

Abstract
We previously found that FoxM1B is overexpressed in human glioblastomas (GBMs) and that forced FoxM1B expression in anaplastic astrocytoma cells leads to the formation of highly angiogenic GBM in nude mice. However, the molecular mechanisms by which FoxM1B enhances glioma angiogenesis are currently unknown. In this study, we found that vascular endothelial growth factor (VEGF) is a direct transcriptional target of FoxM1B. FoxM1B overexpression increased VEGF expression, while blockade of FoxM1 expression suppressed VEGF expression in glioma cells. Transfection of FoxM1 into glioma cells directly activated the VEGF promoter, and inhibition of FoxM1 expression by FoxM1-siRNA suppressed VEGF promoter activation. We identified two FoxM1-binding sites in the VEGF promoter that specifically bound to the FoxM1 protein. Mutation of these FoxM1-binding sites significantly attenuated VEGF promoter activity. Furthermore, FoxM1 overexpression increased and inhibition of FoxM1 expression suppressed the angiogenic ability of glioma cells. Finally, an immunohistochemical analysis of 59 human GBM specimens also showed a significant correlation between FoxM1 overexpression and elevated VEGF expression. Our findings provide both clinical and mechanistic evidence that FoxM1 contributes to glioma progression by enhancing VEGF gene transcription and thus tumor angiogenesis.
Keywords: FoxM1, VEGF, angiogenesis, tumorigenicity, glioblastoma
Introduction
Glioblastoma (GBM) is the most common type of brain tumor and is associated with high morbidity and mortality rates. Patients with GBM routinely undergo surgery followed by adjuvant radiation therapy and chemotherapy. Despite such aggressive treatment approaches, median survival times remain under 1 year in most cases. Brain tumor growth and progression depends on the establishment of an adequate blood supply (i.e., angiogenesis), which confers a tremendous survival and growth advantage on the malignant cells. GBMs are among the most angiogenic of all human tumors, and the level of angiogenesis in GBMs is closely correlated with the degree of malignancy and patient prognosis (1).
The GBM angiogenic phenotype is characterized by an increase in the production of proangiogenic molecules by the tumor cells and organ-specific environments (2). Previous studies have established that GBM cells overexpress many growth factors, including vascular endothelial growth factor (VEGF), and several transcription activators have been identified in the VEGF promoter, including hypoxia inducible factor (HIF)-1 and Stat3. HIF-1 mediates the hypoxia-induced transactivation of the VEGF promoter. However, an elevated level of VEGF production can often be detected in tumor cells located in the extreme periphery of the tumor where there is no apparent hypoxia. Also, many GBM cell lines express high levels of VEGF, even under normoxic conditions (3), suggesting that GBMs can constitutively express VEGF without any apparent external stimuli. Moreover, Stat3 may not be an important transcription factor for VEGF in GBM, as Stat3 plays an opposing role in glial transformation depending on the genetic background of the tumor (4). For example, several studies found that Stat3 promoted the survival of some GBM cell lines in vitro (5, 6), while other studies found that Stat3 activity was absent in most GBMs (7, 8). Given the prominent role of VEGF in glioma angiogenesis, it is critical to understand the molecular basis of constitutive VEGF gene expression.
FoxM1 is a member of the Forkhead box (Fox) transcription factor family, which is a master regulator of the cell cycle (9–12). Previous studies have found that FoxM1 is substantially elevated in several aggressive human carcinomas and can contribute to oncogenesis in many tissue types, including basal cell, breast, hepatocellular, prostate, lung, and colorectal cancers (13–17). Increasing evidence indicates that FoxM1 may also contribute to oncogenesis in gliomas. For example, two studies found that aberrant FoxM1 expression was a common molecular alteration in malignant glioma (18, 19). In our own studies of the functional significance of this factor, we found that FoxM1B was the predominant FoxM1 isoform in human gliomas but not in normal brain tissue (17). We further found that the level of FoxM1 protein expressed in human glioma tissue correlated directly with glioma grade, and the FoxM1 protein level in human GBM tissue correlated inversely with patient survival duration (17). In animal models, increased levels of FoxM1B expression enhanced the tumorigenicity and invasiveness of glioma cells (17, 20). Also, increased FoxM1 expression in anaplastic astrocytoma (AA) cells led to the formation of highly angiogenic GBM (17). In fact, GBMs are defined, in part, by the appearance of proliferating endothelial cells and high blood vessel densities, which histologically distinguish grade IV tumors from lower-grade astrocytomas (21).
These findings underscore the importance of determining the molecular mechanisms by which FoxM1 regulates angiogenesis of glioma. Therefore, we sought to determine the impact of FoxM1 expression on VEGF expression and the angiogenesis of gliomas.
Materials and Methods
Primary human glioma specimens and immunohistochemical (IHC) analysis
We stained tissue sections from paraffin-embedded glioma specimens with an antibody against human FoxM1 (MPP2 K-19; Santa Cruz Biotechnology, Santa Cruz, CA) or an anti-human VEGF antibody (A-20; which slightly cross-reacts with mouse VEGF protein; Santa Cruz Biotechnology). We used tissue sections immunostained with nonspecific immunoglobulin (Ig)G as negative controls. We quantitatively scored the tissue sections according to the percentage of positive cells and staining intensity, as previously defined (22): we assigned a proportion score of 0 if 0% of the tumor cells showed positive staining, 1 if 0% 1% of cells were stained, 2 if 1% 10% stained, 3 if 11% 30% stained, 4 if 31% 70% stained, and 5 if 71% 100% stained. We rated the intensity of staining on a scale of 0 to 3: 0 = negative, 1 = weak, 2 = moderate, and 3 = strong. We then combined the proportion and intensity scores to obtain a total score (range, 0 8) as described previously (22). The use of human glioma specimens and database was approved by the institutional review board at The University of Texas M. D. Anderson Cancer Center.
Immunofluorescent staining of glioma specimens
Tissue sections with 5 m thickness were cut from frozen GBM specimens and processed for immunofluorescence. Sections were immunostained with rabbit anti-FOXM1 and mouse anti-VEGF antibodies (Santa Cruz Biotechnology) and then stained with DAPI, anti-rabbit IgG conjugated with Alexa 488 and anti-mouse IgG conjugated with Texas Red (Molecular Probes). The fluorophore was excited by laser at 405, 488, or 543 nm and detected by a scanning confocal microscope (FV-1000, Olympus).
Cell lines and culture conditions
We purchased the SW1783 (human AA) and U-87MG (GBM) cell lines from American Type Culture Collection (Manassas, VA). We also used the HFU-251MG (GBM) cell line, which we described previously (23). All of the cell lines were maintained in plastic flasks as adherent monolayers in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), sodium pyruvate, nonessential amino acids, L-glutamine, and a vitamin solution (Invitrogen, Carlsbad, CA).
Animals
We purchased female athymic BALB/c nude mice from the Animal Production Area of the National Cancer Institute, Frederick Cancer Research Facility (Frederick, MD). The mice were housed in laminar flow cabinets under specific pathogen-free conditions and used at 8 weeks of age. The animals were maintained according to the Institutional Regulations in Facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care in accordance with current regulations and standards of the U.S. Department of Agriculture, Department of Health and Human Services, and National Institutes of Health.
Northern blot analysis
Cellular mRNA was extracted from glioma cells using the FastTrack mRNA isolation kit (Invitrogen), fractionated on a 1% denaturing formaldehyde agarose gel, electrotransferred to a nylon membrane, and crosslinked with ultraviolet light. We performed Northern hybridization using [32P]dCTP-radiolabeled FoxM1 or VEGF cDNA probes. Equal mRNA loading was monitored by hybridizing the same membrane with a β-actin cDNA probe.
Real-time RT-PCR
We performed real-time reverse transcriptase polymerase chain reaction (RT-PCR) analysis of the FoxM1 and VEGF genes, and we used glyceraldehyde-3-phosphate dehydrogenase as an internal control. Total RNA was isolated with TRIzol reagent (Invitrogen). First-strand cDNA was synthesized from 2 μg of total RNA by using M-MLV Reverse Transcriptase (Invitrogen). We performed real-time PCR using 2 μL of cDNA and the SYBR Green Master Mix (Bio-Rad, Hercules, CA), as recommended by the manufacturer. The forward and reverse primers for FoxM1 and VEGF were as follows: FoxM1 primers (forward: 5′-TGCCCAGCAGTCTCTTACCT-3′; reverse: 5′-CTACCCACCTTCTGGCAGTC-3′), and VEGF165 primers (forward: 5′-CAGATTATGCGGATCAAACCTCA-3′; reverse: 5′-CAAGGCCCACAGGGATTTTC-3′). Each sample was run in triplicate for the target gene and the internal control gene.
Western blot analysis
Whole-cell lysates were prepared from glioma cells as described previously (17). We performed standard Western blot analyses of the whole-cell lysates using an antibody against FoxM1 or anti-VEGF antibody (Santa Cruz Biotechnology) and a second anti-rabbit IgG antibody (Amersham Life Sciences, Arlington Heights, IL). The membranes were then stripped and blotted with an anti-β-actin antibody (Sigma Chemical Co., St. Louis, MO) and used as loading controls. We detected the probe proteins using an enhanced chemiluminescence system (Amersham Life Sciences) according to the manufacturer’s instructions.
Transit or stable transfection of glioma cells
To overexpress FoxM1, we transfected SW1783 and Hs683 cells with 3 mg of pcDNA3.1-FoxM1B or control vector pcDNA3.1 plasmids (17). Stably transfected cell lines were isolated by selection with G418. To inhibit FoxM1 expression, we transfected U-87MG and HFU-251MG cells with a FoxM1-siRNA oligonucleotide (50 nM) with the sequence CUCUUCUCCCUCAGAUAUAdTdT (17) or control siRNA (50 nM). U-87MG and HFU-251MG cells were also transfected with the FoxM1-shRNA expression vector (17) or the control vector for stable transfection. Stably transfected cell lines were isolated by G418 selection. To avoid clonal selection, we pooled all of the G418-resistant colonies to establish stable transfectants.
Promoter reporters and dual-luciferase assay
The full-length VEGF promoter was described previously (24). We generated mutant VEGF promoters by site-specific mutagenesis, as described below. Glioma cells were transfected with the VEGF promoter reporter plasmids. Transfection efficiency was normalized by cotransfection with a phosphorylated β-actin-Renilla luciferase reporter containing a full-length Renilla luciferase gene (20). We quantified both firefly and Renilla luciferase activity using a dual-luciferase assay system (Promega, Madison, WI). We then calculated specific VEGF promoter activity, as described previously (20).
Electrophoretic mobility shift assay
We performed electrophoretic mobility shift assays (EMSA) as described previously (24). We used double-stranded oligonucleotides of putative FoxM1-binding sites in the VEGF promoter as probes. For supershift analyses, the cell extracts were preincubated with specific antibodies against FoxM1 (Santa Cruz Biotechnology).
Chromatin immunoprecipitation assay
We performed chromatin immunoprecipitation (ChIP) assays using the ChIP assay kit from Upstate Biotechnology (Waltham, MA). Briefly, cultured cells were crosslinked with 1% formaldehyde and resuspended in 200 μL of SDS lysis buffer (1% SDS, 10 mM ethylenediaminetetraacetic acid [EDTA], 50 mM Tris-HCl [pH 8.1]) and sonicated on ice to shear the DNA to 200–500 bp. The chromatins were precleared by incubation with protein A-Sepharose beads for 2 h at 4 C. Anti-FoxM1 antibodies were then added, and the samples were incubated overnight at 4 C. We used immunoprecipitation with normal rabbit IgG as a negative control. Immunocomplexes were precipitated for 2 h with protein A-Sepharose beads, and DNA was recovered by means of phenol-chloroform extraction. We then subjected the DNA to PCR to amplify a 215-bp region (-1635 to -1420bp) of the VEGF promoter using the primers 5′-GGAGCGTTTTGGTTAAATTGAG-3′ and 5′-TGCATATAGGAAGCAGCTTGGA-3′ or to amplify a 192-bp region (-634 to -442 bp) of the VEGF promoter using the primers 5′-CCCCTTTCCAAAGCCCATTCC-3′ and 5′-CCTTCTCCCCGCTCCAACACCC-3′. The PCR products were resolved electrophoretically on a 2% agarose gel and visualized by ethidium bromide staining.
Site-specific mutagenesis of the VEGF promoter
We performed site-specific mutagenesis of the VEGF promoter pGL3-V2274 using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer’s instructions. We confirmed the mutations by DNA sequencing.
Endothelial-cell tube formation assay
We performed a tube formation assay as described previously (25). Briefly, 250 μL of growth-factor reduced Matrigel (Collaborative Biomedical Products, Bedford, MA) was pipetted into each well of a 24-well plate and polymerized for 30 min at 37°C. Human umbilical vein endothelial cells (HUVECs) were harvested after trypsin treatment and suspended in conditioned medium from 1 × 106 glioma cells. Next, 2 × 104 HUVECs in 300 μL of conditioned medium were added to each well and incubated at 37°C in 5% CO2 for 20 h. We then photographed the cultures under a bright-field microscope. The quantified results were expressed in number of capillary tubes formed per mm2.
Intracranial human glioma xenograft model
Glioma cells (1 × 106) were injected intracranially into nude mice as described previously (26). We performed two independent experiments with five mice per group in each experiment. Animals showing general or local symptoms were killed; the remaining animals were killed 90 days after glioma-cell injection. Each mouse’s brain was harvested, fixed in 4% formaldehyde, and embedded in paraffin. We then determined the tumor formation and phenotype by histologic analysis of hematoxylin and eosin (H&E)-stained sections.
IHC analysis of xenograft tumors
Brain tissue specimens were fixed by neutral-buffered formalin, embedded in paraffin, and sectioned according to standard protocols. Tissue sections were immunostained with antibodies to FoxM1, VEGF (Santa Cruz Biotechnology), and von Willebrand factor (vWF; factor VIII related antigen; DAKO Carpinteria, CA). For the quantification of the FoxM1 and VEGF expression, we measured immunostaining score of FoxM1 or VEGF in 10 random 0.5 mm2 fields at ×200 magnification of tumor tissues. The score is assigned according to the percentage of positive cells and staining intensity, as defined in the section of “Primary human glioma specimens and immunohistochemical (IHC) analysis”. Two separate individuals who were both blinded to the slides examined and scored each sample. An average value of the two scores was presented in the present study. For quantification of tumor MVD, highly vascular areas were initially identified by scanning tumor sections using light microscopy at low power. Vessels in ten high-power fields (×200 magnification [×20 objective and ×10 ocular]) were counted by two independent investigators without knowledge of the patient outcome (double-blinded) as described previously (25).
Statistical Analysis
We determined the significance of differences in the patient specimen data using the chi-square test, the significance of differences in the in vitro data using the Student’s t-test (two-tailed), and the significance of differences in the in vivo data using the Mann-Whitney U test. A P value of < 0.05 is considered to be significant.
Results
VEGF expression correlates with FoxM1 overexpression in human primary glioma specimens
We first determined the VEGF expression patterns in serial sections of 59 human GBM (grade IV) specimens using IHC analyses. VEGF protein expression was present not only in tumor cells immediately surrounding necrotic areas (Fig. 1A1) but also in the peripheral areas and in the central region of the glioma mass (Fig. 1A1, 1A2, and 1A3). In contrast, VEGF expression was absent from the “normal” brain tissues surrounding the glioma mass (Fig. 1A1 and 1A2). Next, we examined FoxM1 expression in various areas of the glioma specimens. Similar to the pattern we observed for VEGF expression, we found FoxM1 protein expression in tumor cells surrounding necrotic areas, in the peripheral areas, and in the central regions of glioma tissues (Fig. 1A4, 1A5, and 1A6). Colocalization of FoxM1 and VEGF expression was confirmed by double immunoflurorescent assay (Fig. 1B). We then quantified the IHC staining of VEGF and FoxM1 in the glioma specimens on the 0 to 8 scale. We analyzed the scores and found a significant correlation between the FoxM1 and VEGF expression levels (Fig. 1C1; r = 0.79; P < 0.001). We also determined the expression of VEGF and FoxM1 in 25 human AA (anaplastic astrocytoma, grade III) specimens, and observed similar correlation between the expression of FoxM1 and VEGF in AA tumors (Fig. 1C2; r = 0.75; P < 0.001). Furthermore, we performed Western blot analyses using total protein extracts from 3 low-grade astrocytoma (grade II), 3 AA (grade III), and 6 GBM (grade IV) frozen human glioma tissues. As shown in Figure 1D, VEGF expression correlated with FoxM1 expression. Therefore, our results suggest that VEGF expression in human glioma tissues can be detected in tumor cells located in the areas of the tumor where there is no apparent hypoxia as assessed by a lack of hypoxia marker CAIX staining, and VEGF expression is correlated with FoxM1 expression (Supplemental Fig. 1A-D).
Fig. 1.
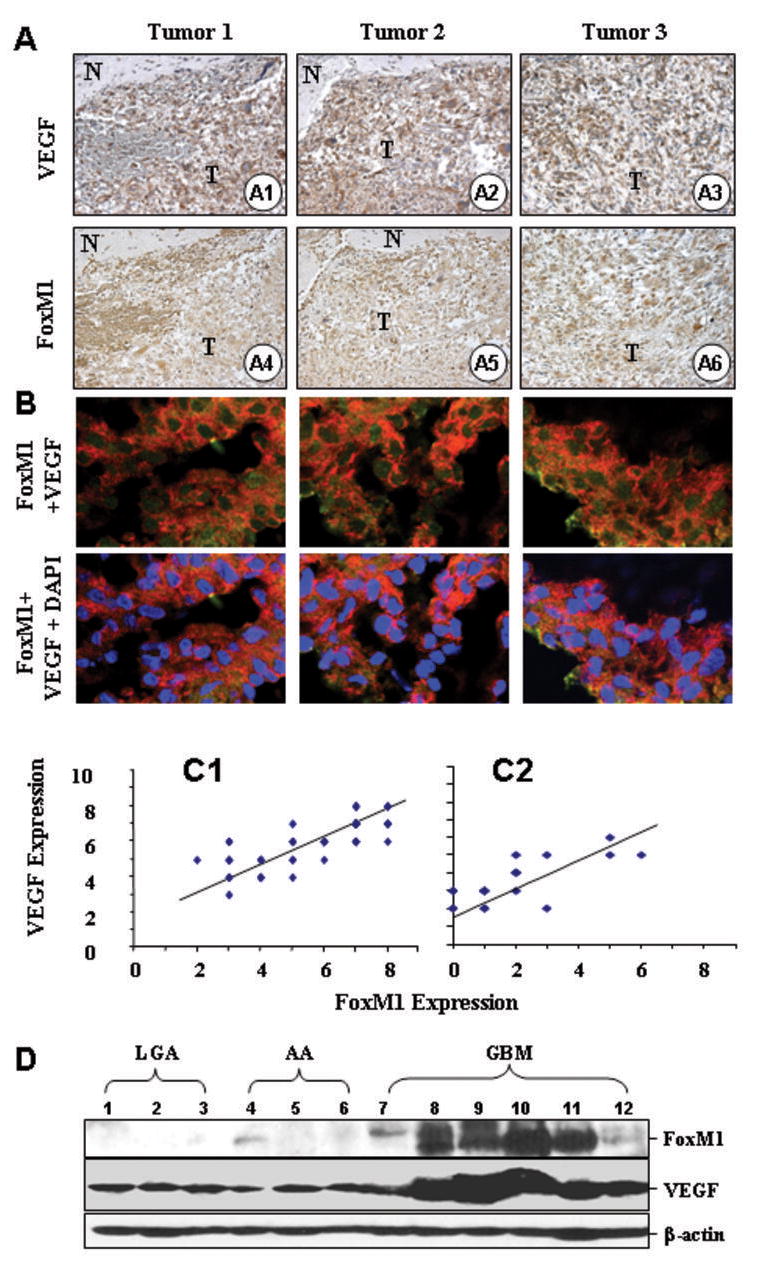
FoxM1 overexpression correlates with upregulation of VEGF expression in human GBM specimens. Immunohistochemical staining using specific anti-FoxM1 and anti-VEGF antibodies were performed on 59 GBM and 25 AA tissues. A, Representative photos of three tumors indicate the pattern of VEGF and FoxM1 expression in GBM sections (A1 ~ A6, “N”, normal, “T”, tumor). B, Colocalization of FoxM1 and VEGF in GBM tissues. Frozen sections of GBMs were processed for triple immunofluorescence staining for FoxM1 (green), VEGF (red) and nuclei (DAPI, blue). These representative photos of three tumors indicate that many cells displayed strong positive nuclei staining, but weak positive cytoplasma staining of FoxM1, whereas VEGF was predominantly in the cytoplasm of the cells. Merged photos indicated that FoxM1 and VEGF colocalized in the cells. C, We quantitatively scored the tissue sections according to the percentage of positive cells and staining intensity as described in “Materials and Methods”. We then combined the percentage and intensity scores to obtain a total score (range, 0 8). FoxM1 expression levels correlated positively with VEGF expression levels in GBM samples (Pearson’s correlation test r = 0.79; P < 0.001), and in AA samples (r = 0.75; P < 0.001). Note that some of the dots on the graphs represented more than one specimen (some scores overlapped). D, FoxM1 and VEGF protein expression levels from 3 LGA, 3 AA, and 6 GBM frozen tissues were determined using Western blot analysis.
Altered FoxM1 expression affects VEGF expression in glioma cells
To determine the effect of increased FoxM1 expression on VEGF expression, we studied two human glioma cell lines, SW1783 and Hs683, that had low levels of the FoxM1 protein (17). We transfected these cells with the FoxM1B expression vector pcDNA3.1-FoxM1B or the control vector pcDNA3.1. To avoid clonal selection and variation, we pooled neomycin (G418)-resistant colonies to establish stable transfectants and designated them FoxM1B-transfected cell lines (SW1783-FoxM1 and Hs683-FoxM1). We found that forced expression of FoxM1B in SW1783 and Hs683 cells significantly increased FoxM1 mRNA and protein expression (Fig. 2A1 and 2A2). These cells also exhibited significantly increased VEGF mRNA and protein expression (Fig. 2A1 and 2A2). Our results indicate that FoxM1 overexpression in glioma cells increased VEGF expression.
Fig. 2.

Effects of altered FoxM1 expression on VEGF expression in human glioma cell lines. A, Upregulation of VEGF mRNA and protein expression by FoxM1B overexpression. FoxM1 and VEGF expression levels in parental, control-vector transfected, and FoxM1 expression-vector transfected SW1783 and Hs683 cells were determined by Northern blot analyses (A1), and Western blot analyses (A2). B, Downregulation of VEGF mRNA and protein expression by knockdown of FoxM1 expression. FoxM1 and VEGF expression in U-87MG and HFU-251MG cells transfected with FoxM1-siRNA or control siRNA were determined by real-time RT-PCR (B1) and Western blot analyses (B2).
Conversely, to determine the effect of decreased FoxM1 expression on VEGF expression, we transfected FoxM1-siRNA (50 nM) into HFU-251MG and U-87MG cells, which typically express high levels of FoxM1 (1). This transfection significantly inhibited FoxM1 mRNA and protein expression in both cell types (Fig. 2B1 and 2B2); the cell transfection also resulted in significantly decreased VEGF mRNA and protein expression in both cell types (Fig. 2B1 and 2B2). These results indicate that blockade of FoxM1 expression suppressed VEGF expression in GBM cells.
FoxM1 regulates VEGF promoter activity in glioma cells
To investigate the role of FoxM1 in regulating VEGF transcription, we studied whether FoxM1 regulates VEGF promoter activity. The VEGF promoter-luciferase construct pGL3-V2274 was cotransfected into SW1783 cells with pcDNA3.1-FoxM1B or the control vector pcDNA 3.1. Cotransfection with pcDNA3.1-FoxM1B activated the luciferase activity driven by the VEGF promoter in the SW1783 cell line (Fig. 3A1). Conversely, to assess the effect of decreased FoxM1 expression on VEGF transcription, we knocked down FoxM1 expression by cotransfecting FoxM1-siRNA and the VEGF promoter into U-87MG cells. FoxM1-siRNA inhibited the luciferase activity driven by the VEGF promoter in the U-87MG cells (Fig. 3A2). Our results suggest that FoxM1 was involved in VEGF promoter activity in glioma cells.
Fig. 3.

The VEGF gene as a transcriptional target of FoxM1. A, Transactivation of the VEGF promoter by FoxM1 and repression of the VEGF promoter by FoxM1-siRNA. A1, Effects of FoxM1 overexpression on VEGF promoter activity. SW1783 and Hs683 cells were cotransfected with 1 μg of the VEGF promoter-luciferase construct pGL3-V2274 and 3 μg of pcDNA 3.1-FoxM1B or pcDNA 3.1. Activation was calculated relative to cells transfected with pcDNA 3.1. A2, Effect of FoxM1-siRNA on VEGF promoter activity. HFU-251MG and U-87MG cells were cotransfected with 1 μg pGL3-V2274 and 50 nM FoxM1-siRNA or control siRNA. Inhibition was calculated as a percentage relative to cells transfected with control siRNA. B, Sequences and positions of putative FoxM1-binding elements on the VEGF promoter. C, Binding of FoxM1 to the VEGF promoter in vitro. We performed EMSA using nuclear protein (NE) extracted from HFU-251MG cells and the oligonucleotide probes of putative FoxM1-binding region 1 (C1) and region 2 (C2) of VEGF promoter. A major shifted band was noted and was competed out by an excess of unlabeled FoxM1-binding region 1 and region 2 oligonucleotides, respectively, and supershifted by an anti-FoxM1 antibody.
Direct interaction of FoxM1 with the VEGF promoter
To determine whether VEGF could be a direct transcriptional target of FoxM1, we analyzed the sequence of the VEGF promoter by using the FoxM1 consensus sequences 5′-AGATTGAGTA-3′ and 5′-TAATCA-3′ (27). We identified two putative FoxM1-binding regions in the VEGF promoter (Fig. 3B). Using EMSA, we found that both binding regions specifically bound to nuclear protein prepared from HFU-251MG cells and resulted in the formation of one major shifted band (Fig. 3C). Moreover, the shifted band could be supershifted by the FoxM1 antibody and competed out by an excess of the unlabeled FoxM1-binding region 1 or 2 probe (Fig. 3C and Supplemental Fig. 1E), indicating that both binding regions were specifically bound to the FoxM1 protein.
Next, to provide direct proof that FoxM1 is recruited to the endogenous VEGF promoter during transcription in vivo, we performed standard ChIP assays with SW1783, SW1783-Neo, SW1783-FoxM1, and U-87MG cells. We found that both of the FoxM1-binding regions of the VEGF promoter bound to endogenous FoxM1 protein in all cell lines (Fig. 4A1 and A2). Moreover, in the SW1783-FoxM1 and U-87MG cells that expressed high FoxM1 levels, five to six times more DNA promoter was bound to FoxM1 than in the SW1783 cells that expressed low FoxM1 levels (Fig. 4A1 and A2). Collectively, our findings demonstrate that FoxM1 bound specifically to FoxM1-binding sites in the VEGF promoter in vitro and in vivo.
Fig. 4.
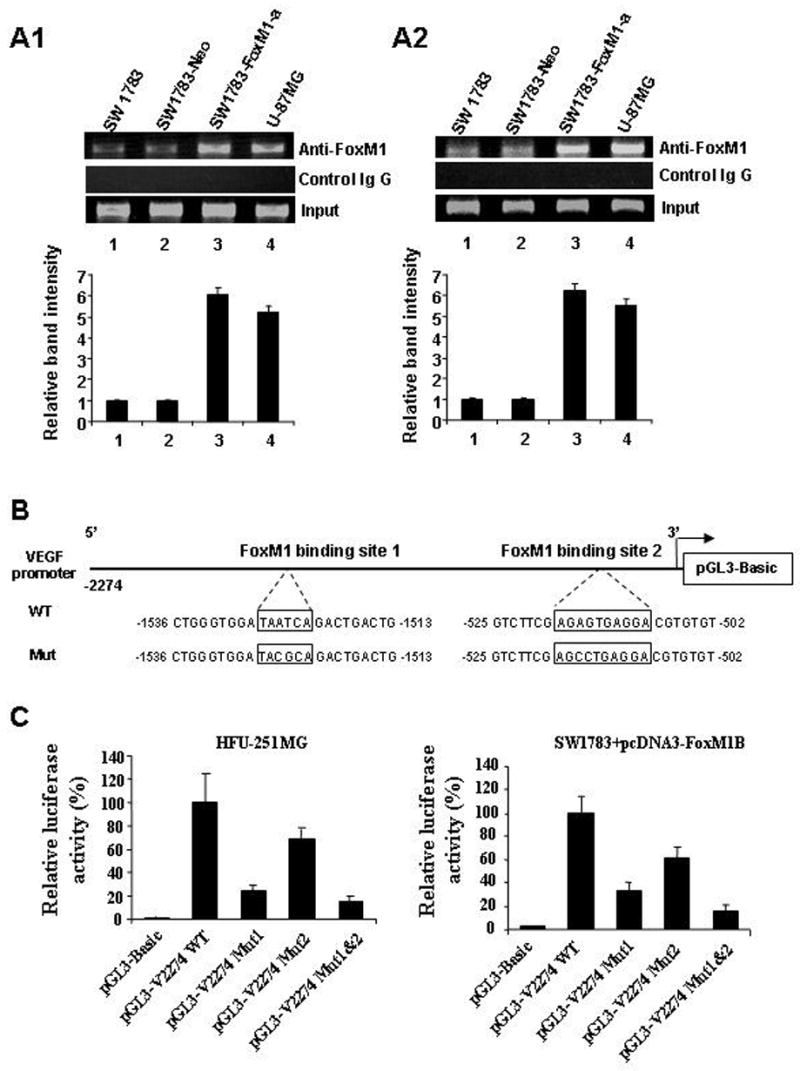
Binding of FoxM1 to the VEGF promoter in vivo and mutational analysis of the FoxM1-binding sites. A, ChIP assays were performed with SW1783, SW1783-Neo, SW1783-FoxM1-a, and U-87MG cells. Chromatin fragments of the cells were immunoprecipitated with an anti-FoxM1 antibody (top panel) or control IgG (middle panel) and subjected to PCR. We subjected 1% of the total cell lysates to PCR before immunoprecipitation as inputs (bottom panel). The relative band intensity was calculated as the ratio between precipitated and input DNA from each cell line. Data shown are the mean values obtained from three experiments. B, Schematic structure of the VEGF promoter. The sequences of the FoxM1-binding sites are shown in both wild-type (WT) and mutant forms. C, VEGF promoter activity with and without mutations in the FoxM1-binding sites. HFU-251MG cells were transfected with the wild-type VEGF promoter or its mutants. Luciferase activity was then determined. Also, SW1783 cells were cotransfected with the wild-type VEGF promoter or its mutants and pcDNA 3.1-FoxM1 (3 g), and luciferase activity was again determined.
FoxM1-binding sites are critical for activation of the VEGF promoter in glioma cells
To assess the functional role of the FoxM1-binding sites in VEGF gene regulation, we performed site-specific mutagenesis within the FoxM1-binding sites of the VEGF promoter pGL3-V2274. As shown in Figure 4B, various mutant reporters were generated from the wild-type VEGF promoter construct, including a FoxM1 binding site 1 mutation only (pGL3-V2274 Mut1), a FoxM1 binding site 2 mutation only (pGL3-V2274 Mut2), and both site 1 and site 2 mutations (pGL3-V2274 Mut1 and -2). We transfected these mutant luciferase reporters into the HFU-251MG cells and compared their activity with that of wild-type VEGF promoter pGL3-V2274 WT (Fig. 4C). Disruption of one or both of the FoxM1 binding sites significantly attenuated VEGF promoter activity in the HFU-251MG cells. Also, disruption of one or both of the FoxM1 binding sites significantly inhibited VEGF promoter transactivation by pcDNA3-FoxM1 in SW1783 cells (Fig. 4C). Our results suggest that the FoxM1-binding sites were critical for VEGF promoter activation in glioma cells.
Inhibition of FoxM1 expression suppresses the growth and angiogenesis of GBM cells
Next, we examined the functional consequence of a FoxM1/VEGF interaction by assessing their roles in glioma growth. We established two stable FoxM1-shRNA transfected U-87MG and HFU-215MG cell lines (U-87MG-FoxM1-shRNA-a and -b and HFU-215MG-FoxM1-shRNA-a and -b) by performing duplicate transfection experiments with each cell line. Western blot analyses showed decreased levels of FoxM1 protein expression in the FoxM1-shRNA-transfected U-87MG and HFU-215MG cell lines (Fig. 5A). We then intracranially injected these cells into nude mice and found that U-87MG, HFU-215MG, and control-shRNA cells produced brain tumors in all of the injected mice (Table 1). The mice became moribund approximately 31 days after the injection. In contrast, except for a small tumor in one of the mice injected with U-87MG-FoxM1-shRNA-b cells, the FoxM1-shRNA-transfected U-87MG and HFU-215MG cells produced no brain tumors, which resulted in a significant increase in overall survival time (P < 0.001). Our results demonstrate that inhibition of FoxM1 expression by FoxM1-shRNA significantly suppresses the tumorigenicity of human GBM cells.
Fig. 5.
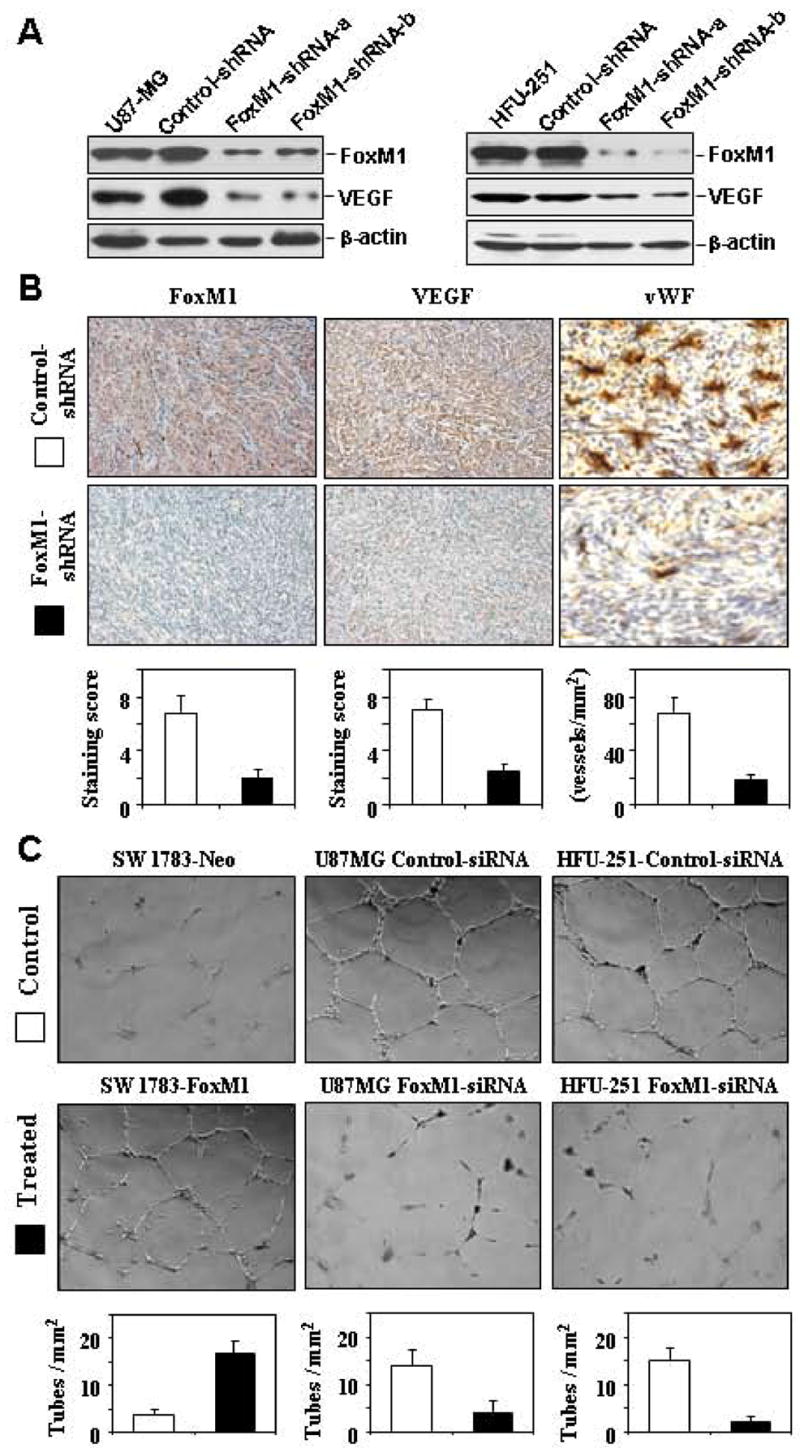
Effects of altered FoxM1 expression on VEGF expression and angiogenic potential of glioma cells. A, Western blot analyses of FoxM1 knockdown and VEGF expression in stable FoxM1-shRNA-transfected U-87MG and HFU-251MG cells (FoxM1-shRNA-a, FoxM1-shRNA-b) and controls (control-shRNA). B, Brain tumors produced by control-shRNA or FoxM1-shRNA-transfected U-87MG cells were processed and sectioned for immunostaining with specific antibodies against FoxM1 and VEGF. The tumor microvessel densities in the samples were determined by staining with an anti-factor VIII antibody. The staining patterns and quantifications shown were representative of those observed in 10 random fields. Note that knocking down FoxM1 expression inhibited VEGF protein expression and decreased microvessel densities in brain tumors. C, The angiogenic potential of glioma cells was determined by an endothelial cell tube formation assay. Samples of conditioned media were prepared from SW1783-Neo, SW1783-FoxM1, and control-shRNA or FoxM1 shRNA-transfected U-87MG or HFU-215MG cells. Human umbilical cord endothelial cells (2 × 104) in 300 μL of conditioned media were then plated on growth factor reduced Matrigel to form a capillary tube. Capillary tube formation in each group was photographed and quantified. The results shown are representative of two experiments.
Table 1.
Effect of FoxM1 knockdown on glioma growth in the brain of nude mice a
| Cell Line | Incidence of tumor formation b | Survival Median Days c (range) |
|---|---|---|
| HFU-251MG | 5/5 | 37 (34 – 53) |
| HFU-251MG-Control-shRNA | 5/5 | 34 (All 34) |
| HFU-251MG-FoxM1-shRNA-a | 0/5 | 90 (All >90) * |
| HFU-251MG-FoxM1-shRNA-b | 0/5 | 90 (All >90) * |
|
| ||
| U-87MG | 5/5 | 31 (25–36) |
| U-87MG-Control-shRNA | 5/5 | 29 (23–34) |
| U-87MG-FoxM1-shRNA-a | 0/5 | 90 (All >90)* |
| U-87MG-FoxM1-shRNA-b | 1/5 | 84 (61 – >90)* |
Glioma cells (1 × 106 cells) were implanted intracranially into nude mice. Mice were killed when they were moribund or on day 90. Brains were harvested, and tumor formation was determined by histology.
Incidence: number of mice with tumor/number of mice injected.
The survival time for brain tumor-free animal was recorded as 90 days.
p < 0.001.
Results were shown for one representative experiment of two with similar findings.
Furthermore, we found that VEGF expression in FoxM1-shRNA-transfected U-87MG and HFU-215MG cells was decreased as compared to control-shRNA cells (Fig. 5A). Thus, we next examined whether inhibition of FoxM1 expression in these tumor cells would affect brain tumors angiogenesis. We assessed glioma vascularization by staining mouse glioma tissue sections with an anti-vWF antibody and found that gliomas produced by U-87MG control-shRNA cells were highly vascularized, whereas gliomas produced by U-87MG-FoxM1-shRNA-b cells had considerably lower microvessel density (Fig. 5B). We evaluated VEGF expression in the brain tumors by means of IHC staining. Consistently, VEGF staining was prominent in the brain tumors formed by U-87MG control-shRNA cells, but not in the brain tumors formed by U-87MG-FoxM1-shRNA-b cells (Fig. 5B).
To provide direct evidence that FoxM1 regulates the angiogenic phenotype of glioma cells, we determined the angiogenic ability of the FoxM1B-transfected and FoxM1-siRNA-transfected glioma cells using an endothelial cell tube formation assay. We found that the conditioned media from SW1783-FoxM1 cultures had greater angiogenic potential than the conditioned media from SW1783-Neo cultures (Fig. 5C). In contrast, the conditioned media from U-87MG or HFU-251MG cells transfected with FoxM1-siRNA had a significantly reduced capillary tube formation compared with the conditioned media from cells transfected with control-siRNA (Fig. 5C). Collectively, our results indicate that FoxM1 regulates the angiogenic ability of glioma cells at least in part through the regulation of VEGF expression.
Discussion
Our study sought to determine the molecular mechanism by which FoxM1 promotes glioma angiogenesis through the upregulation of VEGF expression. We found that human glioma cells constitutively expressed VEGF and that the levels of constitutive FoxM1 expression strongly correlated with the levels of constitutive VEGF expression. Moreover, we found that FoxM1 transactivates VEGF through direct binding to the VEGF gene promoter. FoxM1 overexpression in glioma cells by transfection of a FoxM1B expression vector significantly upregulated VEGF expression and the angiogenic ability of the glioma cells, whereas inhibition of FoxM1 by transfection of FoxM1-siRNA did the opposite. Therefore, FoxM1B overexpression contributes directly to VEGF overexpression in malignant gliomas and appears to be critical for glioma angiogenesis.
Several studies have indicated the importance of FoxM1 in the oncogenesis of several malignancies, including gliomas (13–17). We previously found that FoxM1 protein expression levels in human GBM tissues were inversely correlated with patient survival and that aberrant FoxM1 expression critically regulated the tumorigenicity of human glioma cells (17). Our current study furthered our understanding of the mechanism of action of FoxM1 on glioma oncogenesis in several ways. First, our IHC analyses showed a significant correlation between FoxM1 overexpression and elevated VEGF expression in 59 GBM specimens and provided evidence that FoxM1 regulates VEGF expression and glioma angiogenesis and growth. Our findings suggest that FoxM1 could be a critical pathway in glioma angiogenesis, which is supported by a recent report showing that FoxM1 siRNA reduced VEGF activity in pancreatic cancer cells (28). Second, to our knowledge, this is the first report to demonstrate that VEGF is a direct target of FoxM1. Specifically, we identified two FoxM1-binding sites, mapped between -569 and -560 bp and between -1528 and -1523 bp of the proximal VEGF promoter. FoxM1 appeared to crucially regulate VEGF expression through direct interaction with the VEGF promoter, as mutation of the FoxM1-binding sites significantly downregulated VEGF promoter activity in GBM cells. Our findings using both FoxM1 overexpression and siRNA-knockdown approaches further substantiated this finding. Finally, the levels of FoxM1 expression directly impacted the angiogenic and tumorigenic ability of glioma cells in vitro and in orthotopic animal models. Thus, our study provides both clinical and mechanistic evidence that FoxM1 critically regulates VEGF expression and glioma angiogenesis and describes a novel molecular mechanism by which FoxM1 promotes glioma angiogenesis.
Many studies have also suggested that VEGF expression is regulated by a plethora of external factors in many types of normal and malignant cells (29). Major stimulators of VEGF expression include hypoxia, which occurs frequently within diverse types of expanding tumors, particularly in regions surrounding necrotic areas (30). The role of hypoxia in the angiogenesis switch has been clearly demonstrated in a mouse model (31). Hypoxia-mediated VEGF induction involves transactivation of a VEGF promoter by HIF-1 (32, 33), as well as stabilization of VEGF mRNA expression by proteins that bind to sequences located in the 3’-untranslated region of VEGF mRNA (34–36). Because human GBM is generally hypoxic, the VEGF overexpression we observed in human GBM tissues was due to hypoxia, at least in part (37, 38). Conversely, in human GBMs, elevated VEGF production can often be detected in tumor cells located in the extreme periphery of the tumor where there is no apparent hypoxia. This observation is consistent with the results of previous studies (39–41) that found that exogenous factors, such as hormones, cytokines, and growth factors, modulate VEGF expression through different mechanisms than those operating in hypoxia-induced activation of the VEGF gene. For example, Maity et al (39) found that activation of epidermal growth factor receptor (EGFR) on GBM cells led to VEGF upregulation by a PI3K/Akt-dependent pathway, which was distinct from the signals induced by hypoxia. In another study, fibroblast-derived growth factor and tumor necrosis factor- enhanced VEGF transcription in human glioma cells through the transcription factor Sp1 (40). Also, interleukin-6 induced transcriptional activation of VEGF in GBM cells by means of a direct interaction between Stat3 and Sp1 (41). Nevertheless, previous studies on the regulation of VEGF expression have revealed the diverse complexity of VEGF transcriptional regulation (29, 42). Thus, our current study adds a new mechanism to the body of research on the transcriptional regulation of VEGF expression by providing evidence that VEGF is a direct transcriptional target of FoxM1 and that FoxM1 critically controls constitutive VEGF expression in GBM cells. Whether and how these pathways interact with and impact on glioma angiogenesis warrants further investigations.
Recently, Kim and coworkers reveal elegantly a lack of VEGF expression change in FoxM1-null embryo (43). This interesting finding indicates that in development process VEGF might be regulated by several redundant signaling pathways, whereas in tumor development and progression, tumor cell might predominantly depend on different signaling pathways to regulate VEGF expression. In our current report, we have provided both clinical and experimental evidence that FoxM1 plays an important regulatory role in VEGF expression in and angiogenesis of human glioma.
The mechanisms responsible for FoxM1 overexpression in malignant gliomas are still unknown; however, it is known that FoxM1 expression can be regulated by diverse stimuli, including liver regeneration, keratinocyte growth factor, and oxidative stress (6). The formation and malignant progression of gliomas are widely regarded as multistep processes resulting from complex interplay between multiple genetic and epigenetic events (44–49). Most human gliomas appear to have a set of pathways that are disrupted (e.g., pRB, p53, and PTEN) (44–49), as well as a set of pathways that are abnormally active (e.g., telomerase, EGFR, Akt) (44–49) in both processes. We are currently investigating whether these abnormal pathways lead to aberrant FoxM1B expression in malignant gliomas.
In summary, we found that FoxM1 directly regulated VEGF expression and GBM angiogenesis and growth. Because of the diverse roles of FoxM1 in glioma progression, including regulation of tumor cell proliferation, tumor invasion, and angiogenesis, a better understanding of FoxM1 signaling and function may help identify novel and effective targets for malignant glioma treatment.
Supplementary Material
Acknowledgments
We thank Alyson Todd for her editorial comments.
This work was supported in part by the National Cancer Institute grants R01-CA-116528 and a research grant from the Brain Tumor Society (to S.H.), and National Cancer Institute grant CA-16672.
References
- 1.Kleihues P, Burger PC, Scheithauer BW. The new WHO classification of brain tumours. Brain Pathol. 1993;3:255–68. doi: 10.1111/j.1750-3639.1993.tb00752.x. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 3.Feldkamp MM, Lau N, Rak J, Kerbel RS, Guha A. Normoxic and hypoxic regulation of vascular endothelial growth factor (VEGF) by astrocytoma cells is mediated by Ras. Int J Cancer. 1999;81:118–24. doi: 10.1002/(sici)1097-0215(19990331)81:1<118::aid-ijc20>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 4.de la Iglesia N, Konopka G, Puram SV, et al. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008;22:449–62. doi: 10.1101/gad.1606508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rahaman SO, Harbor PC, Chernova O, et al. Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene. 2002;21:8404–13. doi: 10.1038/sj.onc.1206047. [DOI] [PubMed] [Google Scholar]
- 6.Konnikova L, Kotecki M, Kruger MM, Cochran BH. Knockdown of STAT3 expression by RNAi induces apoptosis in astrocytoma cells. BMC Cancer. 2003;3:23–32. doi: 10.1186/1471-2407-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schaefer LK, Ren Z, Fuller GN, Schaefer TS. Constitutive activation of Stat3alpha in brain tumors: localization to tumor endothelial cells and activation by the endothelial tyrosine kinase receptor (VEGFR-2) Oncogene. 2002;21:2058–65. doi: 10.1038/sj.onc.1205263. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941–51. doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- 9.Ye H, Kelly TF, Samadani U, et al. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol Cell Biol. 1997;17:1626–41. doi: 10.1128/mcb.17.3.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korver W, Roose J, Clevers H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997;25:1715–9. doi: 10.1093/nar/25.9.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laoukili J, Kooistra MR, Bras A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–36. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 12.Wonsey DR, Follettie MT. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005;65:5181–9. doi: 10.1158/0008-5472.CAN-04-4059. [DOI] [PubMed] [Google Scholar]
- 13.Kalinichenko VV, Major ML, Wang X, et al. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004;18:830–50. doi: 10.1101/gad.1200704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalin TV, Wang IC, Ackerson TJ, et al. Increased levels of the FoxM1 transcription factor accelerate development and progression of prostate carcinomas in both TRAMP and LADY transgenic mice. Cancer Res. 2006;66:1712–20. doi: 10.1158/0008-5472.CAN-05-3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim IM, Ackerson T, Ramakrishna S, et al. The Forkhead Box m1 transcription factor stimulates the proliferation of tumor cells during development of lung cancer. Cancer Res. 2006;66:2153–61. doi: 10.1158/0008-5472.CAN-05-3003. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida Y, Wang IC, Yoder HM, Davidson NO, Costa RH. The forkhead box M1 transcription factor contributes to the development and growth of mouse colorectal cancer. Gastroenterology. 2007;132:1420–31. doi: 10.1053/j.gastro.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 17.Liu M, Dai B, Kang SH, et al. FoxM1B is overexpressed in human glioblastomas and critically regulates the tumorigenicity of glioma cells. Cancer Res. 2006;66:3593–602. doi: 10.1158/0008-5472.CAN-05-2912. [DOI] [PubMed] [Google Scholar]
- 18.van den Boom J, Wolter M, Kuick R, et al. Characterization of gene expression profiles associated with glioma progression using oligonucleotide-based microarray analysis and real-time reverse transcription-polymerase chain reaction. Am J Pathol. 2003;163:1033–43. doi: 10.1016/S0002-9440(10)63463-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rickman DS, Bobek MP, Misek DE, et al. Distinctive molecular profiles of high-grade and low-grade gliomas based on oligonucleotide microarray analysis. Cancer Res. 2001;61:6885–91. [PubMed] [Google Scholar]
- 20.Dai B, Kang S-H, Gong W, Liu M, Aldape K, Sawaya R, Huang S. Aberrant FoxM1B expression increases matrix metalloproteinase-2 transcription and enhances the invasion of glioma cells. Oncogene. 2007;26:6212–9. doi: 10.1038/sj.onc.1210443. [DOI] [PubMed] [Google Scholar]
- 21.Kleihues P, Louis DN, Scheithauer BW, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–25. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- 22.Allred DC, Harvey JM, Berardo M. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11:155–68. [PubMed] [Google Scholar]
- 23.Ke LD, Shi YX, Im SA, Chen X, Yung WK. The relevance of cell proliferation, vascular endothelial growth factor, and basic fibroblast growth factor production to angiogenesis and tumorigenicity in human glioma cell lines. Clin Cancer Res. 2000;6:2562–72. [PubMed] [Google Scholar]
- 24.Shi Q, Le X, Abbruzzese JL, Peng Z, Qian CN, Tang H, Xiong Q, Wang B, Li XC, Xie K. Constitutive Sp1 activity is essential for differential constitutive expression of vascular endothelial growth factor in human pancreatic adenocarcinoma. Cancer Res. 2001;61:4143–54. [PubMed] [Google Scholar]
- 25.Huang S, Mills L, Mian B, Tellez C, McCarty M, Yang XD, Gudas JM, Bar-Eli M. Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am J Pathol. 2002;16:125–34. doi: 10.1016/S0002-9440(10)64164-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lal S, Lacroix M, Tofilon P, Fuller GN, Sawaya R, Lang FF. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg. 2000;92:326–33. doi: 10.3171/jns.2000.92.2.0326. [DOI] [PubMed] [Google Scholar]
- 27.Yao KM, Sha M, Lu Z, Wong GG. Molecular analysis of a novel winged helix protein, WIN. Expression pattern, DNA binding property, and alternative splicing within the DNA binding domain. J Biol Chem. 1997;272:19827–36. doi: 10.1074/jbc.272.32.19827. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH. Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells. Cancer Res. 2007;67:8293–300. doi: 10.1158/0008-5472.CAN-07-1265. [DOI] [PubMed] [Google Scholar]
- 29.Xie K, Wei D, Shi Q, Huang S. Constitutive and inducible expression and regulation of vascular endothelial growth factor. Cytokine Growth Factor Rev. 2004;15:297–324. doi: 10.1016/j.cytogfr.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Shweiki D, Itin A, Soffer D, Keshet E. Nature (London) 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 31.Bergers G, Javaherian K, Lo KM, Folkman J, Hanahan D. Science. 1999;284:808–12. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- 32.Levy AP, Levy NS, Wegner S, Goldberg MA. J Biol Chem. 1995;270:13333–40. doi: 10.1074/jbc.270.22.13333. [DOI] [PubMed] [Google Scholar]
- 33.Liu YX, Cox SR, Morita T, Kourembanas S. Circ Res. 1995;77:638–43. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- 34.Stein I, Neeman M, Shweiki D, Itin A, Keshet E. Mol Cell Biol. 1995;15:5363–8. doi: 10.1128/mcb.15.10.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Damert A, Machein M, Breier G, Fujita MQ, Hanahan D, Risau W, Plate KH. Cancer Res. 1997;57:3860–4. [PubMed] [Google Scholar]
- 36.Claffey KP, Shih SC, Mullen A, Dziennis S, Cusick JL, Abrams KR, Lee SW, Detmar M. Mol Biol Cell. 1998;9:469–81. doi: 10.1091/mbc.9.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rong Y, Durden DL, Van Meir EG, Brat DJ. ‘Pseudopalisading’ necrosis in glioblastoma: a familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J Neuropathol Exp Neurol. 2006;65:529–39. doi: 10.1097/00005072-200606000-00001. Review. [DOI] [PubMed] [Google Scholar]
- 38.Kaur B, Khwaja FW, Severson EA, Matheny SL, Brat DJ, Van Meir EG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. 2005;7:134–53. doi: 10.1215/S1152851704001115. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maity A, Pore N, Lee J, Solomon D, O’Rourke DM. Epidermal growth factor receptor transcriptionally upregulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3-kinase and distinct from that induced by hypoxia. Cancer Res. 2000;60:5879–86. [PubMed] [Google Scholar]
- 40.Ryuto M, Ono M, Izumi H, Yoshida S, Weich HA, Kohno K, Kuwano M. Induction of vascular endothelial growth factor by tumor necrosis factor alpha in human glioma cells. Possible roles of SP-1. J Biol Chem. 1996;271:28220–8. doi: 10.1074/jbc.271.45.28220. [DOI] [PubMed] [Google Scholar]
- 41.Loeffler S, Fayard B, Weis J, Weissenberger J. Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int J Cancer. 2005;115:202–13. doi: 10.1002/ijc.20871. [DOI] [PubMed] [Google Scholar]
- 42.Mukhopadhyay D, Datta K. Multiple regulatory pathways of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in tumors. Semin Cancer Biol. 2004;14:123–30. doi: 10.1016/j.semcancer.2003.09.019. Review. [DOI] [PubMed] [Google Scholar]
- 43.Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. The forkhead box m1 transcription factor is essential for embryonic development of pulmonary vasculature. J Biol Chem. 2005;280:22278–86. doi: 10.1074/jbc.M500936200. [DOI] [PubMed] [Google Scholar]
- 44.Masher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, DePinho RA. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 2001;15:1311–33. doi: 10.1101/gad.891601. [DOI] [PubMed] [Google Scholar]
- 45.Furnari FB, Huang HJ, Cavenee WK. Molecular biology of malignant degeneration of astrocytoma. Pediatr Neurosurg. 1996;24:41–9. doi: 10.1159/000121013. [DOI] [PubMed] [Google Scholar]
- 46.Rasheed BK, Wiltshire RN, Bigner SH, Bigner DD. Molecular pathogenesis of malignant gliomas. Curr Opin Oncol. 1999;11:162–7. doi: 10.1097/00001622-199905000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Louis DN. The p53 gene and protein in human brain tumors. J Neuropathol Exp Neurol. 1994;53:11–21. doi: 10.1097/00005072-199401000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Rich JN, Hans C, Jones B, et al. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Res. 2005;65:4051–8. doi: 10.1158/0008-5472.CAN-04-3936. [DOI] [PubMed] [Google Scholar]
- 49.Uhrbom L, Kastemar M, Johansson FK, Westermark B, Holland EC. Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 2005;65:2065–9. doi: 10.1158/0008-5472.CAN-04-3588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.