

Abstract
The mode of action of Δlac-acetogenins, strong inhibitors of bovine heart mitochondrial complex I, is different from that of traditional inhibitors such as rotenone and piericidin A [Murai et al. (2007) Biochemistry 46, 6409−6416]. As further exploration of these unique inhibitors might provide new insights into the terminal electron transfer step of complex I, we drastically modified the structure of Δlac-acetogenins and characterized their inhibitory action. In particular, on the basis of structural similarity between the bis-THF and the piperazine rings, we here synthesized a series of piperazine derivatives. Some of the derivatives exhibited very potent inhibition at nanomolar levels. The hydrophobicity of the side chains and their balance were important structural factors for the inhibition, as is the case for the original Δlac-acetogenins. However, unlike in the case of the original Δlac-acetogenins: (i) the presence of two hydroxy groups is not crucial for the activity, (ii) the level of superoxide production induced by the piperazines is relatively high, (iii) the inhibitory potency for the reverse electron transfer is remarkably weaker than that for the forward event, and (iv) the piperazines efficiently suppressed the specific binding of a photoaffinity probe of natural-type acetogenins ([125I]TDA) to the ND1 subunit. It is therefore concluded that the action mechanism of the piperazine series differs from that of the original Δlac-acetogenins. Photoaffinity labeling study using a newly synthesized photoreactive piperazine ([125I]AFP) revealed that this compound binds to the 49 kDa subunit and an unidentified subunit, not ND1, with a frequency of about 1:3. A variety of traditional complex I inhibitors as well as Δlac-acetogenins suppressed the specific binding of [125I]AFP to the subunits. The apparent competitive behavior of inhibitors that seem to bind to different sites may be due to structural changes at the binding site, rather than occupying the same site. The meaning of the occurrence of diverse inhibitors exhibiting different mechanisms of action is discussed in the light of the functionality of the membrane arm of complex I.
NADH-ubiquinone oxidoreductase (complex I)1 is the first energy-transducing enzyme of the respiratory chains of most mitochondria and many bacteria. The enzyme catalyzes the transfer of two electrons from NADH to ubiquinone, coupled to the translocation of four protons across the inner mitochondrial membrane or bacterial cytosolic membrane (1). The generated electrochemical proton gradient drives energy-consuming processes such as ATP synthesis and flagella movement (1). Complex I is the most complicated multisubunits enzyme in the respiratory chain; e.g., the enzyme from bovine heart mitochondria is composed of 45 different subunits with a total molecular mass of about 1 MDa (2). Recently, the crystal structure of the hydrophilic domain (peripheral arm) of complex I from Thermus thermophilus was solved at 3.3 angstroms resolution, revealing the subunit arrangement and the putative electron transfer pathway (3). However, our knowledge about the functional and structural features of the membrane arm, such as the ubiquinone redox reaction, proton translocation mechanism, and action mechanism of numerous specific inhibitors, is still highly limited (4-6).
Many structurally diverse inhibitors of complex I are known (7-9). With the exception of a few inhibitors that inhibit electron input into complex I (10, 11), all inhibitors are thought to act at the terminal electron transfer step of the enzyme (7, 12). Although these inhibitors are generally believed to act at the ubiquinone reduction site, there is still no hard experimental evidence to verify this possibility. Rather, a photoaffinity labeling study using azidoquinone suggested that the inhibitor binding site is not the same as the ubiquinone binding site (13, 14). On the other hand, photoaffinity-labeling studies with photoreactive derivatives of specific complex I inhibitors (15-19) strongly suggested that a wide variety of inhibitors share a common large binding domain with partially overlapping sites and that the PSST, which is located at the junction of the peripheral and membrane arms (20, 21), ND1, and ND5 subunits may be close to each other and construct a common inhibitor binding domain. It remains, however, to be learned how the binding positions of chemically diverse inhibitors relate to each other.
Acetogenins isolated from the plant family Annonaceae, such as bullatacin (Figure 1), are among the most potent inhibitors of bovine heart mitochondrial complex I (7-9). On the basis of the structure-activity relationship studies of a series of natural and synthetic acetogenins, we have revealed essential structural features of the inhibitors required for the inhibition (22-24). In the course of synthetic study of diverse acetogenin analogs, we synthesized unique mimics by deleting a γ-lactone ring (surrounded by a dashed square in Figure 1), which is a structural feature common to a large number of natural acetogenins, and named the product Δlac-acetogenins (e.g. compounds 1−3) (25). Some Δlac-acetogenins exhibited very potent inhibition of bovine complex I at the nanomolar level (Figure 1). An electron paramagnetic resonance (EPR) spectroscopic study on the redox state of iron-sulfur clusters indicated that the inhibition site of Δlac-acetogenins is downstream of the iron-sulfur cluster N2 (25), as is the case for other ordinary complex I inhibitors such as rotenone and piericidin A. However, the inhibition manner of Δlac-acetogenins differs from that of traditional inhibitors including natural acetogenins in several ways, for instance: i) the inhibitory effect of Δlac-acetogenins on the reverse electron transfer (ubiquinol-NAD+ oxidoreductase activity) in complex I is much weaker than that on the forward event (NADH-ubiquinone oxidoreductase activity) (26) and ii) the level of superoxide production from complex I induced by Δlac-acetogenins is markedly lower than that induced by ordinary inhibitors (26, 27). Using a photoreactive acetogenin derivative, [125I]TDA {[125I](trifluoromethyl)phenyldiazirinyl acetogenin}, we recently showed that various traditional inhibitors suppress the specific binding of [125I]TDA to the ND1 subunit, whereas Δlac-acetogenins do not (19), indicating that Δlac-acetogenins don't share a common binding domain with traditional inhibitors. Taken together, it was unequivocally revealed that Δlac-acetogenins are a new type of inhibitor acting at the terminal electron transfer step of bovine complex I. In view of the proposed dynamic function of the membrane domain (28, 29), it may not be strange that there are diverse chemicals that disturb the function of the membrane domain differently depending on their structural properties.
Figure 1.
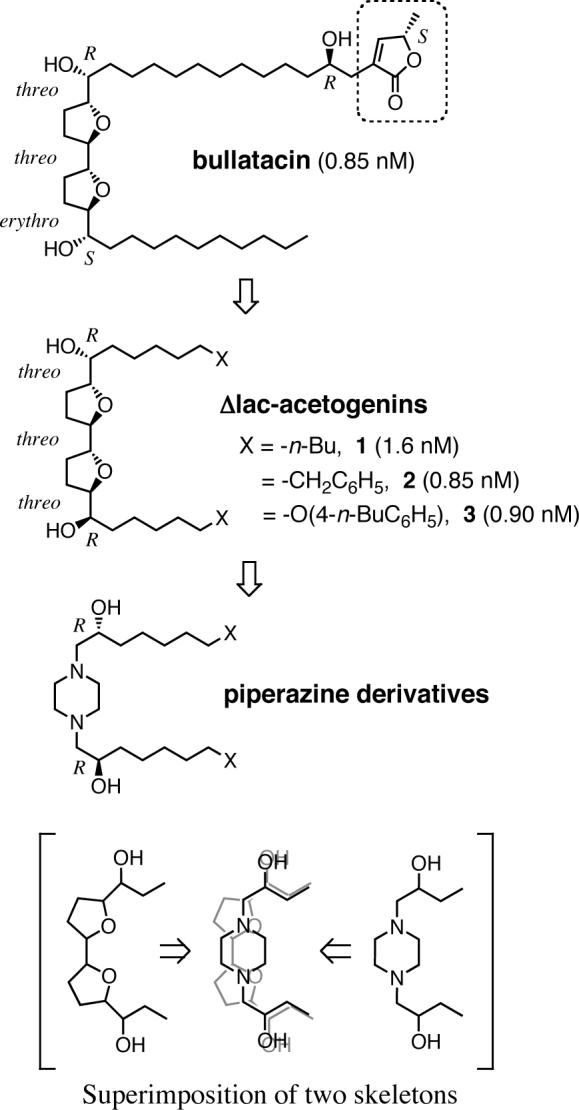
Structures of bullatacin, Δlac-acetogenins (1−3), and piperazine derivatives. The figures in parentheses are IC50 values (nM); cf. Figure 2. Superimposition of the hydroxylated bis-THF and the hydroxylated piperazine skeletons is shown at the bottom.
Since Δlac-acetogenins are a new type of inhibitor of bovine complex I, further exploration of the action manner of these inhibitors might provide new insights into the terminal electron transfer step of the enzyme. To this end, we carried out drastic structural modifications of Δlac-acetogenins to find more unique inhibitors. In particular, on the basis of the structural similarity between the hydroxylated bis-tetrahydrofuran (bis-THF) and piperazine rings, we synthesized a series of piperazine derivatives by replacing the bis-THF ring of Δlac-acetogenin with a piperazine ring, as illustrated in Figure 1. Our results clearly showed that the action manner of the piperazine series differs from that of the original Δlac-acetogenins and that the piperazines are also a new type of inhibitor of complex I. A photoaffinity labeling study using a newly synthesized photoreactive piperazine (4-azido-2,3,5,6-tetrafluorobenzyloxyalkyl piperazine, [125I]AFP) revealed that this compound binds to the 49 kDa subunit and an unidentified subunit with a frequency of about 1:3, but prevents the specific binding of [125I]TDA to the ND1 subunit. Our results strongly suggest that two inhibitors, each of which binds to different sites in complex I, still apparently compete against each other. It is difficult to explain this competitive inhibition based upon the concept that two inhibitors bind to the same site. The binding of one inhibitor to the enzyme may cause structural changes of the binding site for the other inhibitor, rather than simply occupying the same site.
EXPERIMENTAL PROCEDURES
Materials
The series of piperazine-type inhibitors synthesized in this study are shown in Figure 2. The procedures used to produce these compounds (4−23) and a photoreactive piperazine derivative ([125I]AFP) are described in the Supporting Information. The photoreactive acetogenin derivative ([125I]TDA) used for the photoaffinity labeling of the ND1 subunit was synthesized according to a previous procedure (19). Compounds 1−3 are the same samples as used previously (26). Piericidin A and bullatacin were generous gifts from Drs. S. Yoshida (The Institute of Physical and Chemical Research, Saitama, Japan) and J. L. McLaughlin (Purdue University, West Lafayette, USA), respectively.
Figure 2.

Structures of the piperazine derivatives synthesized in this study. The figures in parentheses are IC50 values, which is the molar concentration (nM) needed to reduce the control NADH oxidase activity (0.71 ± 0.06 μmol NADH/min/mg of protein) in SMP by half. Values are means ± SD of three independent experiments.
Measurement of Complex I Activity
Bovine heart submitochondrial particles (SMP) were prepared by the method of Matsuno-Yagi and Hatefi (30) using a sonication medium containing 0.25 M sucrose, 1 mM succinate, 1.5 mM ATP, 10 mM MgCl2, 10 mM MnCl2, and 10 mM Tris-HCl (pH 7.4), and stored in a buffer containing 0.25 M sucrose and 10 mM Tris-HCl (pH 7.4) at −84°C. The NADH oxidase activity in SMP was followed spectrometrically with a Shimadzu UV-3000 (340 nm, ε = 6.2 mM−1cm−1) at 25°C. The reaction medium (2.5 mL) contained 0.25 M sucrose, 1 mM MgCl2, and 50 mM phosphate buffer (pH 7.5). The final mitochondrial protein concentration was 30 μg of protein/mL. The reaction was started by adding 50 μM NADH after the equilibration of SMP with an inhibitor for 4 min.
Measurement of Superoxide Production
Superoxide production was determined at 25°C by monitoring the superoxide-dependent oxidation of epinephrine to adrenochrome (31) with a Shimadzu UV-3000 spectrophotometer (485−575 nm, ε = 3.0 mM−1cm−1) in a dual wavelengths mode. The reaction medium (2.5 mL) contained 0.25 M sucrose, 1 mM epinephrine, 1 mM EDTA, 1 μM catalase, and 10 mM Tris-HCl buffer (pH 7.5). The reaction was started by adding 100 μM NADH after the equilibration of SMP with the test inhibitor for 4 min. The final protein concentration of SMP was 0.25 mg/mL. Superoxide dismutase (bovine liver, Sigma) was used at a final concentration of 60 units/mL to give the assay specificity.
Measurement of Reverse Electron Transfer
Reverse electron transfer (ubiquinol-NAD+ oxidoreductase activity) was generated by the oxidation of succinate and the hydrolysis of ATP (32). The reaction was measured spectrometrically by following the reduction of NAD+ with a Shimadzu UV-3000 (340 nm, ε = 6.2 mM−1cm−1) at 25 °C. The reaction medium (2.5 mL) contained 0.25 M sucrose, 7 mM sodium succinate, 6 mM MgCl2, 1 mM KCN, 1 mM NAD+ and 50 mM Tris-HCl (pH 7.5), and the final protein concentration of SMP was 0.1 mg of protein/mL. The reaction was started by the addition of 2 mM ATP after the equilibration of SMP with an inhibitor for 4 min. The activity was fully sensitive to SF6847 (protonophoric uncoupler) or oligomycin (ATP synthase inhibitor).
Photoaffinity Labeling of the ND1 Subunit by [125I]TDA
Photoaffinity labeling of the ND1 subunit by [125I]TDA was carried out as described previously (19). In brief, SMP (0.3 mg of protein/mL) in 100 μL of buffer containing 250 mM sucrose, 1 mM MgCl2, and 50 mM phosphate (pH 7.4) were treated with [125I]TDA (3 nM) in 1.5 mL eppendorf tubes and incubated for 10 min at room temperature. The samples were then irradiated with a long wavelength UV lamp (Black-Ray model B-100A, UVP) for 10 min on ice at a distance of 10 cm from the light source. When a replacement test was carried out, a competitor (i.e. other inhibitors) was added and incubated for 10 min at room temperature prior to the treatment with [125I]TDA.
SDS-PAGE was performed according to Laemmli (33). Briefly, [125I]TDA-labeled SMP samples were added to 4 x Laemmli's sample buffer and incubated at 35°C for 1 h to prevent protein aggregation. These denatured samples were separated on 10−20% gradient gels (90 × 83 × 1 mm). About 4−6 μg of protein was loaded into each well. Following electrophoresis, gels were fixed, stained with CBB R-250 or silver (silver stain MS kit, Wako Pure Chemical Industries, Osaka, Japan), and exposed to an imaging plate (BAS-MS2040, Fuji Film) for 12−24 h. The radiolabeled ND1 subunit was visualized with a Bio-Imaging Analyzer FLA-5100 (Fuji Film). For quantification of the radioactivity incorporated into the ND1, the digitalized data from autoradiography was quantified using Multi-Gauge software (Fuji), or the dried gels were cut into slices and directly quantified using the γ-counting system (COBRATM II, Packard).
Photoaffinity Labeling of SMP by [125I]AFP
Photoaffinity labeling by [125I]AFP was carried out as described for the labeling study with [125I]TDA, with the exception that 6 nM of [125I]AFP was UV-irradiated in 1.0 mg/mL of SMP for 20 min. Blue Native (BN)-PAGE (analytical scale) was performed using Native PAGETM Novex® Bis-Tris Gel System with a 4−16% pre-cast gel (Invitrogen, Carlsbad, CA), according to manufactures protocol. Electrophoresis was performed at a voltage 150 V with a limited current 15 mA/gel in a cold room. After electrophoresis, complex I was identified by activity staining by NADH/NBT system, and autoradiographed as described above.
[125I]AFP-labeled complex I was isolated by BN-PAGE and electroelution (34). [125I]AFP treated SMP were solubilized by 1% (w/v) DDM (n-dodecyl β-D-maltoside), and added 4x sample buffer (2 M aminocaproic acid, 200 mM Bis-tris/HCl, pH 7.0 at 4°C) and Serva Blue G (CBB R-250, Serva, Heidelberg, Germany). CBB/detergent ratio was adjusted to 1/4 by weight. The sample was separated on 6% isocratic blue native-gels (160 × 180 × 2 mm). The complex I band was excised and electroeluted using Centrilutor® (Millipore) with an elution buffer containing 25 mM tricine and 7.5 mM Bis-tris (adjusted with HCl to pH 7.0) overnight in a cold room. Complex I thus obtained was subjected to SDS-PAGE according to the procedure described above.
Two-dimensional Gel Electrophoresis
First dimensional isoelectoric focusing (IEF) was carried out using IPGphor system (GE healthcare, Buckinghamshire, UK) with Immobiline DryStrip (7 cm, pH 3−10, GE healthcare). Prior to loading the proteins, complex I isolated by BN-PAGE was precipitated by 2D-CleanUp Kit (GE healthcare) according to manufactures protocols. The precipitated proteins were solubilized by rehydration buffer containing 7 M urea, 2 M thiourea, 4% CHAPS, 0.5% IPG buffer, 40 mM DTT, and 0.002% bromophenol blue (BPB). Immobilized pH gradient strips were rehydrated by 125 μL of the rehydration buffer containing mitochondrial proteins for 12−20 h at 20°C and 20 V, followed by successive voltages of 300 V (30 min, step), 1000 V (30 min, gradient), 5000 V (80 min, gradient), and 5000 V (1 h, step). After IEF was completed, the strips were reduced with 5 mL of equilibration buffer (50 mM Tris/HCl, 6 M urea, 1% SDS, 30% glycerol, trace of BPB, pH 8.8) containing 0.25% (w/v) DTT for 10 min, and alkylated with the 5 mL of same equilibration buffer containing 4.5% (w/v) iodoacetamide. The treated strips were subjected to second dimensional analysis using Laemmli's 12.5% SDS gel, and autoradiographed.
Immunoprecipitation
Immunoprecipitation of electroeluted complex I was performed as described previously (17, 35). Briefly, electroeluted complex I was solubilized with 1.25% SDS. To the solubilized complex I solution (60 μL) was added dilution buffer (240 μL, 1.25% Triton X-100, 190 mM NaCl, 6 mM EDTA, 0.1 mM PMSF, 60 mM Tris-HCl, pH 7.4). The resulting mixture (300 μL) was treated with anti-Paracoccus denitrificants NQO9 antibody (for TYKY) or NQO6 (for PSST) antibody (5 μL) and rotated overnight in a cold room. Protein A-Sepharose 4B Fast flow (40 μL, GE healthcare, equilibrated with 50 mM Tris-HCl) was added, and the suspension was rotated at room temperature for 2 h. The incubated beads were washed 4 times with 1 mL of buffer A (0.1% Triton X-100, 0.02% SDS, 150 mM NaCl, 5 mM EDTA, 0.1 mM PMSF, 50 mM Tris-HCl, pH 7.4), then once with 1 mL of buffer B (buffer A without Triton X-100 and SDS). The resulting pellet was treated with SDS-PAGE sample buffer and centrifuged. The supernatant was subjected to electrophoresis and autoradiography.
Western Blotting Analysis
Western blotting analysis was performed according to the reported procedure (36). Complex I isolated from BN-PAGE was subjected to 1D (∼10 μg) or 2D (∼30 μg) gel electrophoresis and then electroblotted onto PVDF membrane (Immn-blot PVDF membrane, 0.2 μm, Bio-Rad) in a buffer contains 10 mM NaHCO3, 3 mM Na2CO3 and 0.025% (w/v) SDS for 16 h at 35 V (100 mA) in a cold room. The blotted membrane was blocked with 1% gelatin in Tween TBS (10 mM Tris/HCl, pH 7.4, 0.9% NaCl, 0.05% Tween 20) for 1 h at room temperature. The membrane was then probed with anti-Pd NQO9 (for TYKY) or NQO6 (for PSST) antibody for 1 h at room temperature, followed by incubation for another 1 h at room temperature with AP-conjugated anti-rabbit secondary antibody (Daiichi Pure Chemicals). The membrane was washed with Tween TBS (10 min × 3 times) and developed with NBT/BCIP chromogenic substrates (AP color development kit, Bio-Rad).
Mass Spectrometry
The photoaffinity labeled proteins were analyzed by MALDI/TOF (matrix-assisted laser desorption ionization/time of flight) MS at APRO Life Science Institute, Inc. (Tokushima, Japan).
RESULTS
Structure-Activity Relationship of Piperazine Derivatives
The original Δlac-acetogenins have two hydrophobic side chains attached to the hydroxylated bis-THF ring. We previously showed that marked hydrophobicity of the side chains is favorable for the inhibition, but the greater the loss of the balance in hydrophobicity, the weaker the inhibitory effect becomes (26, 37). To verify this structural dependency of the activity, the inhibitory effects of compounds 4−7 in terms of IC50 values were compared. These compounds were used as a racemic mixture since the stereochemistry of the hydroxy group was not crucial for the inhibitory effect, as described later. The total number of carbon atoms of the side chains in compounds 5−7 was set to be identical at 22. Compound 4 did not exhibit 50% inhibition of NADH oxidase activity up to a solubility limit (22,000 nM), indicating that hydrophobicity is favorable for the activity. As observed for the original Δlac-acetogenins, the greater the loss of the balance in hydrophobicity of the side chains, the weaker the inhibitory effect became (5 vs. 6 and 7). It is likely that the balance in hydrophobicity decides the precise location of the hydroxylated piperazine ring, which may be at or close to the membrane surface area of the enzyme embedded in the inner mitochondrial membrane.
The simple alkyl side chain of Δlac-acetogenins can be replaced by substituted phenyl groups without any loss of activity (e.g. 1 vs. 2 and 3) (25, 26). We therefore prepared four phenyl derivatives with chains of different lengths (compounds 8−11). These derivatives generally retained the original level of activity, while 10 was the most potent inhibitor among the four compounds. This result means that the hydrophobicity of the side chains is favorable for the activity, but an excess of hydrophobicity is rather adverse to the activity probably due to some sort of trapping in the hydrophobic lipid bilayer of the membrane. A similar tendency was observed for the original Δlac-acetogenins (25, 26). Inversion of the R-configuration of the hydroxy groups in 9 to the S-configuration did not significantly affect the activity (9 vs.12), indicating that the stereochemistry of the hydroxy group is not crucial for the activity. A slight modification of the conformational property of the piperazine ring did not affect the activity (10 vs. 13 and 14). Compound 15 was much less active likely because of a significant increase in the hydrophobicity of the polar head region. We also synthesized a dimethylamine derivative (16) which corresponds to a half of compound 10. This compound had almost no inhibitory effect (IC50 > 30,000 nM), indicating the symmetrical property as well as the cyclic piperazine ring to be essential.
We previously showed that deletion of both hydroxy groups adjacent to the bis-THF ring of Δlac-acetogenin (1) resulted in a remarkable (∼400-fold) loss of activity (25). This finding indicated a crucial role for the hydroxy groups in the inhibition, probably acting as a hydrogen-bond donor. As this notion is important to know the inhibition mechanism of Δlac-acetogenins, we here reexamined this effect using the most potent derivative (2, Figure 1), which was synthesized after the publication of a previous paper (25). Again, the activity decreased by about 1000-fold (2 vs. 17), verifying the crucial role of the hydroxy groups. We also examined the role of the hydroxy groups with the piperazine series. Unexpectedly, the deletion of two hydroxy groups did not significantly affect the activity (8 vs. 18, 9 vs. 19, and 10 vs. 20). This finding strongly suggests that a simple piperazine ring per se is enough for the product to act as a toxophore. The drastic decrease in the activity of 21 indicates that marked hydrophobicity of the side chains is also needed for the inhibitory action of the deoxygenated series.
The Role of the Hydroxy Groups Adjacent to the Piperazine Ring
The deletion of both hydroxy groups in compounds 8−10 did not significantly affect the inhibitory activity. This result was quite unexpected because i) this structural modification drastically changes the physicochemical properties of the inhibitors, resulting in a loss of hydrogen-bonding ability and a decrease in hydrophilicity and ii) the piperazine ring itself seems to be too simple to govern tight binding to the enzyme. Characterization of the action manner, as described later, showed no significant difference between the hydroxylated and deoxygenated piperazines, indicating an equivalency as complex I inhibitors. On the basis of these findings, it is clear that the hydrogen-bonding ability of the hydroxy groups is not essential for tight interaction with the enzyme. This functional group therefore seems to act as a hydrophilic anchor directing the piperazine ring (i.e. a toxophore) at or near the membrane surface. Therefore, at least for the hydroxylated piperazines, the typical amphiphilic nature of the molecule would be crucial to governing the tight interaction between the piperazine ring and the enzyme.
However, this notion raises the question of whether the same holds true for the deoxygenated piperazines which lost hydrophilicity around the piperazine ring. To know the hydrophilic nature of the piperazine ring itself, we estimated the log P value of 1,4-dimethylpiperazine using CLOGP (Ver. 4.0, BioByte Corp., Claremont, CA), taking 1,4-dimethylcyclohexane as a reference (Figure S2). The log P value is widely used as an index of the hydrophobicity/hydrophilicity of low molecular weight chemicals (38); wherein P is a partition coefficient of the chemical in the 1-octanol/water partition system (i.e. P = [chemical]oct/[chemical]water). The calculated log P values of 1,4-dimethylpiperazine and 1,4-dimethylcyclohexane are −0.74 and 4.39, respectively, indicating that the piperazine ring itself is sufficiently hydrophilic compared to the corresponding alkane. It is therefore reasonable to consider that the deoxygenated piperazines also retain an amphiphilic nature, though the amphiphilic nature of the deoxygenated derivatives is less distinct than that of the hydroxylated compounds. In support of this notion, compared to the hydroxylated compounds, a less hydrophobic side chain was favorable for the activity of the deoxygenated piperazines (e.g. 10 vs. 19).
Induction of Superoxide from Complex I
Superoxide is produced from complex I at a relatively high rate in the presence of a specific inhibitor of the enzyme, though the main reductant of oxygen producing superoxide in complex I is not known, and published results are highly conflicting. One possible reason of this is that the superoxide generation from the enzyme varies greatly depending on the tissue, species, and experimental conditions (39-42). The level of superoxide production induced by original Δlac-acetogenins, regardless of whether endogenous or exogenous ubiquinone is used as an electron acceptor, is much lower than that induced by traditional inhibitors (26, 27), indicating the effect of Δlac-acetogenins on the free radical intermediate in complex I to be quite unique.
We compared the rates of superoxide production from complex I between piperazine (10) and Δlac-acetogenin (3) in the NADH oxidase assay, using rotenone, piericidin A, and bullatacin as references. The concentrations of all inhibitors were set high enough to achieve complete inhibition of the NADH oxidase activity. As shown in Figure 3, the rate of production induced by 10 was significantly higher than that induced by an original Δlac-acetogenin (3), though the rate didn't reach the level obtained with the traditional inhibitors. The rate for the deoxygenated derivative 19 was lower than that for 10 (Figure 3).
Figure 3.
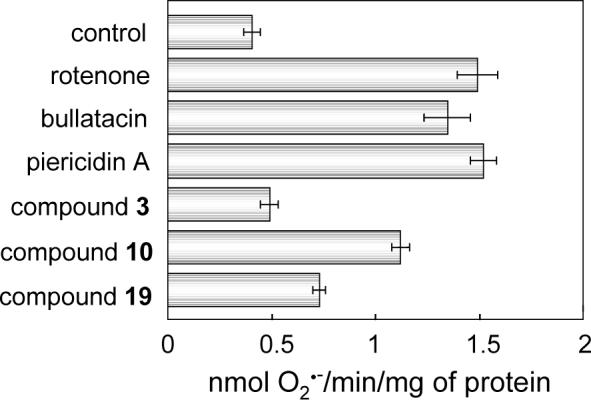
Superoxide production from complex I in the presence of inhibitor. Rates of superoxide production in the NADH oxidase assay were determined with SMP. The concentrations of all inhibitors were set at 1.2 μM, being high enough to achieve complete inhibition of NADH oxidase activity. The final mitochondrial protein concentration is 0.25 mg of protein/mL. Bars show means ± SD of three independent measurements.
Inhibition of Reverse Electron Transfer
The inhibitory effect of Δlac-acetogenins on the reverse electron transfer in complex I in the presence of the proton-motive force is unique. In contrast to ordinary inhibitors, Δlac-acetogenins have a much weaker effect on the reverse than the forward electron transfer (27). Additionally, their inhibition of the reverse electron transfer is markedly time-dependent (see Figure 3 in Ref. 27). The time dependency was eliminated by preincubating SMP with the inhibitor for 4 min before the reverse electron transfer was started (27).
We examined the effect of the piperazines on the reverse electron transfer. For the experiment, SMP were preincubated with the inhibitor for 4 min before the reverse electron transfer was started. The IC50 values (pmol inhibitor/mg of protein) for the forward and reverse electron transfer are listed in Table 1, with rotenone, piericidin A, and bullatacin as references. Since the reverse electron transfer is much slower than the forward electron transfer, a higher concentration of SMP was used in the reverse electron transfer assay (0.1 vs. 0.03 mg of protein/mL), resulting in an increase in volume of the membrane lipid phase as well as the concentration of complex I. Accordingly, as the amount of inhibitor trapped in the hydrophobic lipid bilayer differs between the two assay systems, direct comparison of the IC50 values between the two assays is complicated. Nevertheless, to compare the inhibitor sensitivity of the forward and reverse electron transfer events, the IC50 values were compared as a matter of convenience. Of particular interest, the inhibitory potency of 10 for the reverse electron transfer was about 10-times weaker than that of Δlac-acetogenin (3), and hence the gap in the inhibitory potency of 10 between the forward and the reverse electron transfer events is much more definitive than that observed for 3 (Table 1). About 50-fold higher concentrations of 10 were needed to inhibit the reverse electron transfer. In contrast to 3, no significant time-dependency of the inhibitory effect was observed for 10 (data not shown). Similar results were observed for deoxygenated derivative 19 (Table 1). Thus it turned out that the effect of the piperazines on the reverse electron transfer is significantly different from that of ordinary inhibitors as well as the original Δlac-acetogenins. The piperazines are, to our knowledge, the most definitive “direction-specific” inhibitors among diverse complex I inhibitors. Consequently, it is strongly suggested that the structure of the binding site of piperazines in the forward electron transfer state differ from that in the reverse electron transfer state probably because the two reactions have different rate-limiting steps. A postulate of two different electron transfer pathways is an unlikely explanation for the direction-specific effect of the piperazines.
Table 1.
The inhibitory effects on the forward and reverse electron transfer activities.a
| inhibitors | IC50 values (pmol inhibitor/mg of protein)b | IC50 (R)/IC50 (F)c | |
|---|---|---|---|
| Forward electron transfer | Reverse electron transfer | ||
| compound 3 | 43 ± 4.0 | 180 ± 21 | 4.2 |
| compound 10 | 40 ± 1.7 | 1890 ± 69 | 47 |
| compound 19 | 73 ± 6.7 | 2130 ± 130 | 29 |
| AFP | 32 ± 0.66 | 1070 ± 110 | 33 |
| rotenone | 157 ± 16 | 75 ± 7.3 | 0.48 |
| Bullatacind | 46 ± 5.2 | 28 ± 4.4 | 0.61 |
| piericidin Ad | 81 ± 7.1 | 48 ± 4.3 | 0.59 |
The rates of the forward and reverse electron transfer reactions were 0.71 ± 0.06 and 0.056 ± 0.007 μmol NADH/min/mg of protein, respectively.
Values are means ± SD of three independent experiments.
The ratios of IC50 (reverse)/IC50 (forward).
From ref. 27.
Effect of Ionization at the Nitrogen Atom(s) of Piperazines
The pKA value of the tertiary ammonium cation (R3NH+) in water at 25°C is ∼9.80 (43). It is therefore likely that at least one of the two nitrogen atoms of the piperazines is protonated in the assay buffer (pH 7.4), i.e. carrying a positive charge. Considering that the pKA value changes depending upon the polarity of the surroundings, it is unclear whether the piperazine derivatives have a positive charge at the bound state in the enzyme. To examine the effect of the positive charge formed on the nitrogen atom(s), we prepared two quaternary ammonium cations (R3NCH +3) 22 and 23 (Figure 2) by the methylation of one or both nitrogen atom(s) of compound 8, and examined their inhibitory effect on the enzyme. The IC50 values of 22 and 23 were 1,100 and 4,700 nM, respectively, indicating that a quaternary ammonium ion form is quite unfavorable for the inhibitory action.
Furthermore, to elucidate the inhibitory behavior of positively charged inhibitors of complex I, the case of pyridinium-type cationic inhibitors may be helpful. Pyridinium-type inhibitors, such as N-methyl-p-phenylpyridinium (MPP+), have a positive charge on the pyridine nitrogen atom (44-46). It is well known that the effectiveness of these inhibitors is markedly enhanced, by one to three orders of magnitude, in the presence of a counter-anion such as a tetraphenylboron (TPB−) (44-46). The potentiation effect may be accounted for by an increase in the pyridinium concentration in the membrane lipid phase due to neutral ion-pair formation (44). Therefore we examined the effect of TPB−, from 0.1 to 2.0 μM, on the inhibition by compounds 10 and 19. In contrast to the pyridinium-type inhibitors, no potentiation of the inhibitory effect was observed. As a reference, we confirmed that the inhibitory activities of the quaternary ammonium ion derivatives (22 and 23) are slightly (about two-fold) potentiated by TPB−. The relatively small potentiation may be because the side chains of these compounds are inherently hydrophobic enough to partition into the membrane environment. These results along with the above observation strongly suggest that the piperazine derivatives are not protonated at the bound state in the enzyme; in other words, the active form of the piperazines is neutral (R3N) possessing lone pair electrons on the nitrogen atom.
Suppression of the Specific Binding of [125I]TDA to the ND1 Subunit
Through photoaffinity labeling study using a photoreactive natural-type acetogenin derivative [125I]TDA, we recently showed that various traditional inhibitors such as rotenone, piericidin A, fenpyroximate, and pyridaben completely suppress the specific binding of [125I]TDA to the ND1 subunit, whereas Δlac-acetogenin (3) does not suppress the binding even at largely excess concentrations (over 3000-fold) (19). This finding alone with other observations (27) revealed that Δlac-acetogenins don't share a common binding domain with traditional inhibitors.
To examine whether the piperazine derivatives share a common binding domain with traditional inhibitors, we examined the suppressive effect of the piperazines on the specific binding of [125I]TDA to the ND1 using SMP. In contrast to the original Δlac-acetogenins, compound 10 efficiently suppressed the specific labeling in a concentration-dependent manner and complete suppression was achieved at about 3 μM (Figure 4). A deoxygenated derivative 19 also suppressed the specific binding, though less efficiently than 10 (Figure 4). Bullatacin (IC50 = 0.85 nM), used as a reference compound, suppressed the specific binding more efficiently than the piperazines. It should be noted that the binding affinity of each inhibitor in terms of the IC50 value does not necessarily correlate linearly with the efficiency of suppression since the extent of overlap between each inhibitor and [125I]TDA at bound state in the enzyme may differ depending on the structural nature of the inhibitors. Anyhow, our results indicate that the binding site of the piperazines is not identical to that of the original Δlac-acetogenins and that the piperazines might share a common binding domain with traditional inhibitors.
Figure 4.
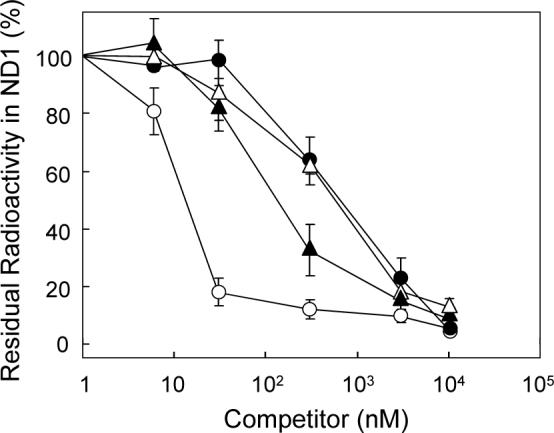
Effects of inhibitors on the specific photoaffinity labeling of the ND1 subunit by [125I]TDA. A competitor was added to SMP (0.3 mg of protein/mL) at given concentrations and incubated for 10 min prior to the treatment with [125I]TDA (3 nM). Key: compound 10 (closed triangles), 19 (closed circles), bullatacin (open circles), and piericidin A (open triangles). Data are the means of three independent measurements ± SD.
Photoaffinity Labeling with Piperazine Derivative [125I]AFP
To identify the binding subunit of the piperazine inhibitor, we carried out photoaffinity labeling study using a newly synthesized photoreactive piperazine derivative [125I]AFP. The IC50 value of AFP (24) was 1.0 ± 0.08 nM, indicating that it retains potent inhibitory activity. We confirmed that the inhibitory effect of AFP on the reverse electron transfer is remarkably weaker than that on the forward event (Table 1). The photoaffinity labeling was carried out according to a previous work (19). SMP were used to ensure the intactness of complex I (17).
UV-irradiation of SMP (1.0 mg of protein/mL) in the presence of [125I]AFP (6 nM) resulted in two labeled regions with an apparent molecular mass of approximately 20 and 50 kDa on a SDS gel at a frequency of about 3:1 (Figure 5). Nearly complete labeling (>90%) was established within 20 min of UV-irradiation. As shown in Figure 6, a good correlation was found between the incorporation of the radioactivity into the two bands and the inhibition of NADH oxidase activity. It is therefore obvious that the photo-cross linking of the two bands is responsible for the inhibition of complex I. Even at a higher concentration of [125I]AFP (120 nM), radioactivity in other regions on the SDS gel was less than 10% of the sum of the two regions, indicating that the extent of nonspecific labeling is negligibly small.
Figure 5.
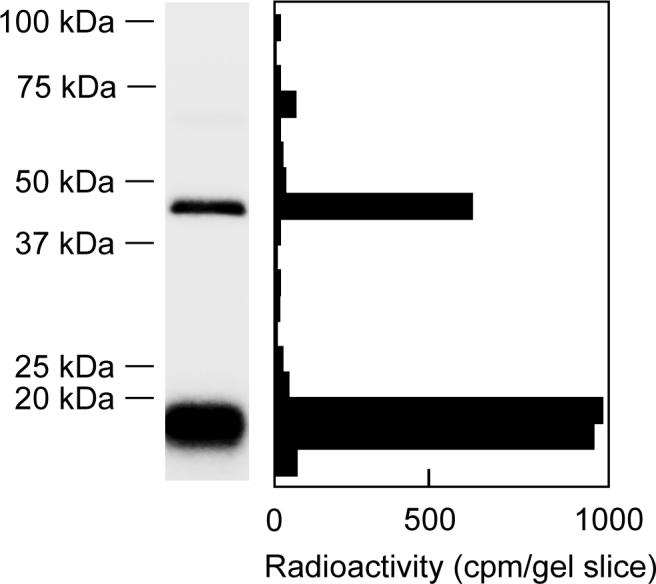
Photoaffinity labeling of bovine SMP with [125I]AFP. SMP (1.0 mg of protein/mL) were photoaffinity labeled with [125I]AFP (6 nM) as described in the Experimental Procedures. Autoradiography of SDS-PAGE analysis (a 10−20% gel) of [125I]AFP-labeled SMP is shown. Data shown are representative of five independent experiments.
Figure 6.
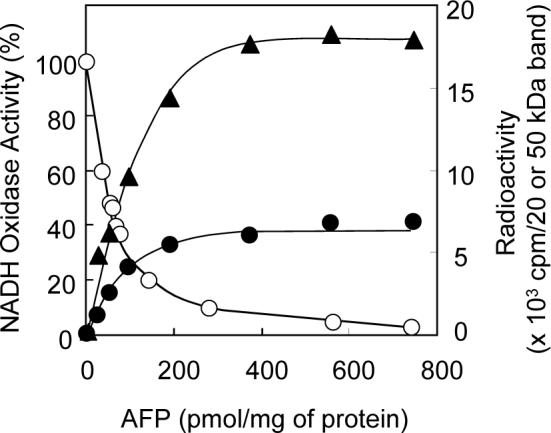
Correlation between the incorporation of radioactivity into the two bands and inhibition of the NADH oxidase activity in SMP. Key: the radioactivity in the 20 (closed triangles) and 50 (closed circles) kDa bands and the inhibition of the NADH oxidase activity (open circles). The values represent the average of two independent measurements.
The structure-activity relationship of a series of piperazines obtained in the present study revealed that the hydrophobicity of the two side chains and their valance are crucial for the strong inhibition and that no specific functional group is needed for the chain moieties. Taking into consideration this finding along with the fact that the hydroxylated piperazine ring moiety is C2-symmetric, two side chains of AFP can be regarded as almost “equivalent” in the binding to the enzyme. It is therefore reasonable that the photolabile group in one side chain resides at and cross-links to different sites (or subunits) under the experimental conditions.
Identification of the Labeled 20 and 50 kDa Proteins
SMP (1.0 mg of protein/mL) cross-linked with 6 nM [125I]AFP were subjected to BN-PAGE and resolved into five oxidative phosphorylation enzyme complexes (Figure 7A). The complex I band was identified by activity staining using a NADH-NBT system (Figure 7B). Major radioactivity was detected in complex I band (Figure 7C), indicating that complex I was predominantly radiolabeled by [125I]AFP. Next, the radiolabeled complex I band was isolated by electroelution and subjected to SDS-PAGE for further separation. As shown in Figure 8, we detected a major peak of radioactivity at around 20 and 50 kDa. The radioactivity of other regions was less than 5% of that of these bands.
Figure 7.

Localization of the labeled 20 and 50 kDa bands in SMP. (A) Separation of the five oxidative phosphorylation enzyme complexes from [125I]AFP-labeled SMP by BN-PAGE (a 4−16% gradient gel). SMP (1.0 mg of protein/mL) were photoaffinity labeled by 6 nM [125I]AFP. (B) Complex I band on the gel was identified by the activity staining with NADH-NBT. (C) Autoradiography of the gel is shown. Data shown are representative of five independent experiments.
Figure 8.

Analysis of the photoaffinity labeled subunit in isolated complex I. (A) SDS-PAGE (a 10−20% gel) analysis of [125I]AFP-labeled complex I isolated by BN-PAGE shown in Figure 7 (CBB staining). (B) Autoradiography of the gel is shown. Data shown are representative of five independent experiments.
To identify the 50 kDa band, the complex I band was isolated from non-crosslinked SMP by electroelution and subjected to SDS-PAGE, and the corresponding gel region was cut into 1−2 mm slice. This band was analyzed by peptide mass fingerprinting of tryptic peptides. The 50 kDa band turned out to be the 49 kDa subunit (Table S1), which located in the proximity of the interface of peripheral and membrane arms (3). Peptides corresponding to 62% of the total sequence (428 a. a.) were identified.
On the other hands, as the gel region corresponding the 20 kDa band contains multiple subunits (36, 47), we carried out immunoprecipitation of the [125I]AFP-labeled complex I with anti-Pd NQO6 and NQO9 antibodies, which strongly recognizes the bovine PSST and TYKY subunits, respectively (18, also see Figure S3). It turned out that the 20 kDa band reacted with neither anti-Pd NQO6 nor NQO9 antibodies (data not shown). To identify the 20 kDa band, we next performed two-dimensional electrophoresis using isolated complex I cross-linked by [125I]AFP (Figure 9A). The position of the 49 kDa subunit on the gel is consistent with calculated isopotential point (Figure 9B, cf. 47). However, as the radioactivity of the 20 kDa subunit was not detected on the gel probably because the subunit is highly hydrophobic and/or aggregated under the experimental conditions, this subunit remains to be identified. As a reference, western blotting analysis using anti-Pd NQO6 (for PSST) was shown (Figure 9C). From core subunits of complex I, a possible candidate for the cross-linked 20 kDa subunit could be the hydrophobic ND6 subunit. We cannot exclude the possibility that AFP cross-linked to one of the hydrophobic accessory subunits since the photolabile group does not necessarily bind to the core subunits as the photolabile group is attached to the side chain where no specific functional group is required for the activity.
Figure 9.

Resolution of the subunits of complex I cross-linked by [125I]AFP on two-dimensional gel. (A) The first dimension: IPG 3−10; second dimension: Laemmli's 12.5% SDS-PAGE. (B) Autoradiography of the gel is shown. (C) Western analysis using anti-Pd NQO6 (for PSST) antibody.
Suppression of Specific Binding of [125I]AFP by Other Complex I Inhibitors
We examined the suppressive effect of other inhibitors on the specific labeling of the two subunits by [125I]AFP (6 nM) over a wide concentration range. A piperazine derivative 10 suppressed the labeling in a concentration-dependent manner (Figure 10). Other traditional inhibitors (rotenone, fenpyroximate, pyridaben and bullatacin) also suppressed the radiolabeling (Figure 10). It is noteworthy that while the mechanism of action was shown to be different between the original Δlac-acetogenins and the piperazines from the above results, Δlac-acetogenin (3) suppressed the specific binding of [125I]AFP. These results seem to mean that [125I]AFP and numerous other inhibitors share the same binding domain in complex I. However careful interpretation of the apparent competitive behavior among the inhibitors is needed, as discussed below.
Figure 10.

Effects of various complex I inhibitors on the specific binding of [125I]AFP. Autoradiography of the two bands is shown. A competitor was added to SMP (1.0 mg of protein/mL) at 0 (a), 60 (b), 600 (c), or 6,000 (d) nM and incubated for 10 min prior to the treatment with [125I]AFP (6 nM). Data shown are representative of at least two independent experiments.
DISCUSSION
The inhibitor-binding domain in complex I is thought to be closely associated with the functions of the membrane arm such as ubiquinone reduction and proton translocation. Therefore, detailed elucidation of the action mechanism of diverse inhibitors including the identification of their binding site(s) is necessary for a full understanding of the functional and structural features of the membrane arm. Photoaffinity labeling and fluorescent-ligand binding studies strongly suggested that a wide variety of complex I inhibitors share a common large binding domain with partially overlapping sites (12, 17-19). If this concept is correct, the occurrence of potent inhibitor Δlac-acetogenins, which don't share a common binding domain with traditional inhibitors (19, 27), is of particular interest, and a thorough elucidation of their action mechanism is desired. Although identification of the binding subunit of Δlac-acetogenins by a photoaffinity labeling technique is currently underway in our laboratory, we planned to carry out drastic structural modifications of Δlac-acetogenins to find further unique inhibitors. We initially designed and synthesized a series of piperazine derivatives as new analogues of Δlac-acetogenins. However, the present results demonstrated that the mode of action of the piperazine series is remarkably different from that of the original Δlac-acetogenins as follows: (i) the presence of two hydroxy groups is not essential for the activity, (ii) the level of superoxide production induced by the piperazines is higher than that by Δlac-acetogenin, (iii) the inhibitory potency for the reverse electron transfer is remarkably weaker than that for the forward event, and (iv) the piperazines efficiently suppress the specific binding of a photoreactive acetogenin ([125I]TDA) to the ND1 subunit. Taken together, we conclude that from a mechanistic point of view, the piperazine series are no longer analogues of Δlac-acetogenins.
The competition test using the photoaffinity probe [125I]AFP as well as [125I]TDA posed an important question: why was an apparent competitive behavior observed between the inhibitors that seem to bind to different sites. For instance, while [125I]AFP does not bind to the ND1 subunit, this compound suppressed the specific binding of [125I]TDA to the ND1 (Figure 4). The same is true for pyridaben and fenpyroximate, which binds to the PSST (17) and ND5 (18) subunits, respectively; these compounds suppressed the specific binding of both [125I]AFP (Figure 10) and [125I]TDA (19). In addition, regardless of remarkable differences in the mode of action between the original Δlac-acetogenins and the piperazine derivatives, competitive behavior was observed between the two series ([125I]AFP vs. 10). To answer this question, it is notable that an X-ray crystallographic study (3) and cross-linking studies (20, 21) showed that the PSST, 49 kDa, TYKY and ND1 subunits are located next to each other at the interface of the peripheral and membrane arms. Several studies suggested that complex I undergoes dynamic conformational changes (48-51). Taking these reports into consideration, we speculate that some, but not all, of the apparent competitive behavior described above is due to structural change at the binding site, rather than the occupation of the same site. The structural changes of the binding site in one subunit induced by inhibitor binding might propagate to the adjacent binding site in another subunit. In this context, it is also likely that the inhibitor binding domain comprised of multiple subunits is certainly larger than is currently supposed (12, 17, 18). It should, however, be realized that the photoaffinity probes used in the earlier works, except [125I]TDA, have a photolabile group at a position remote from the toxophoric moiety of the inhibitor. This fundamental problem with the probe's design makes the interpretation of the results of photoaffinity labeling studies complicated. The complete crystal structure of complex I with inhibitor bound is needed to fully understand the features of the inhibitor binding sites.
Another issue being addressed is whether the piperazine series can be classified as the ordinary inhibitors such as rotenone, piericidin A, and synthetic agrochemicals. The binding site of the piperazines must be located at the interface of the peripheral and membrane arms, as suggested for ordinary inhibitors (52). The competition test showed that all ordinary inhibitors examined suppressed the specific binding of [125I]AFP to the enzyme. However, the 49 kDa subunit identified in this work have never reported in the earlier photoaffinity labeling studies (17-19). In addition, taking into consideration the remarkable direction-specific effect of the piperazines as well as their highly unique structural characteristics, it is unlikely that the binding sites of the piperazines and ordinary inhibitors are identical. While it was suggested that piericidin A and rotenone bind to the Nqo4 (49 kDa in bovine) subunit in P. denitrificants (53), the binding sites of these inhibitors and the piperazines on the 49 kDa subunit must be different. Ordinary inhibitors might suppress the specific binding of [125I]AFP by affecting the structure of its binding site as a secondary effect, as discussed above.
In general, the action site of potent inhibitors of mitochondrial respiratory enzymes, which bind to the site in an almost stoichiometric manner, is mostly responsible for the crucial functionality of the enzyme. For instance, antimycin A and myxothiazol bind to the ubiquinone reduction (Qi) and the ubiquinol oxidation (Qo) sites, respectively, in ubiquinol-cytochrome c oxidoreductase (complex III) with high affinity (54). From a historical point of view, the occurrence of these two inhibitors acting at different sites and the detailed characterization of their action mechanism established the proton-motive Q cycle mechanism of complex III (54, 55). In this sense, the occurrence of new types of inhibitors of complex I, i.e. Δlac-acetogenins and the piperazines synthesized in our laboratory, is quite interesting for elucidating the function of the enzyme. The fact that there are diverse chemicals that disturb the functionality of the interfacial domain of the peripheral and membrane arms in different manners depending on their structural nature suggests that multiple reaction sites or conformations would be involved in the turnover reactions in the enzyme. In this context, on the basis of the markedly different physicochemical properties of two (or three) ubisemiquinones in bovine complex I, Ohnishi and colleagues (56, 57) suggested that the ubisemiquinone species are located at different sites in the enzyme and may play distinct roles. To further elucidate the mode of action of Δlac-acetogenins as well as the piperazine derivatives, identification of their binding sites at the amino acid level is currently underway in our laboratory.
Supplemental Materials
ACKNOWLEDGMENT
We thank Dr. Akemi Matsuno-Yagi (The Scripps research institute, La Jolla) for valuable discussion and Dr. Takahisa Kato (Radioisotope Research Center of Kyoto University) for excellent technical assistance.
ABBREVIATIONS
- AFP
4-azido-2,3,5,6-tetrafluorobenzyloxyalkyl piperazine
- [125I]AFP
125I-labeled AFP
- bis-THF
bis-tetrahydrofuran
- CBB
coomassie brilliant blue
- complex I
mitochondrial proton-pumping NADH-ubiquinone oxidoreductase
- DTT
dithiothreitol
- IC50
the molar concentration (nM) needed to reduce the control NADH oxidase activity in SMP by half
- IPG
immobilized pH gradient
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- SMP
submitochondrial particles
- TDA
(trifluoromethyl)phenyldiazirinyl acetogenin
- [125I]TDA
125I-labeled TDA
- TPB−
tetraphenylboron
Footnotes
This work was supported in part by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (Grant 17380073 to H. M.), a Grant-in-Aid for JSPS Fellows (to N. I. and M. M.), and NIH grant R01GM033712 (to T. Y.).
SUPPORTING INFORMATION AVAILABLE
Syntheses of compounds 4−24 and [125I]AFP, Table S1, and Figures S1-S3. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Walker JE. The NADH-ubiquinone oxidoreductase (complex I) of respiratory chains. Q. Rev. Biophys. 1992;25:253–324. doi: 10.1017/s003358350000425x. [DOI] [PubMed] [Google Scholar]
- 2.Carroll J, Fearnley IM, Skehel JM, Shannon RJ, Hirst J, Walker JE. Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 2006;43:32724–32727. doi: 10.1074/jbc.M607135200. [DOI] [PubMed] [Google Scholar]
- 3.Sazanov LA, Hinchliffe P. Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 4.Yagi T, Matsuno-Yagi A. The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry. 2003;42:2266–2274. doi: 10.1021/bi027158b. [DOI] [PubMed] [Google Scholar]
- 5.Hirst J. Energy transduction by respiratory complex I; an evaluation of current knowledge. Biochem. Soc. Trans. 2005;33:525–529. doi: 10.1042/BST0330525. [DOI] [PubMed] [Google Scholar]
- 6.Brandt U. Energy converting NADH:quinone oxidoreductase (complex I) Annu. Rev. Biochem. 2006;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- 7.Friedrich T, Van Heek P, Leif H, Ohnishi T, Forche E, Kunze B, Jansen R, Trowitzsch-Kienast W, Höfle G, Reichenbach H, Weiss H. Two binding sites of inhibitors in NADH:ubiquinone oxidoreductase (complex I) Eur. J. Biochem. 1994;219:691–698. doi: 10.1111/j.1432-1033.1994.tb19985.x. [DOI] [PubMed] [Google Scholar]
- 8.Degli Esposti M. Inhibitors of NADH-ubiquinone reductase: an overview. Biochim. Biophys. Acta. 1998;1364:222–235. doi: 10.1016/s0005-2728(98)00029-2. [DOI] [PubMed] [Google Scholar]
- 9.Miyoshi H. Structure-activity relationships of some complex I inhibitors. Biochim. Biophys. Acta. 1998;1364:236–244. doi: 10.1016/s0005-2728(98)00030-9. [DOI] [PubMed] [Google Scholar]
- 10.Kean EA, Gutman M, Singer TP. Studies on the respiratory chain-linked nicotinamide adenine dinucleotide dehydrogenase. XXII. Rhein, a competitive inhibitor of the dehydrogenase. J. Biol. Chem. 1971;246:2346–2353. [PubMed] [Google Scholar]
- 11.Majander A, Finel M, Wikström M. Diphenyleneiodonium inhibits reduction of iron-sulfur clusters in the mitochondrial NADH-ubiquinone oxidoreductase (complex I) J. Biol. Chem. 1994;269:21037–12042. [PubMed] [Google Scholar]
- 12.Okun JG, Lümmen P, Brandt U. Three classes of inhibitor share a common binding domain in mitochondrial complex I (NADH-ubiquinone oxidoreductase) J. Biol. Chem. 1999;274:2625–2630. doi: 10.1074/jbc.274.5.2625. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich H, Werner S. Identification of the ubiquinone-binding site of NADH-ubiquinone oxidoreductase (complex I) from Neurospora crassa. Biochemistry. 1992;31:11413–11419. doi: 10.1021/bi00161a020. [DOI] [PubMed] [Google Scholar]
- 14.Gong X, Xie T, Yu L, Hesterberg M, Scheide D, Friedrich T, Yu C-A. The ubiquinone-binding site in NADH-ubiquinone oxidoreductase from Escherichia coli. J. Biol. Chem. 2003;278:25731–25737. doi: 10.1074/jbc.M302361200. [DOI] [PubMed] [Google Scholar]
- 15.Earley FGP, Ragan CI. Photoaffinity labeling of mitochondrial NADH dehydrogenase with arylazidoamorphigenin, an analogue of rotenone. Biochem. J. 1984;224:525–534. doi: 10.1042/bj2240525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Earley FGP, Patel SD, Ragan CI, Attardi G. Photolabeling of a mitochondrially encoded subunit of NADH dehydrogenase with [3H]dihydrorotenone. FEBS Lett. 1987;219:108–113. doi: 10.1016/0014-5793(87)81200-0. [DOI] [PubMed] [Google Scholar]
- 17.Schuler F, Yano T, Bernardo SD, Yagi T, Yankovskaya V, Singer TP, Casida JE. NADH-quinone oxidoreductase: PSST subunit couples electron transfer from iron-sulfur cluster N2 to quinone. Proc. Natl. Acad. Sci. U.S.A. 1999;96:4149–4153. doi: 10.1073/pnas.96.7.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamaru-Ogiso E, Sakamoto K, Matsuno-Yagi A, Miyoshi H, Yagi T. The ND5 subunit was labeled by a photoaffinity analogue of fenpyroximate in bovine mitochondrial complex I. Biochemistry. 2003;42:746–754. doi: 10.1021/bi0269660. [DOI] [PubMed] [Google Scholar]
- 19.Murai M, Ishihara A, Nishioka T, Yagi T, Miyoshi H. The ND1 subunit constructs the inhibitor binding domain in bovine heart mitochondrial complex I. Biochemistry. 2007;46:6409–6416. doi: 10.1021/bi7003697. [DOI] [PubMed] [Google Scholar]
- 20.Di Bernardo S, Yagi T. Direct interaction between a membrane domain subunit and a connector subunit in the H+-translocating NADH-quinone oxidoreductase. FEBS Lett. 2001;508:385–388. doi: 10.1016/s0014-5793(01)03111-8. [DOI] [PubMed] [Google Scholar]
- 21.Kao MC, Matsuno-Yagi A, Yagi T. Subunit proximity in the H+-translocating NADH-quinone oxidoreductase probed by zero-length cross-linking. Biochemistry. 2004;43:3750–3755. doi: 10.1021/bi049927s. [DOI] [PubMed] [Google Scholar]
- 22.Miyoshi H, Ohshima M, Shimada H, Akagi T, Iwamura H, McLaughlin JL. Essential structural factors of Annonaceous acetogenins as potent inhibitors of mitochondrial complex I. Biochim. Biophys. Acta. 1998;1365:443–452. doi: 10.1016/s0005-2728(98)00097-8. [DOI] [PubMed] [Google Scholar]
- 23.Kuwabara K, Takada M, Iwata J, Tatsumoto K, Sakamoto K, Iwamura H, Miyoshi H. Design syntheses and mitochondrial complex I inhibitory activity of novel acetogenin mimics. Eur. J. Biochem. 2000;267:2538–2546. doi: 10.1046/j.1432-1327.2000.01263.x. [DOI] [PubMed] [Google Scholar]
- 24.Abe M, Murai M, Ichimaru N, Kenmochi A, Yoshida T, Kubo A, Kimura Y, Moroda A, Makabe H, Nishioka T, Miyoshi H. Dynamic function of the alkyl spacer of acetogenins in their inhibitory action with mitochondrial complex I (NADH-ubiquinone oxidoreductase) Biochemistry. 2005;44:14898–14906. doi: 10.1021/bi051568t. [DOI] [PubMed] [Google Scholar]
- 25.Hamada T, Ichimaru N, Abe M, Fujita D, Kenmochi A, Nishioka T, Zwicker K, Brandt U, Miyoshi H. Synthesis and inhibitory action of novel acetogenin mimics with bovine heart mitochondrial complex I. Biochemistry. 2004;43:3651–3658. doi: 10.1021/bi030242m. [DOI] [PubMed] [Google Scholar]
- 26.Ichimaru N, Murai M, Abe M, Hamada T, Yamada Y, Makino S, Nishioka T, Makabe H, Makino A, Kobayashi T, Miyoshi H. Synthesis and inhibition mechanism of Δlac-acetogenins: a novel type of inhibitor of bovine heart mitochondrial complex I. Biochemistry. 2005;44:816–825. doi: 10.1021/bi0479003. [DOI] [PubMed] [Google Scholar]
- 27.Murai M, Ichimaru N, Abe M, Nishioka T, Miyoshi H. Mode of inhibitory action of Δlac-acetogenins, a new class of inhibitors of bovine heart mitochondrial complex I. Biochemistry. 2006;45:9778–9787. doi: 10.1021/bi060713f. [DOI] [PubMed] [Google Scholar]
- 28.Holt PJ, Morgan DJ, Sazanov LA. The location of NuoL and NuoM subunits in the membrane domain of the Escherichia coli complex I. J. Biol. Chem. 2003;278:43114–43120. doi: 10.1074/jbc.M308247200. [DOI] [PubMed] [Google Scholar]
- 29.Mamedova AA, Holt PJ, Carroll J, Sazanov LA. Substrate-induced conformational change in bacterial complex I. J. Biol. Chem. 2004;279:23830–23836. doi: 10.1074/jbc.M401539200. [DOI] [PubMed] [Google Scholar]
- 30.Matsuno-Yagi A, Hatefi Y. Studies on the mechanism of oxidative phosphorylation. J. Biol. Chem. 1985;260:14424–14427. [PubMed] [Google Scholar]
- 31.Boveris A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984;105:429–435. doi: 10.1016/s0076-6879(84)05060-6. [DOI] [PubMed] [Google Scholar]
- 32.Ernster L, Lee C-P. Energy-linked reduction of NAD+ by succinate. Methods Enzymol. 1967;10:729–738. [Google Scholar]
- 33.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 34.Schägger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 35.Anderson JD, Blobel G. Immunoprecipitation of proteins from cell-free translations. Methods Enzymol. 1983;96:111–120. doi: 10.1016/s0076-6879(83)96012-3. [DOI] [PubMed] [Google Scholar]
- 36.Sazanov LA, Peak-Chew SY, Fearnley IM, Walker JE. Resolution of the membrane domain of bovine complex I into subcomplexes: implications for the structural organization of the enzyme. Biochemistry. 2000;39:7229–7235. doi: 10.1021/bi000335t. [DOI] [PubMed] [Google Scholar]
- 37.Ichimaru N, Abe M, Murai M, Senoh M, Nishioka T, Miyoshi H. Function of the alkyl side chains of Δlac-acetogenins in the inhibitory effect on mitochondrial complex I (NADH-ubiquinone oxidoreductase) Bioorg. Med. Chem. Lett. 2006;16:3555–3558. doi: 10.1016/j.bmcl.2006.03.082. [DOI] [PubMed] [Google Scholar]
- 38.Hansch C, Leo A. Exploring QSAR: fundamentals and applications in chemistry and biology. ACS Professional Reference Book, American Chemical Society; Washington, DC: 1995. pp. 125–168. [Google Scholar]
- 39.Herrero A, Barja G. Localization of the site of oxygen radical generation inside the complex I of heart and nonsynaptic brain mammalian mitochondria. J. Bioenerg. Biomembr. 2000;32:609–615. doi: 10.1023/a:1005626712319. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002;80:780–787. doi: 10.1046/j.0022-3042.2002.00744.x. [DOI] [PubMed] [Google Scholar]
- 41.Lambert AJ, Brand MD. Superoxide production by NADH:ubiquinone oxidoreductase (complex I) depends on the pH gradient across the mitochondrial inner membrane. Biochem. J. 2004;382:511–517. doi: 10.1042/BJ20040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lambert AJ, Brand MD. Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH-ubiquinone oxidoreductase (complex I) J. Biol. Chem. 2004;279:39414–39420. doi: 10.1074/jbc.M406576200. [DOI] [PubMed] [Google Scholar]
- 43.Issacs NS. Physical Organic Chemistry. John Wiley & Sons, Inc.; New York: 1987. pp. 210–254. [Google Scholar]
- 44.Gluck MR, Krueger MJ, Ramsay RR, Sablin SO, Singer TP, Nicklas WJ. Characterization of the inhibitory mechanism of 1-methyl-4-phenylpyridinium and 4-phenylpyridine analogs in inner membrane preparations. J. Biol. Chem. 1994;269:3167–3174. [PubMed] [Google Scholar]
- 45.Miyoshi H, Inoue M, Okamoto S, Ohshima M, Sakamoto K, Iwamura H. Probing the ubiquinone reduction site of mitochondrial complex I using novel cationic inhibitors. J. Biol. Chem. 1997;272:16176–16183. doi: 10.1074/jbc.272.26.16176. [DOI] [PubMed] [Google Scholar]
- 46.Miyoshi H, Iwata J, Sakamoto K, Furukawa H, Takada M, Iwamura H, Watanabe T, Kodama Y. Specificity of pyridinium inhibitors of the ubiquinone reduction sites in mitochondrial complex I. J. Biol. Chem. 1998;273:17368–17374. doi: 10.1074/jbc.273.28.17368. [DOI] [PubMed] [Google Scholar]
- 47.Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol. Cell. Proteomics. 2003;2:117–126. doi: 10.1074/mcp.M300014-MCP200. [DOI] [PubMed] [Google Scholar]
- 48.Belogrudov GI, Hatefi Y. Catalytic sector of complex I (NADH-ubiquinone oxidoreductase): Subunit stoichiometry and substrate-induced conformation changes. Biochemistry. 1994;33:4571–4576. doi: 10.1021/bi00181a018. [DOI] [PubMed] [Google Scholar]
- 49.Yamaguchi M, Belogrudov GI, Hatefi Y. Mitochondrial NADH-ubiquinone oxidoreductase (complex I): Effects of substrates on the fragmentation of subunits by trypsin. J. Biol. Chem. 1998;273:8094–8098. doi: 10.1074/jbc.273.14.8094. [DOI] [PubMed] [Google Scholar]
- 50.Vinogradov AD, Grivennikova VG. The mitochondrial complex I: Progress in understanding of catalytic properties. IUBMB Life. 2001;52:129–134. doi: 10.1080/15216540152845920. [DOI] [PubMed] [Google Scholar]
- 51.Bottcher B, Scheide D, Hesterberg M, Nagel-Steger L, Friedrich T. A novel, enzymatically active conformation of the Escherichia coli NADH-ubiquinone oxidoreductase (complex I) J. Biol. Chem. 2002;277:17970–17977. doi: 10.1074/jbc.M112357200. [DOI] [PubMed] [Google Scholar]
- 52.Tocilescu MA, Fendel U, Zwicker K, Kerscher S, Brandt U. Exploring the ubiquinone binding cavity of respiratory complex I. J. Biol. Chem. 2007;282:29514–29520. doi: 10.1074/jbc.M704519200. [DOI] [PubMed] [Google Scholar]
- 53.Darrouzet E, Issartel JP, Lunardi J, Dupuis A. The 49-kDa subunit of NADH-ubiquinone oxidoreductase (complex I) is involved in the binding of piericidin and rotenone, two quinone-related inhibitors. FEBS Lett. 1998;431:34–38. doi: 10.1016/s0014-5793(98)00719-4. [DOI] [PubMed] [Google Scholar]
- 54.Trumpower BL, Gennis RB. Energy transduction by cytochrome complexes in mitochondrial and bacterial respiration: the enzymology of coupling electron transfer reactions to transmembrane proton translocation. Annu. Rev. Biochem. 1994;63:675–716. doi: 10.1146/annurev.bi.63.070194.003331. [DOI] [PubMed] [Google Scholar]
- 55.Crofts AR. The cytochrome bc1 complex: function in the context of structure. Annu. Rev. Physiol. 2004;66:689–733. doi: 10.1146/annurev.physiol.66.032102.150251. [DOI] [PubMed] [Google Scholar]
- 56.Magnitsky S, Toulokhonova L, Yano T, Sled VD, Hägerhäll C, Grivennikova VG, Burbaev DS, Vinogradov AD, Ohnishi T. EPR characterization of ubisemiquinones and iron-sulfur cluster N2, central components of the energy coupling in the NADH-ubiquinone oxidoreductase (complex I) in situ. J. Bioenerg. Biomembr. 2002;34:198–208. doi: 10.1023/a:1016083419979. [DOI] [PubMed] [Google Scholar]
- 57.Yano T, Dunham WR, Ohnishi T. Characterization of the ΔμH+-sensitive ubisemiquinone species (SQNf) and the interaction with cluster N2: new insight into the energy-coupled electron transfer in complex I. Biochemistry. 2005;44:1744–1754. doi: 10.1021/bi048132i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.