KE, Lurk M, Kierstead RW. J. Am. Chem. Soc. 1966;88:2079.J. Am. Chem. Soc. 1967;89:5934.Chan TH, Chaly T. Tetrahedron Lett. 1982;23:2935.Rickards RW, Ronneberg H. J. Org. Chem. 1984;49:572.Childers WE, Jr., Pinnick HW. J. Org. Chem. 1984;49:5276.Malkov AV, Kočovsky P. Collect. Czech. Chem. Commun. 2001;66(8):1257.William AD, Kobayashi Y. J. Org. Chem. 2002;67:8771. doi: 10.1021/jo020457m.Org. Lett. 2001;3:2017.
- (4)a).Evans DA, Shaughnessy EA, Barnes DM. Tetrahedron Lett. 1997;38:3193. [Google Scholar]; b) Evans DA, Barnes DM, Johnson JS, Lectka T, Matt PV, Miller SJ, Murry JA, Norcross RD, Shaughnessy EA, Campos KR. J. Am. Chem. Soc. 1999;121:7582. [Google Scholar]
- (5)a).Overman LE. J. Am. Chem. Soc. 1974;96:597. [Google Scholar]; b) Overman LE. Angew. Chem. Int. Ed. Engl. 1984;23:579. [Google Scholar]
- (6).Krapcho AP, Weimaster JF, Eldridge JM, Jahngen GE, Lovey AJ, Stephens WP. J. Org. Chem. 1978;43:138. [Google Scholar]
- (7).Scholl M, Ding S, Lee CW, Grubbs RH. Org. Lett. 1999;1:953. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- (8).Feutrill GI, Mirrington RN. Tetrahedron Lett. 1970:1327. [Google Scholar]
- (9).William AD, Kobayashi Y. J. Org. Chem. 2002;67:8771. doi: 10.1021/jo020457m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.