

Abstract
The ErbB2 receptor tyrosine kinase is overexpressed in ~ 25% of breast tumors and contributes to poor patient prognosis and therapeutic resistance. Here we examine the role of the recently discovered ErbB negative regulator LRIG1 in ErbB2 (+) breast cancer. We observe that LRIG1 protein levels are significantly suppressed in ErbB2-induced mammary tumors in transgenic mice as well as the majority of ErbB2 (+) human breast tumors. These observations raise the possibility that LRIG1 loss could contribute to the initiation or growth of ErbB2 (+) breast tumors. RNAi-mediated knockdown of endogenous LRIG1 in the ErbB2 overexpressing breast tumor cell lines MDA-MB-453 and BT474 further elevates ErbB2 in these cells and augments cellular proliferation. In contrast, ectopic expression of LRIG1 reverses these trends. Interestingly, we observe that LRIG1 protein levels are suppressed in response to ErbB receptor activation in breast tumor cells, but are unaffected by ErbB activation in immortalized non-transformed breast epithelial cells. Our observations indicate that the suppression of LRIG1 protein levels is a common feature of breast tumors. Moreover, our observations point to the existence of a feed-forward regulatory loop in breast tumor cells where aberrant ErbB2 signaling suppresses LRIG1 protein levels, which in turn contributes to ErbB2 overexpression.
Keywords: ErbB2, LRIG1, negative regulator, breast cancer
Introduction
Receptor tyrosine kinases play essential roles in tissue development and homeostasis and their functional inactivation has dramatic consequences on the viability of the organism. In healthy tissue, receptor activation or “positive signaling” is tightly controlled by ligand availability and is counterbalanced by a network of negative regulatory molecules that serve to limit signal output. Disruption of the precise balance between “positive” and “negative” signals is implicated in the development of several diseases, including cancer (1). Ligand-induced down-regulation is the most widely studied attenuation mechanism for receptor tyrosine kinases and uncoupling receptors from this pathway leads to aberrant signaling and cellular transformation (2).
The ErbB family of receptor tyrosine kinases, which includes EGF receptor, ErbB2/Her2/Neu, ErbB3, and ErbB4, forms a complex signaling network driven by ligand stimulated receptor heterodimerization. ErbB2 is overexpressed in 20 – 30% of human breast cancers and correlates with poor patient prognosis and therapeutic resistance (3). The ErbB2/ErbB3 heterodimer functions as an “oncogenic unit” and both receptors are essential for breast tumor cell proliferation (4). In support of this, ErbB2 and ErbB3 overexpression frequently overlap in human breast tumors (5). Moreover, tumors from transgenic mice that express activated ErbB2 in the mammary gland also overexpress ErbB3 (6).
ErbB receptors are subject to strenuous regulation and loss or suppression of negative regulatory proteins unleashes potent growth and survival signals. For example, under-expression of the pan-ErbB inhibitor RALT/Mig-6 in ErbB2 amplified breast cancer cells heightens tumor cell proliferation and favors Herceptin resistance (7). Similarly, loss of the ErbB3 negative regulator Nrdp1 in breast cancer enhances ErbB2/ErbB3 driven tumor cell proliferation and motility (8). Aberrant activation of Src promotes degradation of the ubiquitin ligase c-Cbl, enhancing EGF receptor stability and providing a mechanism by which Src and EGFR cooperate to drive tumorigenesis (9). These studies suggest that full oncogenic signaling by receptor tyrosine kinases requires the suppression of negative regulators and that loss of negative regulatory proteins may cooperate with receptor gene amplification to yield a permissive environment for receptor overexpression. Strategies that augment or restore expression of endogenous negative regulatory molecules may prove effective in the clinic.
LRIG1 is a transmembrane leucine-rich repeat protein and a recently identified negative regulator of all members of the ErbB family as well as the Met receptor tyrosine kinase (10–12). LRIG1 functions by enhancing the proteolytic degradation of its targets and was identified on the basis of its homology with Kekkon-1, a negative regulator of Drosophila EGF receptor (DER). LRIG1 deficient animals develop a psoriasiform epidermal hyperplasia, consistent with deregulated EGF receptor signaling and a growth suppressive role for LRIG1 in the skin (13). Analysis of LRIG1 expression in vivo reveals that it is decreased in several different tumor types including squamous cell and renal cell carcinoma and advanced cervical cancer (14–16). As such, LRIG1 has been proposed to be a tumor suppressor (17). However, LRIG1 expression in certain tumor types has been reported to be heterogeneous; for example, colorectal cancers both under- and overexpress LRIG1 (18). A recent study involving a small number of breast tumors reported that LRIG1 expression was increased in 5/9 tumors, suggesting that LRIG1 may not function as a growth suppressor in the breast (19). As this finding is inconsistent with its role in suppressing ErbB2 function, we sought to clarify the role of LRIG1 in breast cancer.
In this study, we find that LRIG1 is suppressed in human breast tumors and that the majority of ErbB2 overexpressing breast tumors under-express LRIG1. This finding is paralleled in a well characterized transgenic mouse model of ErbB2 (+) breast cancer. RNAi-mediated depletion studies demonstrate that LRIG1 acts to suppress breast tumor cell growth. Interestingly, activation of ErbB2 in breast cancer cells leads to a significant loss of endogenous LRIG1 expression, demonstrating that ErbB2 oncogenic signaling actively contributes to suppression of LRIG1. These data suggest that ErbB2 contributes to its own overexpression by suppressing negative regulators that oppose its action.
Materials and Methods
Reagents and Cell Culture
Human breast cell lines BT474, MCF-7, MDA-MB-453, MDA-MB-361, SKBR-3, and T47D cells were purchased from the ATCC (Manassas, VA) and cultured in the recommended media (Mediatech, Washington, DC) with 10% fetal calf serum (FCS, Invitrogen, Carlsbad, CA) and penicillin-streptomycin antibiotics (Mediatech, Washington, DC). Serum starve media contained only 0.1% FCS. HMEC4 and HMEC6 cells were kind gifts from Dr. Krishna Rao and were grown in DFCI-1 medium as described (20). Neuregulin-1β (Nrg1) was produced and purified as previously described (21). The EGFR/ErbB2 inhibitor 4557W was purchased from EMD Bioscience (San Diego, CA). Antibodies used in these studies include anti-LRIG1–151 (AgriSera, Vännäs, Sweden), anti-ErbB2 Ab3 (EMD Bioscience, San Diego, CA), anti-ErbB3 C-17 (Santa Cruz Biotechnology, Santa Cruz, CA), anti-p-ErbB2 6B12, anti-p-Akt S473, anti-total-Akt, anti-p-Erk1/2, and anti-total-Erk1/2 (Cell Signaling Technologies, Danvers, MA), anti-myc (Invitrogen, Carlsbad, CA), anti-phosphotyrosine (BD Transduction, San Jose, CA), and anti-tubulin and anti-actin AC-15 (Sigma, St. Louis, MO). FuGene6 was purchased from Roche (Palo Alto, CA) and transfections were performed as recommended by the manufacturer.
Human Breast Tissue Analysis
Frozen human tissues from clinical samples were provided by the National Cancer Institute Cooperative Human Tissue Network and the University of California, Davis Cancer Center Specimen Repository. All of the samples were approved for laboratory use by the institutional review board of the University of California, Davis, School of Medicine. Samples were homogenized in 10 ul T-PER (Pierce, Rockford, IL) per mg of tissue in the presence of 4 ug/ml leupeptin, 4 ug/ml pepstatin, 4 ug/ml aprotinin, and 100 nM AEBSF, then centrifuged to remove insoluble products.
Transgenic Mice
NDL mice expressing the constitutively active ErbB2/Neu transgene under the control of the mouse mammary tumor virus promoter (NDL2-5) (6) were bred and maintained at the animal facilities at the University of California, Davis. Following tumor development, mammary fat pad tissue of the tumor and adjacent normal were collected and snap-frozen in liquid nitrogen. Tissue lysates were prepared as described for the human tissues. To produce a pure epithelial NDL2-5 tumor line, one mammary tumor roughly 1 cm in diameter was excised from a 7 month old FvB NDL2-5 mouse. One 2 mm cross section was fixed in formalin, paraffin embedded, and stained with hematoxylin and eosin for histologic validation. The remainder of the tumor was rinsed 2x with PBS, minced with razor blades to form a slurry of cells in 25 ml of lysis buffer [20ml DMEM:F12, 5 ml BSA (7.5% w/v in water), 10ul hydrocortisone, 200ul 1 M HEPES, 10ul cholera toxin (20 ug/ml), 10ul insulin (10 mg/ml), and 3 mg/ml collagenase], then gently agitated overnight at 37ºC. Cells were spun at 80g for 1.5 min, washed 1x with DMEM:F-12, then spun at 200g for 4 min before plating in growth media (DMEM with 10% FBS and penicillin/streptomycin antibiotics). After passaging cells 5x using differential trypsinization, cells were filtered through a 40 um filter to produce a single cell suspension, and labeled with anti-CD49f. Cells were then sorted using FACS to eliminate fibroblast contamination, using a narrow gate for strong CD29f signal, and the resulting epithelial cells were plated in growth media.
Real-Time PCR Analysis
Total RNA was harvested from cells using Trizol followed by the Micro-to-Midi Total RNA Purification System (Invitrogen, Carlsbad, CA). The High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA) was used to convert 5 ug RNA into cDNA. Real-time analysis was carried out with a Bio-Rad iCycler iQ Real Time PCR instrument using Applied Biosystems TaqMan Gene Expression primers and probes that were labeled with FAM and EuroGentec Two Step RT qPCR Master Mix (San Diego, CA).
Cell signaling and growth following transduction
Breast cancer cell lines were virally transduced as described previously (12), and MDA-MB-453 and SKBR-3 cells were selected with 0.75 and 1.75 ug/ml puromycin, respectively. To analyze receptor phosphorylation and downstream signaling, serum starved cells in 12-well plates (Nunc, Rochester, NY) were treated with Nrg1 for various times at 37ºC. Lysates were collected in sample buffer (62.5 mmol/L Tris-HCl, 2% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.05% bromophenol blue), boiled for 5 min at 95ºC, and resolved by 8% SDS-polyacrylamide gel electrophoresis. Following transfer to nitrocellulose (Pall Life Sciences, Pensacola, FL), blots were cut horizontally into strips and immunoblotted with various primary antibodies. Blotted proteins were detected using horseradish peroxidase-conjugated secondary antibodies (Invitrogen, Carlsbad, CA) and developed using the SuperSignal West chemicals (Pierce, Rockford, IL). An Alpha Innotech imaging station with AlphaEase FC software (San Leandro, CA) was used to capture and quantify images. All results are representative of at least 3 independent experiments.
To measure proliferation, MDA-MB-453 and SKBR3 cells were plated into 24-well plates (Corning) at a density of 1x105 and 2.5x104 cells/well, respectively. After 24h, cells were placed in conditioned media and were allowed to proliferate for 48h. During the last 2h of growth, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) was added to the media to measure activity. Crystals formed from the MTT were dissolved in acidic isopropanol and the absorption was measured at 570 nm with a baseline subtraction at 655 nm.
Cell Assays utilizing RNAi
MDA-MB-453 and BT474 cells were plated at a density of 1x105 and 2.5x104 cells/well, respectively, in 24-well plates (Corning, Corning, NY). After settling for 16h, cells were transfected with 100 nM Dharmacon nontargeting or LRIG1 ON-TARGETplus SMARTpool RNAi per manufacturer’s directions (Dharmacon, Lafayette, CO). Proliferation was measured after 48h as described above. For cell signaling experiments, 48h after transfection cells were serum starved overnight. Various Nrg1 concentrations in serum starve media were added to the cells for 15 min, followed by cell lysis in sample buffer. Lysates were then analyzed via western blot as described.
Statistical analysis
Values are expressed as mean +/− standard error (SE) unless noted. P values were determined using the two-tailed Student’s t test with values < 0.05 considered statistically significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Results
LRIG1 is suppressed in ErbB2-overexpressing mouse mammary tumors
Transgenic overexpression of activated ErbB2 (NDL2-5 or Neu deletion mutant) in the mouse mammary epithelium yields focal adenocarcinomas that evolve after a long latency and metastasize to the lung with high frequency (22). Previous studies have demonstrated that endogenous ErbB3 protein levels are elevated 10 to 15 fold in NDL tumor tissue, underscoring the link between ErbB2 and ErbB3 in mammary tumorigenesis (6). Interestingly, this increase in ErbB3 protein expression was accompanied by a rather modest (< 3 fold) elevation in ErbB3 transcript levels suggesting that post-transcriptional mechanisms such as enhanced message translation and/or augmented receptor stability contribute to ErbB3 overexpression in NDL tumors. These mechanisms may also be involved in human breast cancer as ErbB3 gene amplification has not been detected despite ErbB3 protein overexpression. To further explore this, we examined the expression of ErbB2 and ErbB3 in normal mammary gland from non-transgenic FvB littermates, in normal mammary gland from NDL2-5 transgenic mice and in tumor tissue from NDL2-5 transgenic mice by western blotting (Fig. 1A). Endogenous ErbB3 protein expression was significantly elevated in tumor tissue, as previously reported (6). Interestingly, a similar result was observed with ErbB2. ErbB2 was very modestly expressed in non-transgenic normal mammary gland and in normal mammary gland from transgenic animals but was abundantly expressed in tumor tissue. Since the transgene is present in both normal and tumor tissue in transgenic animals, these results suggest that ErbB2 gene amplification is not sufficient for ErbB2 protein overexpression. Rather it suggests that events that occur specifically in the tumor tissue, among them the suppression of negative regulatory molecules, may cooperate with gene amplification to yield maximal ErbB2 protein overexpression.
Figure 1.

LRIG1 is suppressed in tumors from MMTV-ErbB2 mice. A, Normal mammary tissue from non-transgenic FvB littermates was collected along with normal and tumor tissue from MMTV-NDL2-5 mice. Lysates from these tissues were blotted for ErbB2, ErbB3, and actin as a loading control. B, Tumor (T) and adjacent normal (N) tissue were collected from the mammary fat pad of NDL2-5 transgenic mice. Lysates from these tissues were blotted for ErbB2, ErbB3, LRIG1, and actin. C, An epithelial cell line produced from a NDL2-5 tumor was transduced with myc-tagged LRIG1 or control vector pMX. Lysates were immunblotted with antibodies to phosphotyrosine (py20), ErbB2, ErbB3, LRIG1, and actin.
We next examined the expression of LRIG1 in adjacent normal and tumor tissue from the NDL2-5 mice. Tissue from 10 independent mice was examined by western blotting; 4 representative samples are shown in Figure 1B. Both ErbB2 and ErbB3 protein expression were dramatically increased in tumor tissue and this could not be explained by a significant difference in receptor transcript levels as measured by real time PCR analysis. (In each case, transcript levels in normal tissue were normalized to 1.00. Values are an average of 4 mice. Neu: 0.92 +/− 0.19; mErbB3: 1.17 +/− 0.30). Remarkably, all animals examined showed a dramatic loss of LRIG1 expression in tumor tissue. These results raise the possibility that LRIG1 suppression may contribute to ErbB2 overexpression in breast tumors. Real time PCR analysis of LRIG1 transcript levels in normal and tumor tissue revealed a 40% decrease in tumor tissue (Transcript levels in normal tissue were normalized to 1.00. Values are an average of 4 mice. mLRIG1: 0.58 +/− 0.09).
To further examine the role of LRIG1 loss in ErbB2/ErbB3 overexpressing NDL2-5 tumors, an NDL2-5 cell line was established from a primary tumor. This cell line maintained robust expression of ErbB2 and ErbB3 and lacked detectable endogenous LRIG1 by western blotting (Fig. 1C). To examine whether ectopic expression of LRIG1 was sufficient to diminish ErbB2/ErbB3 expression, the NDL2-5 cell line was stably transduced with either control virus (pMX) or virus expressing myc-tagged LRIG1. As shown in Figure 1C, expression of LRIG1 resulted in a dramatic loss of both receptors. These results indicate that the loss of LRIG1 observed in NDL2-5 tumors is functionally relevant.
LRIG1 expression is decreased in human breast cancer
To determine whether the loss of LRIG1 observed in murine mammary tumors was reflected in human breast tumors, LRIG1 expression was examined by western blotting of a panel of normal (n = 42) and tumor (n = 67) tissues. In a comparison of all normal to all tumor, over three-fifths of the tumors (42/67) showed a loss of LRIG1 protein (Fig. 2A), and on average LRIG1 protein levels were decreased by 33% in tumors compared to normal tissues (inset, P < 0.02).
Figure 2.

LRIG1 levels are decreased in human breast tumors. A, Relative LRIG1 expression was compared in breast tumor and normal specimens via western blot analysis. Each of the 67 tumor and 42 normal samples are plotted in order of increasing LRIG1 levels after normalizing to actin levels. Inset, The mean value of LRIG1 protein expression for each subset is plotted with error bars showing SE. B, Using the Richardson-Bloom grade information available from the pathology reports of 58 of the 67 tumors, relative LRIG1 expression of each tumor is plotted as a function of the tumor grade. Low grade represents tumor grades of 1 and 2, with high grade corresponding to grade 3 tumors. Inset, The average LRIG1 level of each grouping is plotted in the right panel with error bars showing the SE. C, Representative plots of normalized LRIG1 transcript in breast tissue from the Oncomine database are pictured. Each study has a P value of < 0.05.
Richardson-Bloom tumor grade information was available for 58 of the 67 tumors in our collection, enabling us to examine whether LRIG1 protein expression varied with tumor grade. Since grade 1 samples were rare in our cohort, tumors were sorted into two classes: low grade tumors, consisting of grades 1 and 2 (n = 26), and high grade (grade 3) tumors (n = 32). Figure 2B shows a plot of relative LRIG1 expression in each of these classes. Strikingly, there is on average a 2-fold decrease in LRIG1 levels in the high grade tumors compared to those of low grade (inset, P < 0.005).
Since a decrease in LRIG1 transcript was observed in tumors from NDL2-5 mice, we were interested in determining whether LRIG1 transcript was also decreased in human breast cancer. We queried the Oncomine database1, a collection of microarray data from more than 18,000 cancer gene expression profiles (23). Analysis of LRIG1 transcript in the breast revealed a statistically significant under-expression of LRIG1 transcript in tumors compared to normal tissue [left panel Fig. 2D; (24)] and an inverse correlation with tumor grade [right panel Fig. 2C; (25, 26)]. Collectively, these observations indicate that LRIG1 protein and transcript are decreased in both murine and human breast tumors relative to normal tissue, suggesting that loss of LRIG1 may be beneficial to tumor growth or survival.
LRIG1 expression is decreased in ErbB2 (+) human breast cancer
Of the 67 tumor specimens in our cohort, 13 were ErbB2 (+) tumors (19.4%) as indicated by their pathology reports. A significant decrease in LRIG1 expression was observed in ErbB2-overexpressing murine mammary tumors (Fig. 1B). To determine whether LRIG1 expression is also decreased in ErbB2 (+) human breast cancer, the level of LRIG1 in each tumor was examined by Western blotting and directly compared to its patient-matched normal tissue control. As shown in Figure 3A, decreased LRIG1 expression was observed in 76% (10 of 13) of ErbB2 (+) breast tumors compared with the matched normal tissue, while two tumors exhibited an increase in LRIG1 expression. To further support these findings, an Oncomine query revealed a loss of LRIG1 transcript in ErbB2 (+) tumors (n = 33) as compared to ErbB2 (−) tumors (n = 99) (27), as shown in Figure 3B (P = 0.039). These findings indicate that LRIG1 protein expression and transcript levels are decreased in the majority of ErbB2 (+) human breast cancers as well as in mammary tumors from ErbB2-overexpressing mice. The consistency in this result suggests that LRIG1 loss may provide ErbB2-overexpressing tumors with a selective advantage.
Figure 3.

ErbB2 (+) human tumors display reduced LRIG1 expression. A, Lysates of ErbB2 (+) tumors and patient-matched normal samples were immunoblotted with antibodies to ErbB2, LRIG1, and actin. A pie chart depicting the frequency of LRIG1 loss in these tumors is shown below. B, The Hess study available through Oncomine (27) was queried for transcript expression of LRIG1 in breast tumors and is plotted on the basis of ErbB2 status of the tumors.
Depletion of LRIG1 in ErbB2-overexpressing breast cancer cells enhances tumor cell growth
To model the loss of LRIG1 observed in ErbB2 (+) breast cancer, LRIG1 was depleted in breast tumor cells by RNAi-mediated silencing and the impact on ErbB2 expression was examined. A survey of several ErbB2-overexpressing breast tumor cells found that MDA-MB-453 cells and BT474 cells express detectable levels of LRIG1 protein (Fig. 4A). As shown in Figure 4A, LRIG1-specific RNAi efficiently reduced LRIG1 protein levels in both cell lines compared to the non-targeting control. This reduction in LRIG1 brought about a > 30% increase in ErbB2 expression (= 0.002 [MDA-MB-453]; p = 0.007 [BT474]) as well as an increase in basal ErbB2 phosphorylation in both cell lines. These findings demonstrate that depletion of LRIG1 is sufficient to further increase ErbB2 expression in ErbB2-overexpressing breast cancer cells.
Figure 4.

Depletion of LRIG1 augments ErbB2 expression and enhances cellular proliferation. A, Lysates from MDA-MB-453 and BT474 cells transfected for 72h with RNAi oligos targeting LRIG1 (LRIG1-KD) or scrambled control (SC) were blotted with antibodies to p-ErbB2, ErbB2, LRIG1, and actin. B, 48h after transfecting with RNAi oligos, MDA-MB-453 cells were serum starved overnight and then treated with increasing concentrations of Nrg1 for 15 min. Whole cell lysates were blotted with antibodies to LRIG1, pAKT, tAKT, pERK, tERK, and tubulin as a loading control. The bottom panel plots the densitometic analysis of the average pAKT signal from three independent experiments for each Nrg1 concentration along with SE. C, 24h after transfecting cells with RNAi oligos as described above, MDA-MB-453 and BT474 cells were placed in serum starve media, in the absence or presence of 2.5 nM Nrg1, or complete media (10% FCS). After 48h, proliferation was measured by an MTT assay. Three independent experiments were conducted with a representative experiment shown. Error bars represent SE of four replicates within this experiment. D, 24h after transfecting cells with RNAi oligos as described above, MDA-MB-453 cells were placed in serum starve or complete media in the absence or presence of the EGFR/ErbB2 inhibitor 4557W. After 48h, proliferation was measured by an MTT assay, with basal proliferation being normalized as 100%. Additionally, whole cell lysates from serum starved cells were collected following treatment and blotted for p-ErbB2 and actin (top panel) to verify inhibition of ErbB2 by 4557W.
We next examined the effect of LRIG1 loss on growth factor-stimulated signal transduction. As shown in Figure 4B, depletion of LRIG1 in MDA-MB-453 cells augmented basal Akt phosphorylation and sensitized cells to Nrg1. Map kinase activation was not similarly effected. Accumulation of ErbB2/ErbB3 at the plasma membrane of breast cancer cells has previously been shown to selectively heighten signaling through the PI-3K/Akt pathway (21). Since depletion of LRIG1 causes accumulation of ErbB receptors at the cell surface (28, 29), this provides a likely mechanism by which Akt signaling is heightened.
To determine whether LRIG1 depletion affected breast tumor cell growth, control and LRIG1-targeted cells were subjected to a MTT proliferation assay. As shown in Figure 4C, depletion of LRIG1 augmented basal (ligand independent) growth in both cell lines. To determine whether the increase in basal growth was due to an increase in constitutive ErbB2 signaling, basal growth of MDA-MB-453 cells was examined in the presence of 4557W, a small molecule inhibitor of EGFR and ErbB2. 4557W efficiently reduced ErbB2 phosphorylation (Fig. 4D, left panel). Depletion of LRIG1 augmented basal cell growth and this advantage was neutralized by 4557W (Fig. 4D, right panel). Since MDA-MB-453 cells do not express detectable amounts of EGFR, these results indicate that LRIG1 loss enhances basal tumor cell growth primarily through an increase in constitutive ErbB2 signaling.
Ectopic expression of LRIG1 in ErbB2-overexpressing breast cancer cells suppresses tumor cell growth
We next sought to determine whether ectopic expression of LRIG1 in ErbB2-overexpressing breast cancer cells was sufficient to affect tumor cell growth. As shown in Figure 5A, MDA-MB-453 cells and SKBR-3 cells were transduced with a myc-tagged version of LRIG1 and puromycin-resistant clones were pooled to negate the effects of clonal variation. SKBR-3 cells express very limited amounts of endogenous LRIG1 but have abundant ErbB2 (see Fig. 6A). Importantly, ectopic expression of LRIG1 in both cell lines diminished ErbB2 protein expression (Fig. 5A) and decreased basal and Nrg1 stimulated tumor cell proliferation (Fig. 5B). Previous studies from our lab and others have demonstrated that LRIG1 decreases ErbB receptor stability (10, 11). However, LRIG1 has also been reported to decrease EGFR protein and transcript levels when ectopically expressed in neuroglioma cells (30). To determine whether ErbB2 transcript was similarly affected in our model systems, we performed real time PCR analysis. pMX was normalized to 1.00 in both cases. Interestingly, ErbB2 transcript levels were suppressed in both lines (Fig. 5A). These results suggest that in addition to destabilizing ErbB2 protein, LRIG1 may suppress ErbB2 transcription or effect ErbB2 transcript stability, perhaps by targeting factors that regulate these processes.
Figure 5.

Ectopic expression of LRIG1 inhibits breast cancer cell proliferation. A, MDA-MB-453 and SKBR-3 cells were transduced with myc-tagged LRIG1 retrovirus or pMX control. Whole cell lysates were immunoblotted with antibodies for ErbB2, myc, and actin. ErbB2 transcript was measured by real-time PCR analysis. pMX transduced cells were normalized to 1.0 B, MDA-MB-453 and SKBR3 cells transduced with LRIG1 or control pMX vector were treated with serum starve media in the presence or absence of 2.5 nM Nrg1, or with complete media, for 48h. Proliferation was then measured via an MTT assay. Each experiment was repeated in triplicate with a representative experiment shown. Error bars represent SE of four replicates. C, MDA-MB-453 cells transduced with LRIG1 or control pMX vector were stimulated with 1.25 nM Nrg1 for various timepoints. Whole cell lysates were then immunoblotted with antibodies to phosphotyrosine (py20), ErbB2, myc, pAKT, tAKT, pERK, tERK, and actin. Densitometric analysis of the pAKT signal normalized to tAKT is plotted for each timepoint along with SE from three independent experiments.
Figure 6.
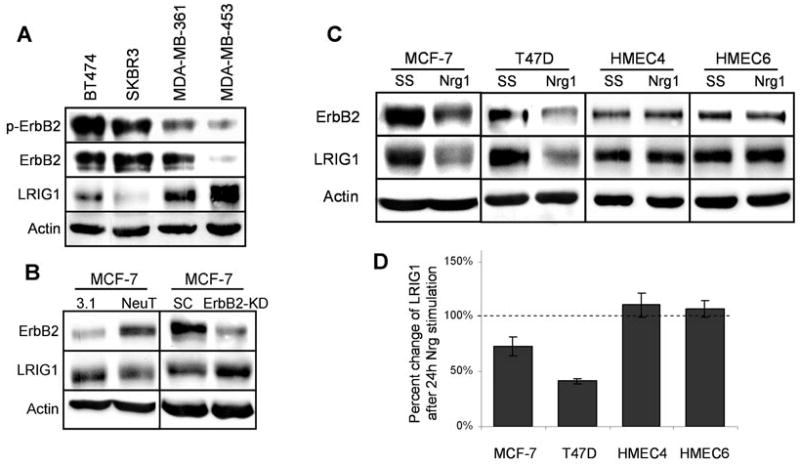
ErbB2-activation reduces LRIG1 expression in breast tumor cells. A, Whole cell lysates collected from BT474, SKBR3, MDA-MB-361 and MDA-MB-453 cells were blotted with antibodies to p-ErbB2, ErbB2, LRIG1, and actin. B, MCF-7 cells were transiently transfected for 48h with NeuT or control vector pcDNA3.1 (3.1) and cell lysates were blotted for ErbB2, LRIG1, and actin. Additionally, MCF-7 cells were treated with 100 nM scrambled control (SC) or ErbB2 RNAi oligos (ErbB2-KD) for 48h. Lysates were collected and blotted for ErbB2, LRIG1, and actin. C, MCF-7, T47D, HMEC4, and HMEC6 cells were treated with SS media in the absence or presence of 10 nM Nrg1. After 24h, whole cell lysates were collected and samples were immunoblotted with antibodies to ErbB2, LRIG1, and actin. D, Densitometric analysis of LRIG1 protein expression from each cell line in C is plotted along with SE from at least four independent experiments.
To examine the mechanism by which LRIG1 suppresses tumor cell growth, downstream signaling was examined in LRIG1-transduced MDA-MB-453 cells (Fig. 5C). Ectopic expression of LRIG1 decreased ErbB2 expression and phosphorylation and diminished Nrg1-stimulated Akt phosphorylation, but did not have a significant effect on Map kinase phosphorylation. These results indicate that the LRIG1 status of breast cancer cells has a significant impact on tumor cell signaling and proliferation.
ErbB2 activation suppresses endogenous LRIG1 expression
Since LRIG1 expression is decreased in ErbB2-overexpressing murine and human breast tumors, we were interested in examining whether ErbB2 activation may contribute to suppression of LRIG1, in effect promoting its own stability. We first examined endogenous LRIG1 expression in a panel of ErbB2 overexpressing breast cancer cell lines with different levels of constitutive ErbB2 activation as shown in Figure 6A. Interestingly, the two cell lines with the highest levels of phospho-ErbB2 expressed the lowest amount of endogenous LRIG1. To examine whether activation of ErbB2 is sufficient to suppress LRIG1 expression, we utilized MCF-7 breast tumor cells which express modest amounts of endogenous ErbB2. MCF-7 cells were transiently transfected with a constitutively active form of ErbB2, NeuT, and endogenous LRIG1 levels were examined by western blotting as shown in Figure 6B (left panel). Expression of NeuT was sufficient to decrease endogenous LRIG1 protein levels by 21% (21 +/− 4; p = 0.0001, n = 8 independent experiments) and this was accompanied by a 15% decrease in LRIG1 transcript levels as determined by real time PCR analysis. This is likely an underestimation of the effect of chronic ErbB2 activation on endogenous LRIG1 expression as these cells were transiently transfected and were not a pure population of NeuT expressing cells. In order to further implicate ErbB2 in negative regulation of LRIG1 expression, endogenous ErbB2 was silenced by siRNA in MCF7 cells and LRIG1 protein expression was examined by western blotting (Fig. 6B, right panel). Following ErbB2 knock down, there was a significant increase in endogenous LRIG1 expression, underscoring the reciprocal relationship between ErbB2 and LRIG1.
We next examined whether activation of endogenous ErbB2 by ligand stimulation was sufficient to impact endogenous LRIG1 expression. Although this means of ErbB2 activation is transient, we were interested in determining its impact on LRIG1 expression. For this experiment, we chose tumor cells that expressed moderate amounts of LRIG1 so that any changes in LRIG1 expression could be easily quantified. MCF-7 and T47D cells were stimulated with Nrg1 and 24 hours later LRIG1 expression was examined by western blotting as shown in Figure 6C. Interestingly, ligand stimulation of both cell lines led to a significant decline in LRIG1 protein expression (Fig. 6C). LRIG1 transcript levels also decreased following ligand stimulation although the magnitude of this decrease varied between the two cell lines (MCF7: 0.89 +/− 0.05; T47D: 0.26 +/− 0.05). These results demonstrate that activation of ErbB2 in breast tumor cells leads to the suppression of the negative regulator LRIG1, at least in part through a decrease in transcript levels. Since LRIG1 depletion by RNAi enhances ErbB2 expression (Fig. 4A), these results uncover a mechanism by which ErbB2 may contribute to or sustain its own overexpression.
To determine whether suppression of LRIG1 was specific to transformed cells, LRIG1 expression was examined in two different human mammary epithelial cell lines, HMEC4 and HMEC6 (20), as shown in Figure 6D, lower panel. Western blotting of these cell lines revealed modest but detectable expression of ErbB2 which could be activated by treatment of cells with Nrg1 (data not shown). Interestingly, stimulation of these cells with Nrg1 for 24 hours did not result in a decrease in LRIG1 expression (quantified in Fig. 6D). A decrease in LRIG1 expression was not observed at any time following Nrg1 treatment of HMEC4/6 cells (data not shown). These results indicate that activation of ErbB receptors in breast tumor cells leads to the suppression of endogenous LRIG1 expression whereas LRIG1 expression is maintained following ErbB activation in normal mammary epithelial cells.
Discussion
ErbB2 gene amplification in breast cancer and its association with poor patient prognosis was first described by Slamon and coworkers in 1987 (31). While gene amplification is typically associated with protein overexpression, this correlation is not absolute. 1+ protein staining (negative or weak) is observed in the presence of high level amplification of the ErbB2 gene (32), suggesting that there are potent mechanisms in place that oppose inappropriate ErbB2 protein expression. Conversely, there is significant evidence of 2+ (moderate) and 3+ (high) protein staining in the absence of gene amplification indicating that gene amplification is not necessary for ErbB2 protein overexpression (33, 34). Our findings with the NDL2-5 transgenic mice demonstrate that gene amplification is not sufficient for ErbB2 protein overexpression in this model. Rather, the loss or compromise of specific negative regulatory molecules such as LRIG1 in breast tumor cells appears to cooperate with increased gene dosage to yield the most robust ErbB2 protein expression. In agreement with this, LRIG1 is poorly expressed in ErbB2 (+) human breast tumors and RNAi-mediated depletion of LRIG1 in ErbB2-amplified breast cancer cells further enhances ErbB2 expression and tumor cell growth.
Recent studies have begun to provide some insight into endogenous mechanisms that limit ErbB2 expression. Foxp3, a member of the forkhead/winged helix transcription factor family, is an X-linked transcriptional repressor of ErbB2 and loss or mutational inactivation of Foxp3 is strongly correlated with ErbB2 overexpression in breast cancer. Importantly, in ErbB2 amplified breast cancers, Foxp3 (+) tumors express less ErbB2 protein compared to Foxp3 (−) tumors, indicating that Foxp3 resists ErbB2 overexpression even in the amplified setting (35). Loss of the 14-3-3σ tumor suppressor may provide a distinct mechanism for ErbB2 overexpression. The EGR2/Krox20 transcription factor and its coactivator CITED1 are upregulated in the MMTVCre/Flox-Neo-NeuNT mammary tumor model, a model which employs activated ErbB2 under the control of its endogenous promoter (36). Interestingly, 14-3-3σ sequesters EGR2 in the cytoplasm and ectopic expression of 14-3-3σ decreases ErbB2 expression in mammary tumor cells.
Post-transcriptional regulators of ErbB2 include LRIG1, RALT/Mig6 and the chaperone dependent ubiquitin ligase CHIP (37). The expression of CHIP in breast cancer has not yet been examined. RALT/Mig-6, a feedback inhibitor of the ErbB family, suppresses receptor catalytic activity through interaction with the kinase domain. ErbB2 overexpressing breast cancer cells are defective in their transcriptional induction of RALT/Mig-6 (7) and the RALT/Mig6 gene is downregulated in breast cancer patients with shortened survival (38). Such studies illustrate the diversity of mechanisms employed by cells in their strict regulation of ErbB2 signaling and the consequences of the loss of these endogenous regulators.
In this study, we provide evidence that LRIG1 is a growth suppressor in breast cancer. We demonstrate that LRIG1 expression is decreased in human breast cancer and that the majority of ErbB2 (+) tumors underexpress LRIG1. This is reflected in a transgenic mouse model of ErbB2 (+) breast cancer. Depletion of residual LRIG1 in ErbB2 overexpressing breast cancer cells further augments ErbB2 expression and tumor cell proliferation while ectopic expression of LRIG1 decreases ErbB2 expression and attenuates tumor cell proliferation. We demonstrate that activation of ErbB2 in breast tumor cells is sufficient to decrease LRIG1 expression, at least in part through decreases in LRIG1 transcript, suggesting that ErbB2 takes an active role in its own overexpression through the marginalization of negative regulatory pathways. Suppression of negative regulatory pathways would create an environment in which inappropriate expression of ErbB2 would not be effectively policed. Depending on timing and cellular context, this could contribute to tumor initiation, progression or metastasis.
Acknowledgments
We thank Dr. Krishna Rao of the Division of Hematology and Oncology, Department of Internal Medicine at Southern Illinois University School of Medicine for the HMEC4 and HMEC6 cell lines.
This work was supported by NIH grants CA118384 (C.S.) and GM068994 (K.C.). J.M. and D.S. are recipients of Department of Defense Breast Cancer Research Program Pre-doctoral fellowships: W81XWH-06-1-0402 (J.M.) and W81XWH-06-1-0772 (D.S.).
Footnotes
References
- 1.Sweeney C, Miller JK, Shattuck DL, Carraway KL., 3rd ErbB receptor negative regulatory mechanisms: implications in cancer. J Mammary Gland Biol Neoplasia. 2006;11:89–99. doi: 10.1007/s10911-006-9015-3. [DOI] [PubMed] [Google Scholar]
- 2.Peschard P, Park M. Escape from Cbl-mediated downregulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell. 2003;3:519–23. doi: 10.1016/s1535-6108(03)00136-3. [DOI] [PubMed] [Google Scholar]
- 3.Moasser MM. Targeting the function of the HER2 oncogene in human cancer therapeutics. Oncogene. 2007;26:6577–92. doi: 10.1038/sj.onc.1210478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, 3rd, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003;100:8933–8. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naidu R, Yadav M, Nair S, Kutty MK. Expression of c-erbB3 protein in primary breast carcinomas. Br J Cancer. 1998;78:1385–90. doi: 10.1038/bjc.1998.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siegel PM, Ryan ED, Cardiff RD, Muller WJ. Elevated expression of activated forms of Neu/ErbB-2 and ErbB-3 are involved in the induction of mammary tumors in transgenic mice: implications for human breast cancer. Embo J. 1999;18:2149–64. doi: 10.1093/emboj/18.8.2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anastasi S, Sala G, Huiping C, et al. Loss of RALT/MIG-6 expression in ERBB2-amplified breast carcinomas enhances ErbB-2 oncogenic potency and favors resistance to Herceptin. Oncogene. 2005;24:4540–8. doi: 10.1038/sj.onc.1208658. [DOI] [PubMed] [Google Scholar]
- 8.Yen L, Cao Z, Wu X, et al. Loss of Nrdp1 enhances ErbB2/ErbB3-dependent breast tumor cell growth. Cancer Res. 2006;66:11279–86. doi: 10.1158/0008-5472.CAN-06-2319. [DOI] [PubMed] [Google Scholar]
- 9.Bao J, Gur G, Yarden Y. Src promotes destruction of c-Cbl: implications for oncogenic synergy between Src and growth factor receptors. Proc Natl Acad Sci U S A. 2003;100:2438–43. doi: 10.1073/pnas.0437945100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gur G, Rubin C, Katz M, et al. LRIG1 restricts growth factor signaling by enhancing receptor ubiquitylation and degradation. Embo J. 2004;23:3270–81. doi: 10.1038/sj.emboj.7600342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laederich MB, Funes-Duran M, Yen L, et al. The leucine-rich repeat protein LRIG1 is a negative regulator of ErbB family receptor tyrosine kinases. J Biol Chem. 2004;279:47050–6. doi: 10.1074/jbc.M409703200. [DOI] [PubMed] [Google Scholar]
- 12.Shattuck DL, Miller JK, Laederich M, et al. LRIG1 is a novel negative regulator of the Met receptor and opposes Met and Her2 synergy. Mol Cell Biol. 2007;27:1934–46. doi: 10.1128/MCB.00757-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suzuki Y, Miura H, Tanemura A, et al. Targeted disruption of LIG-1 gene results in psoriasiform epidermal hyperplasia. FEBS Lett. 2002;521:67–71. doi: 10.1016/s0014-5793(02)02824-7. [DOI] [PubMed] [Google Scholar]
- 14.Lindstrom AK, Ekman K, Stendahl U, et al. LRIG1 and squamous epithelial uterine cervical cancer: correlation to prognosis, other tumor markers, sex steroid hormones, and smoking. Int J Gynecol Cancer. 2008;18:312–7. doi: 10.1111/j.1525-1438.2007.01021.x. [DOI] [PubMed] [Google Scholar]
- 15.Tanemura A, Nagasawa T, Inui S, Itami S. LRIG-1 provides a novel prognostic predictor in squamous cell carcinoma of the skin: immunohistochemical analysis for 38 cases. Dermatol Surg. 2005;31:423–30. doi: 10.1111/j.1524-4725.2005.31108. [DOI] [PubMed] [Google Scholar]
- 16.Thomasson M, Hedman H, Guo D, Ljungberg B, Henriksson R. LRIG1 and epidermal growth factor receptor in renal cell carcinoma: a quantitative RT--PCR and immunohistochemical analysis. Br J Cancer. 2003;89:1285–9. doi: 10.1038/sj.bjc.6601208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hedman H, Nilsson J, Guo D, Henriksson R. Is LRIG1 a tumour suppressor gene at chromosome 3p14.3? Acta Oncol. 2002;41:352–4. doi: 10.1080/028418602760169398. [DOI] [PubMed] [Google Scholar]
- 18.Ljuslinder I, Golovleva I, Palmqvist R, et al. LRIG1 expression in colorectal cancer. Acta Oncol. 2007;46:1118–22. doi: 10.1080/02841860701426823. [DOI] [PubMed] [Google Scholar]
- 19.Ljuslinder I, Malmer B, Golovleva I, et al. Increased copy number at 3p14 in breast cancer. Breast Cancer Res. 2005;7:R719–27. doi: 10.1186/bcr1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng JM, Ding M, Aribi A, Shah P, Rao K. Loss of RAB25 expression in breast cancer. Int J Cancer. 2006;118:2957–64. doi: 10.1002/ijc.21739. [DOI] [PubMed] [Google Scholar]
- 21.Funes M, Miller JK, Lai C, Carraway KL, 3rd, Sweeney C. The mucin Muc4 potentiates neuregulin signaling by increasing the cell-surface populations of ErbB2 and ErbB3. J Biol Chem. 2006;281:19310–9. doi: 10.1074/jbc.M603225200. [DOI] [PubMed] [Google Scholar]
- 22.Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ. Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007;7:389–97. doi: 10.1038/nrc2127. [DOI] [PubMed] [Google Scholar]
- 23.Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–80. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Richardson AL, Wang ZC, De Nicolo A, et al. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006;9:121–32. doi: 10.1016/j.ccr.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 25.Miller LD, Smeds J, George J, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci U S A. 2005;102:13550–5. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van ‘t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 27.Hess KR, Anderson K, Symmans WF, et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006;24:4236–44. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- 28.Jensen KB, Watt FM. Single-cell expression profiling of human epidermal stem and transit-amplifying cells: Lrig1 is a regulator of stem cell quiescence. Proc Natl Acad Sci U S A. 2006;103:11958–63. doi: 10.1073/pnas.0601886103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stutz MA, Shattuck DL, Laederich MB, Carraway KL, 3rd, Sweeney C. LRIG1 negatively regulates the oncogenic EGF receptor mutant EGFRvIII. Oncogene. 2008 doi: 10.1038/onc.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye F, Guo DS, Niu HQ, et al. [Molecular mechanism of LRIG1 cDNA-induced apoptosis in human glioma cell line H4] Ai Zheng. 2004;23:1149–54. [PubMed] [Google Scholar]
- 31.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 32.Todorovic-Rakovic N, Jovanovic D, Neskovic-Konstantinovic Z, Nikolic-Vukosavljevic D. Comparison between immunohistochemistry and chromogenic in situ hybridization in assessing HER-2 status in breast cancer. Pathol Int. 2005;55:318–23. doi: 10.1111/j.1440-1827.2005.01831.x. [DOI] [PubMed] [Google Scholar]
- 33.Bofin AM, Ytterhus B, Martin C, O’Leary JJ, Hagmar BM. Detection and quantitation of HER-2 gene amplification and protein expression in breast carcinoma. Am J Clin Pathol. 2004;122:110–9. doi: 10.1309/8A2D-JFT0-7NE6-EWHE. [DOI] [PubMed] [Google Scholar]
- 34.Jimenez RE, Wallis T, Tabasczka P, Visscher DW. Determination of Her-2/Neu status in breast carcinoma: comparative analysis of immunohistochemistry and fluorescent in situ hybridization. Mod Pathol. 2000;13:37–45. doi: 10.1038/modpathol.3880007. [DOI] [PubMed] [Google Scholar]
- 35.Zuo T, Wang L, Morrison C, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell. 2007;129:1275–86. doi: 10.1016/j.cell.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dillon RL, Brown ST, Ling C, Shioda T, Muller WJ. An EGR2/CITED1 transcription factor complex and the 14-3-3sigma tumor suppressor are involved in regulating ErbB2 expression in a transgenic-mouse model of human breast cancer. Mol Cell Biol. 2007;27:8648–57. doi: 10.1128/MCB.00866-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A. 2002;99:12847–52. doi: 10.1073/pnas.202365899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amatschek S, Koenig U, Auer H, et al. Tissue-wide expression profiling using cDNA subtraction and microarrays to identify tumor-specific genes. Cancer Res. 2004;64:844–56. doi: 10.1158/0008-5472.can-03-2361. [DOI] [PubMed] [Google Scholar]