

Abstract
Total synthesis of the eighteen-membered ring cyclodepsipeptide believed to be respirantin (1b) has been achieved. The key step in the synthesis is an intramolecular transesterification of the β-ketoester alcohol 6 to afford the protected macrocycle 5. The synthetic product was shown to be identical to a natural product presumed to be respirantin (1b) and the absolute stereochemistry of 6 of the 7 asymmetric centers of cyclodepsipeptide 1b was unequivocally established. Respirantin (1b) was found to be a remarkable inhibitor of cancer cell growth and related to the antimycin family of antibiotics.
In the preceding report, we summarized the isolation and structures of three exceptional cancer cell growth inhibitory cyclodepsipeptides from the bacterium Kitasatospora sp. found on the Beaufort Sea coast of the Alaska North Slope.1a One of these corresponded to a unique structure and was designated kitastatin 1 (1a) (Figure 1) while the other two cyclodepsipeptides on the basis of reported NMR assignments were presumed to be respirantin (1b) and a valeryl modification (1c).1b
Figure 1.

Kitastatin, respirantin and valeryl modification. (+)-Antimycin A3b.
Respirantin (1b) was first reported in 19931b as an insecticidal antibiotic isolated from a Streptomyces species found in a soil sample from Japan and shown to have cyclodepsipeptide structure 1b based on analysis of its spectroscopic properties. The stereochemistry was not determined. Kitastatin 1 (1a) and respirantin (1b) contain a blastmycic acid unit also found in the antimycins2 such as 2 and neoantimycin3. An unusual structural feature is the β-ketoester linkage (carbons 6-8) in the 18-membered depsipeptide macrocycle. In order to obtain sufficient material for more extensive biological evaluation as well as overall to determine the stereochemistry and absolute configuration of kitastatin 1 (1a) and respirantin (1b), we undertook research to develop a total synthesis of 1b with flexibility to enable future SAR development. Herein we report the successful results.
Results and Discussion
Inspection of the kitastatin 1 (1a) and respirantin (1b) macrocycle revealed that they are composed of common amino acids, or α-hydroxycarboxylic acids derived from them, along with the β-ketoester unit. Since the absolute stereochemistry of 1a and 1b was undetermined at the onset of this study, our initial target 1b was selected by assuming the most common S-configuration for the constituent amino acids and their presumed α-hydroxy derivatives. Fortunately, that proved to be the correct choice among the 256 possible optical isomers. A retrosynthetic analysis of the ultimately successful route to respirantin (1b) is presented in Scheme 1. Prior antimycin syntheses2 offered good precedent for appending the protected benzoic acid 3 to amino substituted macrocycles. However other issues that needed to be addressed in developing our approach to 1b included introduction of the β-ketoester unit, selection of appropriate esterification and peptide bond forming methods, protecting-group strategy, and the method and site of its macrocyclic lactonization.
Scheme 1. Retrosynthetic Analysis of Respirantin.

Our initial approach is outlined in Scheme 2 where β-ketoester 74 represented a good starting material for incorporating carbons 6-9. Introduction of the gem-dimethyl groups was not trivial, but after some experimentation α, α-dimethyl ester 11 was obtained in reasonable yield. While β-ketoacids are well known to be susceptible to decarboxylation, carboxylic acid 12 was acquired via carefully controlled saponification. However all attempts to esterify 12 with alcohol 135 were deflected either by decarboxylation to ketone 15 or intramolecular cyclization to the pyrrolidine-2,4-dione 16. The method of choice for the preparation of complex β-ketoesters is via transesterification. However, as this reaction proceeds through a ketene intermediate,6,7 ester 11 is not a suitable substrate. Nevertheless this approach was pursued in the belief that the introduction of the gem-dimethyl groups could be postponed until the needed β-ketoester linkage was formed. After extensive experimentation we were able to obtain ester 14 via reaction of ester 7 with excess alcohol 13 in refluxing cyclohexane8 in the presence of a catalytic amount of activated zinc.9 However the modest yield and the need for excess alcohol 13 limited this approach. A timely report10 describing a high yield intramolecular β-ketoester transesterification used to form a 15-membered macrocycle seemed to not only address the troublesome formation of the required β-ketoester linkage but also to simplify projected functional group manipulations needed for macrocycle formation.
Scheme 2.

a
aReagents and conditions: (a) K2CO3, MeI, DMSO, 23 °C, 48 h, 65%. (b) KOH, aq CH3OH, 23 °C, 0.25 h, 81%. (c) 13, Zn, cyclohexane, 80 °C, 33%.
The initial approach designed to utilize the intramolecular β-ketoester transesterification method of macrocyclization is outlined in Scheme 3. The diester 20 was obtained via reaction of the acid chloride derived from silyl ester 18 under neutral conditions11,12 with alcohol 19.5 Selective hydrolytic cleavage of methyl ester 20 could not be achieved as extensive cleavage of the internal ester linkage occurred. The desired carboxylic acid 21 was obtained via nucleophilic alkyl cleavage with LiI in pyridine.13 Formation of the amide linkage leading to amide 22 proved to be problematic. Reaction of carboxylic acid 21 with the amine derived from TFA deprotection of Boc protected 7 under a variety of peptide coupling procedures (BOP14, PyBroP15, DEPC16) afforded at best low yields of amide 22 along with the pyrazine 24. The formation of 24 can be explained by dimerization of the amine free base via Schiff base formation followed by oxidative aromatization. Subsequent experimentation revealed that while the trifluoroacetate salt of the parent amine from 7 could be isolated we were not able to isolate the corresponding free base. In attempts to prepare amide 22, dimerization of the free base occurred in preference to reaction with the activated carboxylic acid 21. To avoid this problem a solution of the TFA salt in DCM was added to a solution of 21, PyBroP, and three equivalents of DIPEA in DCM. By this method, the free base was only generated in the presence of excess activated carboxylic acid and a reasonable yield of amide 22 was reproducibly obtained.
Scheme 3.

a
aReagents and conditions: (a) (i) Oxalyl chloride, catalytic DMF, DCM, 0-23 °C, 2h. (ii) 19, pyridine, 23°C, 16 h, 75%. (b) LiI, pyridine, 110°C, 40 h, 89%. (c) (i) 7, 1:1 TFA-DCM, 0.5 h. (ii) PyBroP, DIPEA, DCM, product from (i), 4 h, 65%. (d) BF3·Et2O, DCM, 0.5 h, 87% for 23, 83% for 27-28. (e) 25, MNBA, DMAP, TEA, DCM, 23 °C, 16 h, 77%.
Deprotection of amide 22 was also nontrivial. Standard TBAF treatment afforded a fairly complex mixture of which the desired alcohol 23 was the major component. Better results were obtained using BF3·Et2O17 which provided alcohol 23 cleanly and in high yield. Condensation of 23 with carboxylic acid 2518 mediated with 2-methyl-6-nitrobenzoic anhydride (MNBA)19 afforded ester 26 in a reasonable yield. Desilylation of 26 using the BF3·Et2O procedure cleanly provided a 1:1 mixture of isomeric alcohols 27-28. The spectroscopic and analytical properties of both were consistent with the expected product and characterized as 27-28, a mixture of diastereomers arising from racemization of the carbon bearing the terminal hydroxyl.
An explanation for the epimerization evident during the deprotection of silyl ether 26 remains obscure. Model studies (Scheme 4) did not indicate evidence of any obvious problem. The epimerization problem and the somewhat variable results in the presence of the β-ketoester suggested that presence of this potentially base labile moiety could be problematic and that delaying its introduction should be beneficial. Concurrently additional model studies indicated another area of concern. The C1-5 fragment 34 was prepared by condensation of the acid chloride derived from silyl ester 3220 and alcohol 3321 with a view toward increasing the convergency of the synthesis. However, efforts to deprotect either the carboxyl (mild base or LiI/pyridine) or the hydroxyl (TBAF) groups led to β-elimination of the leucic acid moiety leading to olefin 36 as the major product (Scheme 5). These results introduced additional constraints upon the reagents available for this synthetic approach. The desired desilylated alcohol 35 was eventually obtained by BF3·Et2O deprotection.
Scheme 4.
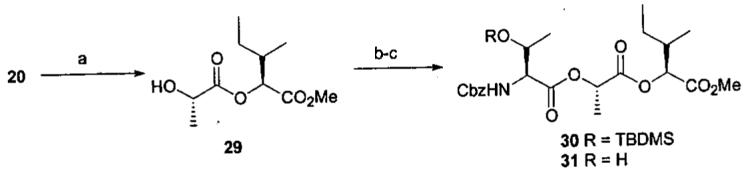
a
aReagents and conditions: (a) TBAF, THF, 0 °C, 1 h, 85%. (b) 25, MNBA, DMAP, TEA, DCM, 23°C, 16 h, 75%. (c) BF3·Et2O, DCM, 23 °C, 1.5 h, 92%.
Scheme 5.

a
aReagents and conditions: (a) (i) Oxalyl chloride, catalytic DMF, DCM, 0-23 °C, 2h. (ii) 33, pyridine, 23 °C, 16 h, 55%. (b) BF3·Et2O, DCM, 23 °C, 1 h, 51%. (c) K2CO3, aqueous CH3OH or TBAF.
With these results in mind we embarked on the ultimately successful route to respirantin. Scheme 6 outlines our approach to the respirantin macrocyclic lactone 5. Condensation of the acid chloride derived from 18 with alcohol 3722 provided ester 38. The t-butyl ester was chosen for carboxyl protection due to the lability of the leucic acid portion to the conditions required for methyl ester cleavage. Desilylation with TBAF followed by MNBA mediated condensation of the resulting alcohol 10 with carboxylic acid 25 provided ester 39 and ultimately alcohol 9 following TBAF deprotection. Anticipating the need for acidic conditions to achieve deprotection of the t-butyl ester it was considered prudent to utilize the more stable TBDPS protecting group rather than the usual TBDMS group for the terminal hydroxyl protection. Condensation of carboxylic acid 40, (available from ester 13),5 with alcohol 9 using the MNBA procedure provided tetraester 42. Cleavage of the t-butyl group was achieved with ZnBr2 in DCM23 providing carboxylic acid 44, which was coupled with the amine derived from β-ketoester 7 employing the PyBroP coupling procedure previously described to afford amide 45. At this point we were challenged by attempts to remove the TBDPS group which resulted in eliminating the leucic acid unit. For example, amide 45 was inert to BF3·Et2O at ambient temperature, as well as several other acidic reagents, and TBAF caused the expected elimination of the leucic acid residue. Consequently, we chose to proceed with a MNBA promoted coupling of alcohol 9 with carboxylic acid 41 to provide ester 43. To achieve good results with this esterfication, it was necessary to use freshly prepared acid 41. Apparently the acidity of 41 is sufficient to cause decomposition to the corresponding α-hydroxyacid. As anticipated, cleavage of the t-butyl ester in the presence of the TBDMS group proved to be problematic. The ZnBr2 procedure successful with silyl ether 42, resulted in simultaneous cleavage of the TBDMS group and the t-butyl ester. Selective carboxyl deprotection was achieved by treatment of t-butyl ester 43 with flash silica gel24 in refluxing toluene to afford 8. PyBroP promoted condensation of 8 with the amine derived from β-ketoester 7 provided the key intermediate ester 46. Desilylation once again proved to be a nontrivial operation. Similar to the results observed with silyl ether 26, reaction of silyl ether 46 with BF3·Et2O afforded a 1:1 mixture of compounds with spectral and analytical properties consistent with epimeric alcohols 6 and 47. Better results were obtained by effecting desilylation using acetyl chloride in CH3OH25 which provided predominantly a single product. While we were unable to unequivocally distinguish between epimers 6 and 47 for the desilylation product, we tentatively assigned isomer 6 as the structure for the predominant product. The stage was now set for the key macrocyclization step. Gratifyingly, treatment of alcohol 6 in refluxing toluene10 in the presence of catalytic anhydrous CuSO426 smoothly afforded macrocyclic lactone 5.
Scheme 6.

a
aReagents and conditions: (a) (i) Oxalyl chloride, catalytic DMF, DCM, 0-23 °C, 2h. (ii) 37, pyridine, 23 °C, 16 h, 81%. (b) TBAF, THF, 23 °C, 1 h, 100%. (c) 25, MNBA, DMAP, TEA, DCM, 23 °C, 16 h, 87%. (d) TBAF, THF, 23 °C, 1 h, 100%. (e) (i) TBDPSCl or TBDMSCl, imidazole, DMF, 23 °C, 16 h. (ii) LiOH, aq THF/CH3OH, 0 - 23 °C, 24 h, 80% for 40, 85% for 41. (f) 40 or 41, MNBA, DMAP, TEA, DCM, 23 °C, 16 h, 85% for 42, 83% for 43. (g) ZnBr2, DCM, 23 °C, 24 h, 80% for 44; SiO2, toluene, 110 °C, 4 h, 59% for 8. (h) (i) 7, 1:1 TFA-DCM, 0.5 h. (ii) PyBroP, DIPEA, DCM, product from (i), 4 h, 52% for 45, 65% for 46. (i) CH3OH, AcCl, 0.5 h, 63%. (j) Toluene, anhydrous CuSO4, 125 °C, 4 h, 80%.
The synthesis of the aromatic synthon 3 was achieved in four steps from the commercially available intermediate 48 as outlined in Scheme 7. The completion of the synthesis is outlined in Scheme 8. Introduction of the gem-dimethyl groups at C7 (5 → 52) was problematic. Insertion of one methyl group occurred readily, while addition of the second methyl group to afford lactone 52 was more difficult and occurred in only a modest yield. Hydrogenolysis of the Cbz protecting group afforded amine 4 which was condensed with benzoic acid 3 employing EDCI to provide amide 53. Removal (hydrogenolysis) of the benzyl ether protecting group provided the cyclodepsipeptide presumed on the basis of spectroscopic data to be respirantin (1b). The synthetic specimen of cyclodepsipeptide (1b) was found to be identical with the natural product 1b. The spectral properties (1H, 13C NMR, IR, HRMS) of 1b matched perfectly with the published values for respirantin.1b
Scheme 7.

a
aReagents and conditions: (a) Formamide, 150 °C, 0.5 h, 100%.27 (b) MeI, NaHCO3, DMF, 23 °C, 18 h, 83%.28 (c) BzlBr, K2CO3, DMF, 60 °C, 18 h, 95%.29 (d) LiOH, aq THF/CH3OH, 23 °C, 18 h, 79%.
Scheme 8.

a
aReagents and conditions: (a) MeI, K2CO3, DMSO, 23 °C, 3 h, 28%. (b) H2, Pd/C, EtOAc, 23 °C, 2 h, 73%. (c) 3, EDCI, HOBt, NMM, DMF, 23 °C, 11 h, 61%. (d) H2, Pd/C, EtOAc, 23 °C, 2 h, 82%.
The absolute stereochemistry of depsipeptide 1b at carbons 2, 3, 9, 11, and 13 follows from the chirality of the starting materials. Presumably, the 2(S), 3(R)-stereochemistry of natural threonine and the 2(S), 3(S)-stereochemistry of natural isoleucine have been retained in the biosynthesis of kitastatin 1 (1a) and respirantin (1b). In accord with that assumption, C-5 was tentatively assigned the R configuration (cf. 1b) as the synthetic and natural specimens were identical. The modular nature of this approach should offer ready access to the scaleup synthesis of respirantin, kitastatin, and a variety of structural modifications to develop structure-activity relationships in this interesting class of powerful cancer cell growth inhibitors.
Kitastatin 1 (1a), respirantin (1b), and the valeryl analog 1c were evaluated as inhibitors of cancer cell growth versus the murine P388 leukemia cell line30 and a panel of human cancer cell lines.31 The data are reported in Table 1. All three compounds displayed an impressive spectrum of activity. An interesting observation was the substantially better activity of kitastatin 1 (1a) against the pancreas BXPC-3 human cancer cell line relative to the other panel members. Whether this indicates a special selectivity against this cancer is a question which must be explored. Pancreatic cancer is one of the most deadly types and is notoriously refractory to current modes of treatment. In addition to the human cancer cell line activity cyclodepsipeptide 1b had activity against the pathogenic fungus Cryptococcus neoformans (minimum inhibitory activity, MIC = 2)1a.
Table 1. Comparison of the Cancer Cell Growth Inhibition (GI50, μg/mL) of Kitastatin 1 (1a), Respirantin (1b), and the Valeryl Analog 1c against a Panel of Murine (P388, Lymphocytic Leukemia) and Human Cancer Cell Lines.
| Compound and Cancer Cell Types | Leukemia P388 | Pancreas BXPC-3 | Breast MCF-7 | CNS SF268 | Lung-NSC NCI-H460 | Colon KM20L2 | Prostate DU-145 |
|---|---|---|---|---|---|---|---|
| 1a | 0.045 | 0.0066 | 0.004 | 0.0035 | <0.001 | 0.0024 | 0.0026 |
| 1b | 0.0037 | 0.47 | 0.0006 | 0.0016 | 0.0006 | 0.0006 | 0.00018 |
| 1c | 0.033 | 1.2 | 0.00062 | 0.016 | 0.00063 | 0.00058 | <0.0001 |
Experimental Section
Solvents were redistilled prior to use. Reagents were used as received. MNBA was obtained from TCI America. Thin layer chromatography (TLC) was carried out with Analtech 250 μ thick silica gel GHLF plates and visualized with H2SO4, phosphomolybdic acid, iodine, or UV. Organic extracts were dried over anhydrous Na2SO4 and evaporated under reduced pressure using a rotary evaporator. The crude products were separated by flash column chromatography on flash (230-400 mesh ASTM) silica from E. Merck.
Melting points are uncorrected and were determined employing an Electrothermal Mel-Temp apparatus. Optical rotations were measured using a Perkin-Elmer 241 polarimeter. The [α]D values are given in 10-1 deg cm2 g-1. IR spectra were obtained with a Thermo Nicolet Avatar 360 FT-IR instrument equipped with a Single Reflection Horizontal ATR sampling device from PIKE Technologies. HRMS data were recorded with a JEOL LCmate mass spectrometer. The 1H and 13C spectra were recorded employing Varian Gemini 300, Varian Unity 400, or Varian Unity 500 instruments in CDCl3 unless otherwise noted and were referenced to either TMS or the solvent. Elemental analyses were determined by Galbraith Laboratories, Inc., Knoxville, TN.
Methyl 4-(t-butoxycarbonyl)amino-2,2,6-trimethyl-3-oxoheptanoate (11)
Ketone 7 (0.47 g, 1.63 mmol), K2CO3 (2.26 g, 16.3 mmol) and MeI (0.31 mL, 0.71 g, 4.98 mmol) were placed in DMSO (7 mL) under N2 and stirred at ambient temperature for 48 h. The reaction mixture was diluted with H2O (30 mL) and extracted with Et2O (3 × 20 mL). The extracts were combined, washed with H2O (10 mL), 5 M NaCl (5 mL), dried and evaporated. The residue was flash chromatographed (15 g, SiO2, 95:5 hexane-EtOAc) to afford 0.34g (65%) of ketone 11 as a colorless oil: tlc Rf 0.63 (4:1 hexane-EtOAc); IR 3377, 1705 cm-1; 1H NMR δ (4.78 1H, br d), 4.65 (1H, m), 3.73 (3H, s), 1.70 (1H, m), 1.43 (17H, m), 0.95 and 0.92 (6H, 2 d, J = 6.6 Hz); 13C NMR δ 208.7, 173.5, 155.0, 79.7, 54.7, 53.8, 52.5, 41.8, 28.3, 24.6, 23.5, 22.2, 22.0, 21.33.
4-(t-Butoxycarbonyl)amino-2,2,6-trimethyl-3-oxoheptanoic acid (12)
To ester 11 (65.7 mg, 0.21 mmol) in CH3OH (0.35 mL) under N2 was added 3.5 N KOH (0.25 mL, 0.88 mmol) and the solution stirred at ambient for 15 min. The reaction mixture was diluted with H2O (15 mL) and washed with Et2O (2 × 15 mL). The aqueous layer was acidified (pH 2) with 1 N H2SO4 and extracted with Et2O (3 × 15 mL). The combined extract was washed with H2O (5 mL), 5 M NaCl (5 mL), dried and evaporated to afford 50.8 mg (81%) of 12 as a viscous oil which solidified on standing: mp 121°C; tlc Rf 0.52 (95:5:1 DCM-CH3OH-HOAc); IR 3268, 1714, 1655 cm-1; 1H NMR (d6-DMSO) δ 7.05 (1H, d, J = 9 Hz), 4.51 (1H, td, J = 9, 7 Hz), 1.87 (1H, m), 1.38 (9H, s), 1.34 (3H, s), 1.26 (4H, s and m), 0.99 (1H, dd, J = 9, 7 Hz), 0.87 (6H, d); 13C NMR (d6-DMSO) δ 214.1, 155.5, 78.0, 56.8, 38.3, 35.3, 28.1, 23.0, 21.1, 18.7, 18.2. Anal. C 60.12%, H 9.27%, N 4.68%, calcd for C15H27NO5, C 59.78%, H 9.03%, N 4.65%.
1-Methoxycarbonyl-3-methylbutyl 4-t-butoxycarbonylamino-6-methyl-3-oxoheptanoate (14)
Ketone 7 (0.274 g, 0.95 mmol), alcohol 13 (0.17 g, 1.13 mmol), and activated Zn (30 mg, 0.46 mmol) were placed in cyclohexane (4 mL) under N2 and heated at reflux for 16 h with a Dean-Stark separator. Additional 13 (0.17 g, 1.13 mmol) in cyclohexane (1 mL) was added and heating at reflux continued for 24 h. The solution was diluted with EtOAc (40 mL), filtered through Celite, washed with 6% NaHCO3 (3 × 10 mL), H2O (10 mL), 5 M NaCl (10 mL), dried, and evaporated to give 0.38 g of a pale yellow oil. This was flash chromatographed (10 g, SiO2, 93:7 hexane-EtOAc) to afford 0.127 g (33%) of ester 14 as a colorless oil: tlc Rf 0.50 (4:1 hexane-EtOAc); IR 3368, 1751, 1712 cm-1; 1H NMR δ 5.48 (1H, br d), 5.08 (1H, m), 4.28 (1H, br t), 3.77 and 3.46-3.80 (5H, s and m), 1.56-1.82 (6H, m), 1.45 (9H, s), 0.95 (12H, m); 13C NMR δ 203.3, 171.1, 166.2, 155.8, 79.8, 71.6, 58.5, 52.4, 46.1, 39.7, 39.6, 28.2, 24.7, 24.4, 23.2, 22.8, 21.4, 21.3. Anal. C 59.88%, H 9.02%, N 3.49%, calcd for C20H35NO7, C 59.83%, H 8.79%, N 3.49%.
Methyl 2-[2-(t-butyldimethylsilyl)oxypropionyloxy]-3-methylpentanoate (20)
Silyl ether 18 (3.23 g, 10.16 mmol) was dissolved in CH2Cl2 (10 mL) containing DMF (280 μL, 0.26 g, 3.62 mmol) under N2 and cooled to 0 °C. Oxalyl chloride (5.6 mL of 2M solution in CH2Cl2, 11.2 mmol) was added dropwise over 5 min. The solution was stirred at 0 °C for 1.5 h and at ambient temperature for 0.5 h. The solvent was evaporated. To the residue was added dropwise a solution of alcohol 19 (1.27 g, 8.71 mmol) in pyridine (5 mL). The solution was stirred under N2 for 16 h, diluted with THF (100 mL) and filtered through celite. The filtrate was evaporated and the residue was partitioned between EtOAc (200 mL) and H2O (20 mL). The organic phase was separated, washed with H2O (20 mL), 6% NaHCO3 (2 × 30 mL), H2O (20 mL), and 5M NaCl (10 mL), dried and evaporated. The residue was flash chromatographed (90 g, SiO2, 96:4 hexane-EtOAc) to yield 2.16 g (75%) of ester 20: TLC Rf 0.37 (95:5 hexane-EtOAc); IR 1757 cm-1; 1H NMR δ 4.93 (1H, d, J = 4.8 Hz), 4.41 (1H, q, J = 6.6 Hz), 3.72 (3H, s), 2.01 (1H, m), 1.45 (3H, d), 1.33 (2H, m), 0.97 (3H, d), 0.92 (3H, t), 0.91 (9H, s), 0.11 and 0.09 (6H, 2s); 13C NMR δ 179.1, 175.2, 81.7, 73.4, 57.3, 41.9, 31.0, 29.9, 26.7, 23.6, 20.6, 16.8, 0.4, 0.0. Anal. C 58.07%, H 9.87%, calcd for C16H32O5Si, C 57.79%, H 9.70%.
2-[2-(t-Butyldimethylsilanyloxy)propionyloxy]-3-methylpentanoic acid (21)
Ester 20 (1.01 g, 3.04 mmol) and LiI (1.24 g, 9.25 mmol) were placed in pyridine (8.0 mL) under N2 and stirred at 105 °C for 40 h. The reaction mixture was allowed to cool, diluted with toluene (30 mL), evaporated, and coevaporated with toluene (20 mL). The residue was diluted with H2O (50 mL), acidified (pH 4) with KHSO4, and extracted with EtOAc (3 × 30 mL). The extracts were combined, washed with 10% Na2S2O3 (10 mL), H2O (10 mL), and 5 M NaCl (10 mL), dried, and evaporated. The residue was flash chromatographed (30 g, SiO2, 99:1:0.5 DCM-CH3OH-HOAc) to provide 0.86g (89%) of carboxylic acid 21 as a pale yellow oil: TLC Rf 0.58 (95:5:1 DCM-CH3OH-HOAc); IR 1726 cm-1; 1H NMR δ 4.98 (1H, d, J = 4.4 Hz), 4.42 (1H, q, J = 7.6 Hz), 2.04 (1H, m), 1.56 (1H, m), 1.45 (3H, d, J = 6.6 Hz), 1.37 (1H, m), 1.01 (3H, d, J = 7.2 Hz), 0.94 (3H, t, J = 7.1 Hz), 0.91 (9H, s), 0.11 and 0.08 (6H, 2s); 13C NMR δ 175.3, 173.8, 75.9, 68.1, 36.5, 25.7, 24.4, 21.3, 18.2, 15.3, 11.5, -5.0, -5.4.
Methyl 4-{2-[2-(t-butyldimethylsilyloxy)propionyloxy]-3-methylpentanoylamino}-6-methyl-3-oxoheptanoate (22)
Ketone 7 (0.88g, 3.05 mmol) was placed in 1:1 TFA DCM (12.0 mL) under N2 and stirred at ambient temperature for 1 h. The solvent was removed and the residue coevaporated with toluene (2 × 10 mL). Carboxylic acid 21 (0.88g, 2.76 mmol) and PyBroP (1.29 g, 2.76 mmol) in DCM (6.0 mL) under N2 was cooled to 0 °C.
Diisopropylethylamine (1.07 g, 1.4 mL, 8.28 mmol) was added over 5 min. The residue from the TFA cleavage reaction was dissolved in DCM (10 mL) and added over 15 min. The solution was stirred at 0 °C for 4.5 h. The reaction mixture was diluted with EtOAc (100 mL), washed with 5% citric acid (2 × 10 mL), H2O (10 mL), 6% NaHCO3 (2 × 10 mL), H2O (10 mL), and 5 M NaCl (10 mL), dried and evaporated. The residue was flash chromatographed (60 g, SiO2, 90:10 → 80:20 hexane-EtOAc) to afford 0.88g (65%) of amide 22 as a yellow oil: TLC Rf 0.39 (4:1 hexane- EtOAc); IR 3341, 1750, 1665 cm-1; 1H NMR δ 11.98 (0.1H, s), 6.46 (1H, d, J = 7.5 Hz), 5.14 (1H, d, J = 4.5 Hz), 4.72 (1H, m), 4.43 (1H, q), 3.86 (0.5H, s), 3.72 (3H, s), 3.57 (1H, d, J = 16.5 Hz), 3.49 (1H, d, J = 15.3 Hz), 2.04 (1H, m), 1.65 (2H, m), 1.46 (6H, m and d), 1.26 (1H, m), 0.91 (21H, m), 0.12 and 0.10 (6H, 2 d); 13C NMR δ 201.5, 172.8, 169.1, 167.2, 89.5, 77.6, 68.3, 56.3, 52.4, 46.1, 39.7, 37.0, 25.7, 24.8, 24.2, 23.2, 22.4, 21.4, 21.3, 18.1, 14.9, 11.4, -4.9, -5.2; MS APCI+ 488.30444 [M+H]+ Calcd 488.3044. Anal. C 58.94%, H 9.54%, N 2.94%, calcd for C24H45NO7Si, C 59.11%, H 9.30%, N 2.87%.
Methyl 4-[2-(2-hyroxypropionyloxy)-3-methylpentanoylamino]-6-methyl-3-oxo-heptanoate (23)
To amide 22 (0.51g, 1.04 mmol) in DCM (30 mL) under N2 was added BF3·Et2O (1.42 g, 1.27 mL, 10 mmol) and the solution stirred at ambient for 2 h. The solution was poured into 6% NaHCO3-ice (100 mL). The organic phase was separated and the aqueous phase extracted with DCM (40 mL). The combined organic extract was washed with 6% NaHCO3 (30 mL), H2O (20 mL), 5 M NaCl (20 mL), dried and evaporated. The residue was flash chromatographed (15g, SiO2, 60:40 hexane-EtOAc) to give 0.34g (87%) of carboxylic acid 23 as a colorless oil: TLC Rf 0.34 (50:50 hexane-EtOAc); 1H NMR δ 12.02 (0.1H, enolic H, s), 6.53 (1H, d, J = 8.2 Hz), 5.14 (1H, d, J = 4.9 Hz), 4.72 (1H, m), 4.40 (1H, m), 3.74 (3H, s), 3.60 (1H, d, J = 16 Hz), 3.50 (1H, d, J = 16 Hz), 2.95 (1H, d, J = 5.5 Hz), 2.05 (1H, m), 1.61 (2H, m), 1.50 (4H, d and m, J = 7.1 Hz), 1.28 (1H, m), 0.95 (12H, m); 13C NMR δ 201.6, 174.4, 168.7, 167.4, 78.5, 67.1, 56.5, 52.6, 46.2, 40.0, 37.0, 25.0, 24.3, 23.7, 21.5, 20.2, 15.0, 11.4; FAB MS 374.2189 [M + H]+. Calcd 374.2179. Anal. C 57.16%, H 8.56%, N 3.70%, calcd for C18H31NO7·0.2H2O, C 57.33%, H 8.41%, N 3.71%.
Methyl (3,6-diisobutyl-5-methoxycarbonylmethyl-pyrazin-2-yl)acetate (24)
Ketone 7 (0.28g, 0.97 mmol) was placed in 1:1 TFA-DCM (4.0 mL) under N2 and stirred at ambient for 45 min. The solvent was evaporated and the residue coevaporated with toluene (2 × 10 mL). The residue was dissolved in DCM (3.0 mL) and cooled to 0 °C. TEA (0.41 mL, 293.3 mg, 2.91 mmol) was added dropwise and the solution stirred at 0 °C for 4 h. The reaction mixture was diluted with EtOAc (50 mL), washed with 5% citric acid (2 × 10 mL), H2O (10 mL), 6% NaHCO3 (2 × 10 mL), H2O (10 mL), 5 M NaCl (10 mL), dried and evaporated. The residue was flash chromatographed (10 g, SiO2, 95:5 → 90:10 hexane-EtOAc) to afford 78.2 mg (49%) of 24 as a pale yellow solid, which was recrystallyzed from hexane (1 mL): TLC Rf 0.34 (4:1 hexane-EtOAc); mp 72-74 °C; IR 1734 cm-1; 1H NMR δ 3.87 (4H, s), 3.70 (6H, s), 2.61 (4H, d, J = 7.1 Hz), 2.14 (2H, m), 0.92 (12H, d); 13C NMR δ 170.6, 151.7, 146.2, 52.1, 42.6, 40.6, 28.2, 22.4. Anal. C 63.82%, H 8.52%, N 8.20%, calcd for C18H28N2O4, C 64.26%, H 8.39%, N 8.33%.
Methyl 4-(2-{2-[2-benyloxycarbonylamino-3-(t-butyldimethylsilyloxy)butyryloxy]propionyloxy}-3-methylpentanoylamino)6-methyl-3-oxoheptanoate (26)
Carboxylic acid 25 (0.26g, 0.72 mmol), alcohol 23 (241.2 mg, 0.65 mmol), MNBA (0.25g, 0.73 mmol), DMAP (20.0 mg, 0.16 mmol) and TEA (0.30 mL, 0.22 g, 2.13 mmol) were placed in DCM (3.5 mL) under N2 and stirred at ambient for 16 h. The reaction mixture was diluted with EtOAc (50 mL), washed with H2O (10 mL), 6% NaHCO3 (2 × 10 mL), H2O (10 mL), 5% citric acid (2 × 10 mL), H2O (10 mL), 5 M NaCl (10 mL), dried, and evaporated. The residue was flash chromatographed (15 g, SiO2, 85:15 hexane-EtOAc) to yield 0.36g (77%) of ester 26 as a colorless oil which crystallized on standing: mp 84-86 °C; TLC Rf 0.22 (4:1 hexane-EtOAc); 1H NMR δ 7.37 (5H, m), 6.94 (1H, d, J = 8.3 Hz), 5.47 (1H, d, J = 9.4 Hz), 5.16 (4H, m), 4.67 (1H, m), 4.48 (1H, q), 4.27 (1H, m), 3.72 (3H, s), 3.52-3.62 (2H, m), 2.00 (1H, m), 1.63 (2H, m), 1.53 (4H, d and m, J = 7.1 Hz), 1.25 (5H, m), 0.93 (12H, m), 0.84 (9H, s), 0.06 and -0.01 (6H, 2 s); 13C NMR δ 202.0, 171.7, 168.9, 168.8, 167.5, 156.7, 136.2, 128.6, 128.3, 128.0, 78.5, 70.0, 68.7, 67.1, 60.2, 56.4, 52.3, 45.9, 38.8, 37.1, 25.6, 24.7, 24.2, 23.3, 21.2, 21.1, 17.8, 16.9, 14.8, 11.4, -4.4, -5.4; MS FAB+ 723.3890 (M + H). Calcd 723.3889. Anal. C 59.95%, H 8.44%, N 3.91%, calcd for C36H58N2O11Si, C 59.81%, H 8.09%, N 3.87%.
Methyl 4-{2-[2-(2-benzyloxycarbonylamino-3-hydroxybutyryloxy)propionyloxy]-3-methylpentanoylamino}-6-methyl-3-oxo-heptanoate (27-28)
To silyl ether 26 (0.73g, 1.02 mmol) in DCM (30 mL) under N2 was added BF3·Et2O (1.44 g, 1.3 mL, 10.2 mmol) and the solution stirred at ambient for 1.5 h. The solution was poured into 6% NaHCO3-ice (100 mL). The organic phase was separated and the aqueous phase extracted with DCM (50 mL). The combined extract was washed with 6% NaHCO3 (30 mL), H2O (20 mL), and 5 M NaCl (20 mL), dried and evaporated. The residue was flash chromatographed (20 g, SiO2, 70:30 hexane-EtOAc) to afford 0.155g, (25%) of alcohol 27 as a single isomer: TLC Rf 0.62 (50:50 hexane-EtOAc); 1H NMR δ 12.11 (0.1H, enolic H, s), 7.35 (5H, m), 6.72 (1H, d, J = 8.2 Hz), 5.60 (1H, d, 9.9 Hz), 5.18 and 5.14 (4H, m and s), 4.67 (1H, m), 4.53 (1H, m), 4.39 (1H, d, J = 9.3 Hz), 3.72 (3H, s), 3.64 (1H, d, J = 16.2 Hz), 3.50 (1H, d, J = 16.5 Hz), 3.13 (1H, d, J = 5.8 Hz), 2.05 (1H, m), 1.59 and 1.46-1.64 (7H, d and m), 1.32 (5H, m), 0.92 (12H, m); 13C NMR δ 202.5, 171.5, 169.9, 168.9, 167.9, 156.9, 136.2, 128.5, 128.2, 128.0, 78.5, 69.9, 67.6, 67.2, 58.8, 56.4, 52.6, 46.2, 38.8, 37.1, 24.7, 24.0, 23.2, 21.3, 20.1, 17.2, 15.0, 11.4. Anal. C 59.09%, H 7.45%, N 4.49%, calcd for C30H44N2O11, C 59.20%, H 7.29%, N 4.60%. Continued elution led to 0.16g (31%) of alcohols 27-28 as a mixture of isomers. Further elution provided 0.20g, (39%) of alcohol 28 as a single isomer: TLC Rf 0.58 (50:50 hexane-EtOAc); 1H NMR δ 12.04 (0.1 H, enolic H, s), 7.35 (5H, m), 6.50 (1H, d, J = 8.3 Hz), 5.65 (1H, d, J = 9.3 Hz), 5.18 and 5.14 (3H, m and s), 5.03 (1H, m), 4.72 (1H, m), 4.53 (1H, m), 4.42 (1H, d, J = 9.4 Hz), 3.72 (3H, s), 3.55 (2H, m), 3.03 (1H, d, J = 5.5 Hz), 1.99 (1H, m), 1.61 (6H, d and m, J = 7.2 Hz), 1.29 (5H, m and d, J = 6.6 Hz), 0.93 (12 H, m), 13C NMR δ 201.7, 171.1, 170.2, 168.5, 167.3, 156.8, 136.2, 128.5, 128.2, 128.0, 78.8, 69.7, 67.8, 67.1, 59.2, 56.5, 52.5, 46.2, 39.6, 36.9, 24.9, 24.4, 23.2, 21.6, 21.3, 19.7, 16.8, 14.8, 11.2. Anal. C 59.28%, H 7.58%, N 4.24%, calcd for C30H44N2O11, C 59.20%, H 7.29%, N 4.60%.
Methyl 2-(2-hydroxypropionyloxy)-3-methylpentanoate (29)
To a solution cooled to 0 °C of silyl ether 20 (0.52 g, 1.56 mmol) in THF (10 mL) under N2 was added a 1M THF solution (3.2 mL) of TBAF dropwise and the resulting solution stirred at 0 °C for 20 min. The solution was poured into H2O and extracted with EtOAc (3 × 25 mL). The combined extract was washed with H2O (10 mL), and 5 M NaCl (10 mL), dried and evaporated. The residue was flash chromatographed (15 g, SiO2, 85:15 hexane-EtOAc) to afford 0.29 g (85%) of alcohol 29 as a colorless oil: TLC Rf 0.29 (80:20 hexane-EtOAc); IR 3488, 1744 cm-1; 1H NMR δ 5.00 (1H, d, J = 4.4 Hz), 4.37 (1H, q, J = 6.6 Hz), 3.75 (3H, s), 2.82 (1H, d, J = 6.0 Hz), 2.04 (1H, m), 1.50 (4H, d and m, J = 6.0 Hz), 1.33 (1H, m), 0.98 (3H, d, J = 7.1 Hz), 0.93 (3H, t, J = 7.7 Hz); 13C NMR δ 175.44, 169.58, 66.66, 52.13, 36.51, 24.41, 20.50, 15.31, 11.49.
Methyl 2-{2-[2-benyloxycarbonylamino-3-(t-butyldimethylsilyloxy)butyryloxy]-propionyloxy}-3-methyl-pentanoate (30)
Alcohol 29 (0.115 g, 0.53 mmol) and carboxylic acid 25 (0.213 g, 0.58 mmol) were allowed to react using the MNBA esterification procedure described for 26 to afford 0.23g (75%) of ester 30 as a colorless oil: TLC Rf 0.43 (80:20 hexane-EtOAc); 1H NMR δ 7.37 (5H, m), 5.47 (1H, d, J = 9.3 Hz), 5.24 (1H, q, J = 7.1 Hz), 5.14 (2H, s), 4.98 (1H, d, J = 4.4 Hz), 4.47 (1H, m), 4.30 (1H, dd, J = 9.3, 1.6 Hz), 3.73 (3H, s), 2.01 (1H, m), 1.57 (3H, d, J = 6.6 Hz), 1.50 (1H, m), 1.30 (1H, m), 1.26 (3H, d, J = 6.0 Hz), 0.97 (3H, d, J = 6.5 Hz), 0.91 (3H, t, J = 7.7 Hz), 0.83 (9H, s), 0.05 and 0.00 (6H, 2 s); 13C NMR δ 170.35, 169.85, 169.63, 159.61, 136.32, 128.56, 128.19, 68.91, 68.63, 67.13, 60.39, 59.71, 52.10, 36.52, 25.70, 24.43, 21.25, 17.88, 17.10, 15.31, 11.50, -4.31, -5.34.
Methyl 2-{2-[2-benyloxycarbonylamino-3-hydroxybutyryloxy]-propionyloxy}-3-methyl-pentanoate (31)
Ester 30 (0.60g, 1.05 mmol) was converted employing the BF3·Et2O desilylation procedure described for 23 to provide 0.44g (92%) of alcohol 31 as a colorless oil: TLC Rf 0.13 (80:20 hexane-EtOAc); 1H NMR δ 7.35 (5H, m), 5.56 (1H, d, J = 9.9 Hz,), 5.27 (1H, q, J = 7.1 Hz), 5.13 (2H, s), 5.02 (1H, d, J = 4.4 Hz), 4.58 (1H, m), 4.43 (1H, dd, J = 9.0, 1.6 Hz), 3.74 (3H, s), 3.17 (1H, d, J = 4.4 Hz), 2.02 (1H, m), 1.63 (3H, d), 1.47 (1H, m), 1.32 (1H, m), 1.26 (3H, d, J = 6.0 Hz), 0.98 (3H, d, J = 6.5 Hz), 0.92 (3H, t, J = 7.7 Hz); 13C NMR δ 171.14, 170.88, 169.59, 156.78, 136.25, 128.49, 128.08, 127.91, 77.07, 69.14, 67.73, 67.05, 59.38, 52.31, 36.59, 24.41, 18.97, 16.58, 15.20, 11.46.
2-Benzyloxycarbonylamino-2-methoxycarbonyl-1-methylethyl 2-(t-butyldimethylsilyloxy)-4-methylpentanoate (34)
Silyl ester 32 (4.5 g, 12.5 mmol) and DMF (300 μL, 3.7 mmol) were placed in DCM (20 mL) under N2 and cooled to 0 °C. Oxalyl chloride (12.5 mL of a 2 M solution in DCM, 25 mmol) was added dropwise. The mixture was warmed to ambient temperature, stirred for 4.5 h and solvent evaporated. To the residue under N2 was added a solution of alcohol 33 (2.02 g, 8 mmol), DMAP (2.9 g, 24 mmol), and TEA (2.3 mL, 20 mmol) in DCM (15 mL) at 0 °C. The mixture was stirred at ambient temperature for 2 h, the reaction was terminated with 6% NaHCO3 (50 mL), and extracted with Et2O (3 × 50 mL). The extracts were combined, dried, and evaporated. The residue was flash chromatographed (100 g, SiO2, 6:1 hexane-EtOAc) to afford 2.17 g (56%) of ester 34 as a colorless oil: TLC Rf 0.33 (80:20 hexane-EtOAc); 1H NMR δ 7.37 (5H, m), 5.44 (2H, m), 5.15 (2H, s), 4.56 (1H, d, J = 8.1 Hz), 4.15 (1H, dd, J = 4.2, 8.1 Hz), 3.72 (3H, s), 1.78 (1H, m), 1.59 (1H, m), 1.40 (1H, m), 1.32 (3H, d, J = 6.6 Hz), 0.87 (18H, m), 0.22 (6H, m); FAB MS [M + H]+ 496.2735. Calcd for C25H42NO7Si: 496.2731.
2-Benzyloxycarbonyl-2-methoxycarbonyl-1-methylethyl 2-hydroxy-4-methylpentanoate (35)
The BF3·Et2O desilylation procedure described for 23 was applied to silyl ester 34 (265.3 mg, 0.54 mmol) to provide 0.11 g (51%) of alcohol 35 as a colorless oil: TLC Rf 0.18 (80:20 hexane-EtOAc); 1H NMR δ 7.37 (5H, m), 5.56 (1H, d, J = 9.3 Hz), 5.49 (1H, qd, J = 6.6, 2.2 Hz), 5.14 (2H, s), 4.55 (1H, dd, J = 9.3, 2.5 Hz), 4.12 (1H, q, J = 6.6 Hz), 3.73 (3H, s), 2.71 (1H, d, J = 6.1 Hz), 1.83 (1H, hept, J = 6.6 Hz), 1.49 (2H, t, J = 6.6 Hz), 1.33 (3H, d, J = 6.6 Hz), 0.94 and 0.92 (6H, 2 d); 13C NMR δ 174.5, 170.2, 156.5, 128.6, 128.3, 128.2, 71.6, 69.0, 67.4, 57.4, 52.8, 43.2, 24.3, 23.1, 21.5, 16.8.
t-Butyl 2-[2-(t-butyldimethylsilyloxy)propionyloxy]-3-methylpentanoate (38)
The acid chloride derivative of silyl ester 18 (8.73g, 27.4 mmol) and alcohol 37 (3.90 g, 20.7 mmol) were allowed to react using the catalytic DMF, oxalyl chloride esterification procedure described for ester 20 to give 6.24 g (81%) of ester 38 as a colorless oil: TLC Rf 0.60 (4:1 hexane-EtOAc); IR 1738, 1651 cm-1; 1H NMR δ 4.78 (1H, d, J = 4.5 Hz), 4.38 (1H, dd, J = 13.8, 6.6 Hz), 1.95 (1H, m), 1.24-1.54 (15H, m), 0.84-0.97 (15H, m), 0.10 (6H, m); MS APCI+ 375.2567 [M + H]+. Calcd for C19H38O5Si: 375.2567. Anal. C 61.42%, H 10.36%, calcd for C19H38O5Si, C 60.92%, H 10.23%.
t-Butyl 2-(2-hydroxypropionyloxy)-3-methylpentanoate (10)
Ester 38 (0.77 g, 2.05 mmol) was transformed employing the TBAF desilylation procedure described for alcohol 29 to provide 0.54 g (100%) of alcohol 10 as a colorless oil: TLC Rf 0.41 (80:20 hexane-EtOAc); IR 1745 cm-1; 1H NMR δ 4.85 (1H, d, J = 4.5 Hz), 4.35 (1H, dd, J = 13.8, 6.6 Hz), 2.72 (1H, m), 2.01 (1H, m), 1.23-1.68 (15H, m), 0.95 (6H, m). Anal. C 59.93%, H 9.35%, calcd for C13H24O5, C 59.98%, H 9.29%.
t-Butyl 2-{2-[2-benzyloxycarbonylamino-3-(t-butyldimethylsilyloxy)butyryloxy] propionyloxy}-3-methylpentanoate (39)
Alcohol 10 (0.50 g, 1.92 mmol) was esterfied with carboxylic acid 25 (768 mg, 2.09 mmol) by means of the MNBA procedure described for ester 26 to yield 1.02 g (87%) of ester 39 as a colorless oil: TLC Rf 0.55 (80:20 hexane-EtOAc); IR 3453, 1745, 1625 cm-1; 1H NMR δ 7.36 (5H, m), 5.46 (1H, m), 5.24 (1H, m), 5.13 (2H, s), 4.81 (1H, d, J = 4.2 Hz), 4.46 (1H, d, J = 6.0 Hz), 1.96 (1H, m), 1.22-1.55 (18H, m), 0.95 (6H, m), 0.82 (9H, s), 0.02 (6H, m); MS APCI+ 610.3456 [M + H]+. Calcd for C31H52NO9Si: 610.3411.
t-Butyl 2-[2-(2-benzyloxycarbonylamino-3-hydroxybutyryloxy)propionyloxy]-3-methylpentanoate (9)
Ester 39 (1.02 g, 1.68 mmol) was converted using the TBAF desilylation procedure described for alcohol 29 to afford 0.83 g (100%) of alcohol 9 as a colorless oil: TLC Rf 0.46 (6:1 hexane-EtOAc); IR 1745 cm-1; 1H NMR δ 7.32 (5H, m), 5.54 (1H, d, J = 9.9 Hz), 5.16-5.23 (2H, m), 5.12 (2H, s), 4.86 (1H, d, J = 4.5 Hz), 4.58 (1H, m), 4.41 (1H, dd, J = 9.3, 3.0), 3.29 (1H, d, J = 4.2 Hz), 1.98 (1H, m), 1.21-1.60 (18H, m), 0.84-0.99 (9H, m); MS APCI+ 496.2541 [M + H]+. Calcd for C25H38NO9: 496.2547. Anal. C 60.50%, H 7.67%, N 2.66%, calcd for C25H37NO9, C 60.59%, H 7.53%, N 2.83%.
2-(t-Butyldiphenylsilyloxy)-4-methylpentanoic acid (40)
Alcohol 13 (3.00 g, 20.5 mmol), imidazole (2.79 g, 41.0 mmol), and TBDPSC1 (7.93 g, 28.8 mmol) were dissolved in DMF (30 mL under N2) and the solution stirred at ambient temperature for 18 h. The reaction was terminated with 5 M NaCl (100 mL) and extracted with EtOAc (2 × 100 mL). The extracts were combined, washed with cold 5% citric acid (50 mL), H2O (20 mL), 5 M NaCl (20 mL), dried and evaporated, and the residue coevaporated with toluene (2 × 75 mL). The residue was flash chromatographed (270 g, SiO2, 95:5 hexane-EtOAc) to afford 6.92 g (88%) of the methyl ester: TLC Rf 0.37 (95:5 hexane-EtOAc); IR 1750, 1649 cm-1; 1H NMR δ 7.67 (4H, m), 7.40 (6H, m), 4.23 (1H, dd, J = 4.2, 7.2 Hz), 3.44 (3H, s), 1.43-1.76 (3H, m), 1.09 (9H, s), 0.81 (6H, dd, J = 6.0, 16.5 Hz); 13C NMR δ 174.0, 136.0, 135.9, 133.9, 133.3, 129.73, 129.66, 127.6, 127.4, 71.5, 51.3, 44.3, 26.9, 24.1, 22.9, 22.2, 19.4.
A portion of this material (1.76 g, 4.58 mmol) was placed in 1:1 THF- CH3OH (40 mL) at 0 °C under N2 and LiOH (14 mL of 0.5 M cold solution, 7.0 mmol) was added over 20 min. The mixture was stirred at ambient temperature for 28 h, cooled to 0 °C, acidified (pH 3) with 1 M KHSO4, and extracted with EtOAc (2 × 50 mL). The combined extract was washed with H2O (20 mL), 5 M NaCl (20 mL), dried, and solvent evaporated. The residue was flash chromatographed (60 g, SiO2, 8:1 hexane-EtOAc) to provide 1.73 g (98%) of carboxylic acid 40 as a colorless oil: TLC Rf 0.67 (95:5:1 DCM-CH3OH-HOAc); IR 1721 cm-1; 1H NMR δ 7.64 (4H, m), 7.41 (6H, m), 4.26 (1H, t, J = 6.0 Hz), 1.48-1.74 (3H, m), 1.08 (9H, s), 0.69 (6H, dd, J = 6.6, 9.3 Hz).
2-(t-Butyldimethylsilyloxy)-4-methylpentanoic acid (41)
Alcohol 13 (1.06 g, 6.84 mmol) was treated with TBDMSC1 (1.61 g, 10.26 mmol) according to the procedure described for obtaining 40 and silyl ester that lead to 1.74 g (98%) of the TBDMS ether as a colorless oil: IR 1761 cm-1; 1H NMR δ 4.22 (1H, dd, J = 3.9, 8.4 Hz), 3.70 (3H, s), 1.76 (1H, m), 1.55 (2H, m), 0.93-0.98 (15H, m), 0.04 (6H, dd, J = 4.2, 18.6). A portion of this methyl ester (1.30 g, 5.0 mmol) was hydrolyzed as described for carboxylic acid 40 and that led to 1.07 g (87%) of carboxylic acid 41 as a somewhat unstable colorless oil: 1H NMR δ 4.27 (1H, dd, J = 4.2, 7.2 Hz), 1.84 (1H, m), 1.62 (2H, m), 0.82-0.95 (15H, m), 0.06-0.12 (6H, m).
2-Benzyloxycarbonylamino-2-[1-(1-t-butoxycarbonyl-2-methylbutoxycarbonyl)ethoxycarbonyl]-1-methylethyl 2-(t-butyldiphenylsilyloxy)-4-methylpentanoate (42)
Alcohol 9 (0.83 g, 1.68 mmol) was esterified with carboxylic acid 40 (0.78 g, 2.1 mmol) employing the MNBA procedure described for diester 26 to afford 1.21 g (85%) of ester 42 as a colorless oil: IR 1745 cm-1; 1H NMR δ 7.61 (4H, m), 7.35 (11H, m), 5.22 (2H, m), 5.09 (3H, m), 4.80 (1H, d, J = 4.5 Hz), 4.43 (1H, dd, J = 3.3, 9.3 Hz), 4.30 (1H, t, J = 6.1 Hz), 1.94 (1H, m), 1.24-1.69 (16H, m), 1.05 (9H, s), 0.94 (6H, m), 0.74 (6H, dd, J = 4.2, 15.3 Hz); MS APCI+ 848.4402 [M + H]+. Calcd for C47H66NO11Si: 848.4406.
2-Benzyloxycarbonylamino-2-[1-(1-t-butoxycarbonyl-2-methylbutoxycarbonyl)ethoxycarbonyl]-1-methylethyl 2-(t-butyldimethylsilyloxy)-4-methylpentanoate (43)
By applying the preceding method (cf. 26 and 42) alcohol 9 (3.17 g, 6.40 mmol) was esterified with carboxylic acid 41 (1.84 g, 7.60 mmol) using MNBA and that reaction led to 3.85 g (83%) of ester 43 as a colorless oil: TLC Rf 0.38 (6:1 hexane-EtOAc); IR 3446, 3336, 1747 cm-1; 1H NMR δ 7.35 (5H, m), 5.43 (2H, m), 5.11 (3H, m), 4.82 (1H, d, J = 4.5 Hz), 4.55 (1H, m), 4.22 (1H, m), 1.94 (1H, m), 1.24-1.59 (21H, m), 0.85-0.98 (21H, m), 0.11 (6H, m); 13C NMR δ 173.0, 169.2, 169.1, 128.5, 128.2, 128.1, 82.2, 76.9, 70.5, 69.5, 67.3, 57.6, 43.9, 36.6, 28.0, 25.7, 24.5, 23.4, 16.9, 15.3, 11.6, -4.8, -.5.5. Anal. C 61.57%, H 8.62%, N 1.87%, calcd for C37H61NO11Si, C 61.38%, H 8.49%, N 1.93%.
2-Benzyloxycarbonylamino-2-[1-(1-carboxy-2-methylbutoxycarbonyl) ethoxycarbonyl]-1-methylethyl 2-(t-butyldiphenylsilyloxy)-4-methylpentanoate (44)
To t-butyl ester 42 (1.09 g, 1.29 mmol) in DCM (5 mL) was added ZnBr2 (1.45 g, 6.43 mmol), the solution was stirred for 48 h, H2O (20 mL) was added and stirring continued for 2 h. The organic phase was separated and the aqueous phase extracted with DCM (2 × 20 mL). The organic solutions were combined, dried and evaporated to furnish 0.82 g (80%) of carboxylic acid 44 as a colorless oil: TLC Rf 0.50 (50:1 DCM-CH3OH); IR 1752 cm-1; 1H NMR δ 7.62 (4H, s), 7.30 (11H, m), 4.91-5.21 (5H, m), 4.44 (1H, m), 4.30 (2H, m), 1.98 (1H, m), 0.73-1.66 (33H, m); FAB MS 792.3786 [M + H]+. Calcd for C43H58NO11Si: 792.3780.
2-Benzyloxycarbonylamino-2-[1-(1-carboxy-2-methylbutoxycarbonyl)ethoxycarbonyl]-1-methylethyl 2-(t-butyldimethylsilyloxy)-4-methylpentanoate (8)
To t-butyl ester 43 (2.52 g, 3.10 mmol) in toluene (70 mL) was added 230-400 mesh silica gel (5 g). The mixture was heated at reflux under N2 for 6 h, allowed to cool, and diluted with 4:1 DCM-CH3OH (200 mL). The solution was filtered and the solid phase washed with 4:1 DCM-CH3OH (50 mL). The combined DCM filtrate and washings were evaporated to dryness. The residue was flash chromatographed (60g, SiO2, 50:1 DCM-CH3OH to afford 1.54 g (66%) of carboxylic acid 8 as a colorless oil: TLC Rf 0.51 (50:1 DCM-CH3OH); [α]26D -33.9 (c 1.1, CHCl3); IR 3319, 1755 cm-1; 1H NMR δ 7.34 (5H, m), 5.31 (2H, m), 4.98-5.13 (5H, m), 4.57 (1H, dd, J = 3.3, 9.9 Hz), 4.20 (2H, dd, J = 3.6, 8.7 Hz), 2.01 (1H, m), 1.33-1.76 (9H, m), 0.81-1.24 (22H, m), 0.01-0.05 (6H, m); 13C NMR δ 173.1, 169.3, 156.5, 136.0, 128.6, 128.3, 128.1, 76.2, 70.7, 70.5, 69.5, 67.4, 57.6, 43.9, 36.5, 25.7, 24.4, 24.0, 23.4, 21.5, 18.1, 16.9, 16.7, 15.3, 11.5, -4.8, -5.5. Anal. C 59.49%, H 8.32%, N 1.97%, calcd for C33H53NO11Si, C 59.35%, H 8.00%, N 2.10%.
Methyl 4-[2-(2-{2-Benzyloxycarbonylamino-3-[2-(t-butyldiphenylsilyloxy)-4-methylpentanoyloxy]butyryloxy}propionyloxy)-3-methylpentanoylamino]-6-methyl-3-oxoheptanoate (45)
Boc-protected ketone 7 (0.287 g, 1.0 mmol) was deprotected (TFA-DCM) and allowed to react with carboxylic acid 44 (0.640 g, 0.81 mmol) were reacted employing the PyBroP mediated amide formation procedure described for 22 to afford 0.375 g (52%) of amide 45 as a colorless oil: tlc Rf 0.45 (80:20 hexane-EtOAc); IR 3367, 1755, 1682 cm-1; 1H NMR δ 7.59 (4H, m), 7.33 (11H, m), 6.70 (1H, d, J = 7.8 Hz), 5.00-5.13 (6H, m), 4.70 (1H, m), 4.41 (1H, m), 4.27 (1H, m), 3.69 (3H, d, J = 3.0 Hz), 3.52 (2H, d, J = 2.7 Hz); 13C NMR δ 201.7, 172.4, 169.7, 168.7, 167.3, 156.3, 136.0, 135.7, 133.1, 129.9, 128.6, 128.3, 128.1, 127.7, 78.6, 71.7, 70.7, 70.1, 67.3, 57.9, 56.4, 52.4, 46.0, 44.5, 39.2, 37.0, 29.7, 26.8, 24.8, 24.2, 24.1, 23.3, 22.9, 22.3, 21.4, 19.4, 16.9, 14.9, 11.4; FAB MS 961.4854 [M + H]+. Calcd for C52H73N2O13Si: 961.4882. Anal. C 64.76%, H 7.77%, N 2.72%, calcd for C52H72N2O13Si, C 64.98%, H 7.55%, N 2.91%.
Methyl 4-[2-(2-{2-benzyloxycarbonylamino-3-[2-(t-butylmethylsilyloxy)-4-methylpentanoyloxy]butyryloxy}propionyloxy)-3-methylpentanoylamino]-6-methyl-3-oxoheptanoate (46)
Ketone 7 (1.16 g, 4.00 mmol) following cleavage of the Boc group and carboxylic acid 8 (2.25 g, 3.37 mmol) were coupled by PyBroP promoted amide formation as described for amide 22 to supply 1.83 g (65%) of amide 46 as a colorless oil: TLC Rf 0.46 (80:20 hexane-EtOAc); [α]25D -38.5 (c 0.98, CHCl3); IR 3359, 1753, 1682 cm-1; 1H NMR δ 7.35 (5H, m), 6.70 (1H, d, J = 7.8 Hz), 5.43 (2H, d, J = 9.3 Hz), 5.13 (4H, m), 4.68 (1H, m), 4.54 (1H, m), 4.18 (1H, dd, J = 3.6, 9.3 Hz), 3.70 (3H, t, J = 3.0 Hz), 3.52 (2H, s), 2.00 (1H, s), 1.27-1.74 (18H, m), 0.88-0.94 (24H, m), 0.02 (6H, m). Anal. C 60.40%, H 8.52%, N 3.31%, calcd for C42H68N2O13Si, C 60.26%, H 8.19%, N 3.35%.
Methyl 4-(2-{2-[2-Benzyloxycarbonylamino-3-(2-hydroxy-4-methylpentanoloxy)butyryloxy]propionyloxy}-3-methylpentanoylamino)-6-methyl-3-oxoheptanoate (6)
To amide 46 (0.367 g, 0.44 mmol) in CH3OH (5 mL) under N2 at 0 °C was added acetyl chloride (50 μL, 69.5 mg, 0.88 mmol). The solution was stirred at ambient temperature for 30 min, diluted with DCM (40 mL), washed with 6% NaHCO3 (20 mL) and H2O (10 mL), dried and evaporated. The residue was flash chromatographed (10 g, SiO2, 2:1 hexane-EtOAc) to afford 0.198 g (63%) of epimer 6 as a colorless oil: TLC Rf 0.40 (50:50 hexane-EtOAc); [α]25D -30.4 (c 0.92, CHCl3); 1H NMR δ 7.37 (5H, m), 6.63 (1H, d, J = 8.4 Hz), 5.45 (2H, m), 5.53 (2H, s), 5.03 (1H, d, J = 4.8 Hz), 4.65 (2H, m), 4.10 (1H, m), 3.72 (3H, d, J = 2.7 Hz), 3.50 (2H, m), 1.86-2.02 (2H, m), 0.68-1.83 (33H, m); 13C NMR δ 201.7, 174.8, 169.3, 169.2, 168.6, 135.8, 128.6, 128.4, 128.2, 79.0, 78.8, 71.2, 69.9, 69.0, 68.9, 67.5, 57.5, 56.5, 56.4, 46.1, 42.8, 39.7, 36.8, 29.7, 24.9, 24.3, 24.3, 23.2, 21.5, 16.7, 14.8, 11.2; FAB MS 723.3735 [M + H]+. Calcd for C36H55N2O13: 723.3704. Anal. C 59.64%, H 7.81%, N 3.79%, calcd for C36H54N2O13, C 59.82%, H 7.53%, N 3.88%.
Benzyl (5-s-butyl-8,13-diisobutyl-2,16-dimethyl-3,6,9,11,14,18-hexaoxo-1,4,12,15-tetraoxa-7-azacyclooctadec-17-yl)carbamate (5)
A mixture of alcohol 6 (0.12 g, 0.17 mmol) and anhydrous CuSO4 (0.60 g, 3.75 mmol) in toluene (150 mL, under N2) was stirred at 120 °C for 12 h. The mixture was allowed to cool, the mixture filtered and the solvent evaporated. The residue was flash chromatographed (10 g, SiO2, to afford 92 mg (80%) of lactone 5 as a colorless solid: TLC Rf 0.61 (75:25 hexane-EtOAc); [α]25D +13.5 (c 0.68, CHCl3); IR 3336, 1735, 1717, 1684 cm-1; 1H NMR δ 7.46 (1H, d, J = 8.7 Hz), 7.38 (5H, m), 5.91 (1H, m), 5.70 (1H, dd, J = 7.2, 13.8 Hz), 5.54 (1H, d, J = 9.6 Hz), 5.20 (3H, m), 4.82 (1H, d, J = 9.0 Hz), 4.71 (3H, m), 3.45 (1H, d, J = 15.9 Hz), 3.23 (1H, d, J = 15.6 Hz), 2.03 (1H, m), 1.42-1.86 (10H, m), 1.36 (6H, d, J = 6.9 Hz), 0.87-1.02 (15H, m); 13C NMR δ 204.5, 171.7, 170.1, 169.8, 167.7, 166.4, 156.7, 135.8, 128.6, 128.4, 128.2, 81.2, 72.4, 72.2, 71.3, 67.6, 57.8, 57.2, 47.0, 41.2, 39.2, 36.7, 25.3, 24.9, 24.4, 23.5, 22.8, 21.8, 20.8, 18.5, 16.4, 14.4, 10.6; FAB MS 691.3450 [M + H]+. Calcd for C35H51N2O12: 691.3442. Anal. C 61.23%, H 7.30%, N 4.06%, calcd for C35H50N2O12, C 60.86%, H 7.30%, N 4.06%.
2-Hydroxy-3-formylaminobenzoic acid (49)
Aniline 48 (0.51 g, 3.31 mmol) was suspended in formamide (3.0 mL under N2) and the mixture stirred at 150 °C for 0.5 h. The resulting solution was allowed to cool, dissolved in 6% NaHCO3 (50 mL), acidified with 1 M KHSO4, and extracted with EtOAc (3 × 50 mL). The combined extract was washed with 5 M NaCl (10 mL), dried, evaporated, and the residue coevaporated with toluene (10 mL) to furnish 90% of phenol 49 as a greenish gray solid: mp 168-169 °C; TLC Rf 0.20 (95:5:1 DCM-MeOH-HOAc); 1H NMR (d6-DMSO) δ 9.82 (1H, s), 8.38 (1H, d, J = 9.3 Hz), 7.55 (1H, d, J = 7.7 Hz), 6.92 (1H, t, J = 7.7 Hz); 13C NMR (d6-DMSO) δ 172.3, 160.3, 151.2, 126.5, 125.7, 124.5, 118.6, 112.6.
Methyl 2-hydroxy 3-formylaminobenzoate (50)
Benzoic acid derivative 49 (1.37 g, 7.57 mmol) and NaHCO3 (1.40 g, 16.65 mmol) were placed in DMF (20 mL) under N2. MeI (5.37 g, 2.36 mL, 37.85 mmol) in DMF (20 mL) was added and the mixture stirred at ambient temperature for 15 h. The mixture was diluted with EtOAc (250 mL), washed with H2O (50 mL), 6% NaHCO3 (50 mL), H2O (20 mL), 5 M NaCl (20 mL), dried, solvent evaporated and the residue coevaporated with toluene (50 mL). The residue was flash chromatographed (36 g, SiO2, 70:30 hexane-EtOAc) to supply 1.17 g (79%) of ester 50 as an off-white solid: mp 99 °C; TLC Rf 0.66 (95:5 DCM-MeOH); IR 3248, 1693, 1651 cm-1; 1H NMR δ 11.29 and 11.18 (1H, 2 s), 8.76 and 8.56 (1H, 2 dd, J = 7.9, 1.7 Hz), 8.51 (1H, d, J = 1.7 Hz), 7.97 (1H, br s), 7.65 and 7.57 (1H, 2 dd, J = 8.2, 1.6 Hz), 6.90 and 6.88 (1, 2 t, J = 8.2 Hz), 3.97 (3H, s); 13C NMR δ 170.8, 170.4, 161.2, 158.9, 151.2, 150.3, 126.5, 125.8, 125.4, 124.3, 121.7, 119.2, 113.1, 111.9, 52.7, 52.6.
Methyl 2-benzyloxy-3-formylaminobenzoate (51)
To methyl ester 50 (1.01 g, 5.20 mmol) and benzyl bromide (1.44 g, 1 mL, 8.42 mmol) in DMF (20 mL under N2) was added K2CO3 (1.44 g, 10.40 mmol) and the mixture stirred at 60 °C for 15 h. The mixture was diluted with EtOAc (100 mL), washed with H2O (2 × 20 mL), 5 M NaCl (10 mL), dried, solvent evaporated and the residue coevaporated with toluene (20 mL). The residue was separated by flash chromatography (50 g, SiO2, 80:20 hexane-EtOAc) to afford 1.41 g (95%) of benzyl ester 51 as a pinkish oil which solidified on standing. A portion was recrystallized from toluene-hexane: mp 52-53 °C; TLC Rf 0.59 (50:50 hexane-EtOAc); IR 3284, 1720, 1674 cm-1; 1H NMR δ 8.53 (1H, dd, J = 8.3, 1.6 Hz), 8.19 (1H, d, J = 1.7 Hz), 7.65 (2H, dd, J = 8.2, 1.6 Hz), 7.41 (5H, m), 7.18 (1H, t, J = 8.3 Hz), 5.03 (2H, s), 3.93 (3H, s); 13C NMR δ 165.7, 158.7, 147.7, 136.4, 132.0, 128.9, 128.9, 128.5, 126.7, 124.9, 124.4, 77.8, 52.4. Anal. C 67.51%, H 5.42%, N 4.88%, calcd for C16H15NO4, C 67.36%, H 5.30%, N 4.91%.
2-Benzyloxy-3-formylaminobenzoic acid (3)
To methyl ester 51 (1.28 g, 4.48 mmol) in 3:1 THF-MeOH (20 mL) under N2 was added LiOH (11.6 mL of 0.5 M aqueous solution, 5.8 mmol) and the mixture stirred at ambient temperature for 18 h. The reaction mixture was acidified (pH 3) with 1 M KHSO4, diluted with H2O (200 mL), and extracted with EtOAc (3 × 65 mL). The combined extract was washed with H2O (10 mL), and 5 M NaCl (20 mL), dried, and evaporated. The residue was crystallized from EtOAc-hexane to afford 0.96 g (79%) of benzoic acid 3 as an off-white solid: mp 133 °C; TLC Rf 0.51 (95:5:1 DCM-CH3OH-HOAc); IR 3343, 1697, 1636 cm-1; 1H NMR (d6-DMSO) δ 13.09 (1H, s), 9.76 (1H, s), 8.34 (1H, s), 8.30 (1H, d, J = 8.2 Hz), 7.25-7.60 (6H, m), 7.18 (1H, t, J = 7.7 Hz), 4.95 (2H, s); 13C NMR (d6-DMSO) δ 167.0, 160.5, 147.3, 136.8, 132.3, 128.4, 128.0, 127.9, 126.4, 125.6, 124.7, 124.0, 75.7. Anal. C 65.84%, H 4.91%, N 5.06%, calcd for C15H13NO4·0.1 H2O, C 65.97%, H 4.88%, N 5.12%.
Benzyl (5-s-butyl-8,13-diisobutyl-2,10,10,16-tetramethyl-3,6,9,11,14,18-hexaoxo-1,4,12,15-tetraoxa-7-azacyclooctadec-17-yl)carbamate (52)
To a stirred solution of lactone 5 (76 mg, 0.11 mmol) in DMSO (3 mL) under N2 were added K2CO3 (153 mg, 1.01 mmol) and MeI (20 μL, 0.33 mmol). The mixture was stirred at ambient temperature for 3 h, diluted with H2O (12 mL), and extracted with EtOAc (3 × 20 mL). The extracts were combined, washed with H2O (10 mL), and 5 M NaCl (5 mL), dried and evaporated. The residue was flash chromatographed (10 g, SiO2, 3:1 hexane-EtOAc) to afford 22 mg (28%) of Cbz-protected lactone 52 as an oil: TLC Rf 0.55 (75:25 hexane-EtOAc); [α]25D -17.7 (c 0.90, CHCl3); IR 3330, 1738, 1718 cm-1; 1H NMR δ 7.50 (1H, d, J = 9.3 Hz), 7.37 (5H, m), 5.91 (1H, m), 5.80 (1H, dd, J = 6.6, 13.8 Hz), 5.56 (1H, d, J = 9.0 Hz), 5.18 (2H, dd, J = 12.0, 17.1 Hz), 4.86 (2H, m), 4.72 (1H, d, J = 9.0 Hz), 4.59 (1H, dd, J = 4.2, 10.2 Hz), 2.06 (1H, m), 1.13-1.83 (20H, m), 0.77-0.98 (19H, m); 13C NMR δ 208.3, 173.1, 171.8, 170.0, 169.7, 167.8, 156.6, 135.8, 128.7, 128.5, 128.2, 80.8, 72.3, 71.9, 71.1, 67.6, 57.7, 56.4, 53.0, 43.0, 39.4, 36.6, 31.9; MS APCI+ 719.3767 [M + H]+. Calcd for C37H55N2O12: 719.3755.
17-Amino-5-s-butyl-8,13-diisobutyl-2,10,10,16-tetramethyl-1,4,12,15-tetraoxa-7-azacyclooctadecane-3,6,9,11,14,18-hexaone (4)
Benzyl carbamate 52 (16 mg, 0.022 mmol) and 10% Pd/C (15 mg) in EtOAc (3 mL) was stirred under a H2 atmosphere at ambient temperature for 2 h. The solution phase was filtered through Celite and the solid phase washed with CH3OH (20 mL). The combined solvent filtrate and washings were evaporated and the residue flash chromatographed (10 g, SiO2, 3:1 hexane-EtOAc) to furnish 9.5 mg (73%) of amine 4 as a colorless oil: TLC Rf 0.40 (75:25 EtOAc-hexane); [α]27D -55.1 (c 0.67, CHCl3); IR 3222, 1749, 1712, 1686 cm-1; 1H NMR δ 7.60 (1H, d, J = 9.3 Hz), 5.90 (1H, d, J = 6.6 Hz), 5.78 (1H, dd, J = 6.3, 13.5 Hz), 4.86 (2H, m), 4.63 (1H, dd, J = 4.2, 9.9 Hz), 3.62 (1H, br s), 2.08 (1H, m), 1.26-1.82 (22H, m), 0.86-1.18 (18H, m); 13C NMR δ 208.5, 173.1, 172.1, 171.7, 170.2, 170.1, 80.7, 72.8, 72.1, 70.6, 58.3, 56.5, 53.1, 43.0, 39.5, 36.6, 29.7, 25.3, 24.7, 24.5, 24.0, 23.6, 22.9, 21.4, 21.1, 19.8, 18.2, 16.5, 14.5, 10.5; FAB MS 585.3360 [M + H]+. Calcd for C29H49N2O10: 585.3387.
2-Benzyloxy-N-(5-s-butyl-8,13-diisobutyl-2,10,10,16-tetramethyl-3,6,9,11,14,18-hexaoxo-1,4,12,15-tetraoxa-7-azacyclooctadec-17-y1)-3-formylaminobenzamide (53)
Benzoic acid 3 (14.0 mg, 0.051 mmol), 1-hydroxybenzotriazole (7.0 mg, 0.051 mmol), EDCI (7.4 mg, 0.038 mmol), and N-methylmorpholine (20μL, 0.18 mmol) were added successively to a solution of amine 4 (15.0 mg, 0.026 mmol) in DMF (1.5 mL) under N2. The reaction mixture was stirred at ambient temperature for 11 h, the reaction was terminated by addition of saturated NaHSO4 (20 mL), and extracted with EtOAc (30 mL). The extract was dried, evaporated, and the residue flash chromatographed (10 g, SiO2, 2.2:1 hexane-EtOAc) to provide 13 mg (61%) of amide 53 as a colorless oil: TLC Rf 0.42 (2:1 hexane-EtOAc); [α]25D -45.7 (c 0.65, CHCl3); IR 3321, 1745, 1678 cm-1; 1H NMR δ 8.45 (1H, d, J = 8.1 Hz), 8.20 (1H, d, J = 9.3 Hz), 8.10 (1H, s), 7.79 (1H, d, J = 6.0 Hz), 7.52 (1H, d, J = 9.3 Hz), 7.26-7.38 (7H, m), 6.05 (1H, d, J = 6.6 Hz), 5.86 (1H, dd, J = 8.2, 13.8 Hz), 5.45 (1H, d, J = 11.7), 5.36 (1H, m), 4.88 (3H, m), 4.56 (1H, d, J = 9.9 Hz), 2.10 (1H, m), 1.47-1.85 (8H, m), 1.13-1.42 (12H, m), 0.75-1.02 (18H, m); 13C NMR δ 208.2, 173.3, 171.8, 170.0, 169.8, 167.9, 165.8, 146.0, 135.3, 131.5, 129.5, 129.2, 129.1, 126.5, 126.2, 125.5, 124.9, 80.9, 79.1, 72.6, 72.0, 71.2, 56.4, 55.9, 53.1, 43.1, 39.5, 36.6, 25.3, 24.6, 24.4, 24.1, 21.0, 19.8, 18.3, 16.6, 14.5, 10.4; MS APCI+ 838.4131 [M + H]+. Calcd for C44H60N3O13: 838.4126.
N-(5-s-Butyl-8,13-diisobutyl-2,10,10,16-tetramethyl-3,6,9,11,14,18-hexaoxo-1,4,12,15-tetraoxa-7-azacyclooctadec-17-yl)-3-formylamino-2-hydroxybenzamide (Respirantin 1b)
Amide 53 (15 mg, 0.018 mmol) and 10% Pd/C (17 mg) in EtOAc (3 mL) was stirred under a H2 atmosphere at ambient temperature for 2 h. The solution phase was filtered through Celite and the solid phase washed with 1:1 EtOAc-CH3OH (20 mL). The combined solvent filtrate and washings was evaporated and the residue flash chromatographed (10 g, SiO2, 1:1 hexane-EtOAc) to afford 11 mg (82%) of 1b as a glassy solid: TLC Rf 0.38 (50:50 hexane-EtOAc); [α]25D -6.0 (c 0.53, CH3OH); IR 3325, 1749, 1708, 1687 cm-1; 1H NMR δ 12.51 (1H, br), 8.58 (1H, d, J = 8.0 Hz), 8.52 (1H, d, J = 1.5 Hz), 7.94 (1H, s), 7.47 (1H, d, J = 9.5 Hz), 7.36 (1H, d, J = 9.5 Hz), 7.15 (1H, d, J = 9.0 Hz), 6.97 (1H, t, J = 8.0 Hz), 6.03 (1H, dd, J = 2.7, 6.6 Hz), 5.86 (1H, q, J = 6.8 Hz), 5.21 (1H, dd, J = 2.7, 8.7 Hz), 4.94 (1H, ddd, J = 3.8, 9.9, 11.0 Hz), 4.86 (1H, d, J = 9.6 Hz), 4.68 (1H, dd, J = 4.5, 9.9 Hz), 2.12 (1H, m), 1.50-1.90 (10H, m), 1.24-1.44 (12H, m), 0.90-1.02 (16H, m); 13C NMR δ 208.1, 173.4, 171.8, 170.4, 169.9, 169.5, 167.5, 159.0, 150.6, 127.5, 125.0, 120.3, 119.1, 112.8, 80.9, 72.3, 72.0, 71.5, 56.4, 55.6, 53.0, 43.1, 39.4, 36.5, 25.3, 24.7, 24.5, 24.1, 23.6, 22.8, 21.4, 21.0, 19.8, 18.2, 16.6, 14.4, 10.4; MS APCI+ 748.3631 [M + H]+. Calcd for C37H54N3O13: 748.3657.
Acknowledgment
We are pleased to acknowledge financial support provided by Outstanding Investigator Grant CA44344-10-12 and RO1 CA90441-01-05 awarded by the Division of Cancer Treatment and Diagnosis, National Cancer Institute, DHHS; the Arizona Disease Control Research Commission; the Robert B. Dalton Endowment Fund; Dr. Alec D. Keith; Dr. William Crisp; Mrs. Anita Crisp; Gary L. and Diane R. Tooker; Dr. John C. Budzinski. Other helpful assistance was provided by Drs. D. L. Doubek, F. Hogan, V. J. R. V. Mukku, and J. Chapuis, and L. Williams. We thank Dr. Zbigniew A. Cichacz for helpful discussions. We also thank Daniel Jensen for technical support in the preparation of starting materials and intermediates.
References and Notes
- 1(a).Contributon 561 of the series Antineoplastic Agents. For the preceding part, see:Pettit GR, Tan R, Pettit RK, Smith TH, Feng S, Doubek DL. J. Nat. Prod. in press.Urushibata I, Isogai A, Matsumoto S, Suzuki A. J. Antibiotics. 1993;46:701–703. doi: 10.7164/antibiotics.46.701.
- 2.Nishii T, Suzuki S, Yoshida K, Tsunoda T. Tetrahedron Lett. 2003;44:7829–7832. [Google Scholar]
- 3.Cagliotti L, Misiti D, Mondelli R, Selva A, Arcamone F, Cassinelli G. Tetrahedron. 1969;25:2193–2221. doi: 10.1016/s0040-4020(01)82768-x. [DOI] [PubMed] [Google Scholar]
- 4.Mansour TS, Evans CA. Syn. Commun. 1990;20:773–781. [Google Scholar]
- 5.Hoffman RV, Tao J. J. Org. Chem. 1997;62:2292–2297. doi: 10.1021/jo961836g. [DOI] [PubMed] [Google Scholar]
- 6.Campbell DS, Lawrie CW. J. Chem. Soc., Chem. Commun. 1971:355–356. [Google Scholar]
- 7.Witzeman JS. Tetrahedron Lett. 1990;31:1401–1404. [Google Scholar]
- 8.Christoffers J, Onal N. Eur. J. Org. Chem. 2000:1633–1635. [Google Scholar]
- 9.Bandgar BP, Sadavarte VS, Uppalla LS. J. Chem. Research (S) 2001:16–17. [Google Scholar]
- 10.Williams DR, Myers BJ, Mi L. Org. Lett. 2000;2:945–948. doi: 10.1021/ol0000197. [DOI] [PubMed] [Google Scholar]
- 11.Wissner A, Grudzinskas CV. J. Org. Chem. 1978;43:3972–3974. [Google Scholar]
- 12.Less SL, Handa S, Millburn K, Leadlay PF, Dutton CJ, Staunton J. Tetrahedron Lett. 1996;37:3515–3518. [Google Scholar]
- 13.McMurry JE, Wong GB. Syn. Commun. 1972;2:389–394. [Google Scholar]
- 14.Fehrentz J-A, Castro B. Synthesis. 1983:676–678. [Google Scholar]
- 15.Coste J, Frerot E, Jouin P. J. Org. Chem. 1994;59:2437–2446. [Google Scholar]
- 16.Yamada S, Kasai Y, Shioro T. Tetrahedron Lett. 1973:1595–1598. [Google Scholar]
- 17.Kelly DR, Roberts SM. Syn. Commun. 1979;9:295–299. [Google Scholar]
- 18.Kurokawa N, Ohfune Y. Tetrahedron. 1993;49:6195–6222. [Google Scholar]
- 19.Shiina I, Kubota M, Oshiumi H, Hashizume M. J. Org. Chem. 2004;69:1822–1830. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]
- 20.Vidya R, Eggen M, Nair SK, Georg GI, Himes RH. J. Org. Chem. 2003;68:9687–9693. doi: 10.1021/jo0302197. [DOI] [PubMed] [Google Scholar]
- 21.Golebiowski A, Jurczak J. Tetrahedron. 1991;47:1037–1044. [Google Scholar]
- 22.Yang D, Li B, Ng F-F, Yan Y-L, Qu J, Wu Y-D. J. Org. Chem. 2001;66:7303–7312. doi: 10.1021/jo010376a. [DOI] [PubMed] [Google Scholar]
- 23.Wu Y, Limburg DC, Wilkinson DE, Vaal MJ, Hamilton GS. Tetrahedron Lett. 2000;41:2847–2849. [Google Scholar]
- 24.Jackson RW. Tetrahedron Lett. 2001;42:5163–5165. [Google Scholar]
- 25.Khan AT, Mondal E. Synlett. 2003:694–698. [Google Scholar]
- 26.Bandgar BP, Sadavarte VS, Uppala LS. Syn. Commun. 2001;31:2063–2066. [Google Scholar]
- 27.Skibo EK, Gilchrist JH. J. Org. Chem. 1988;53:4209–4218. [Google Scholar]
- 28.Bocchi V, Casnati G, Dossena A, Marchelli R. Synthesis. 1979:961–962. [Google Scholar]
- 29.Venuti MC, Loe BE, Jones GH, Young JM. J. Med. Chem. 1988;31:2132–2136. doi: 10.1021/jm00119a013. [DOI] [PubMed] [Google Scholar]
- 30.Suffness M, Douros J. Drugs of Plant Origin. In: DeVita VT, Bush H, editors. Methods in Cancer Research. Academic Press; 1979. pp. 73–126. [Google Scholar]
- 31.Monks A, Scudiero D, Skehan P, Shoemaker R, Paul K, Vestica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A. J. Natl. Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]