

The waters of the “red tide” are awash with noxious agents, the most infamous of which are the paralytic shellfish poisons (PSPs).1 Small molecule, bis-guanidinium structures — saxitoxin, neosaxitoxin, and the gonyautoxins — unique in both their form and function, represent the principle constituents of PSPs.2 These highly polar, heteroatom-rich compounds are exquisitely designed corks that act to stopper ion flux through voltage-gated Na+ channels (NaV), thus inhibiting electrical conduction in cells.3 The intricate molecular shape common to these toxins coupled with their importance as pharmacological tools for ion channel study have inspired efforts aimed at their de novo assembly. Three prior works have described preparations of saxitoxin (STX) and one a decarbamoyloxy form.4,5 The first synthetic path to any member of the more than 20 known sulfated poisons, gonyautoxin 3 (GTX 3), is outlined in this report (Figure 1).6,7
Figure 1.
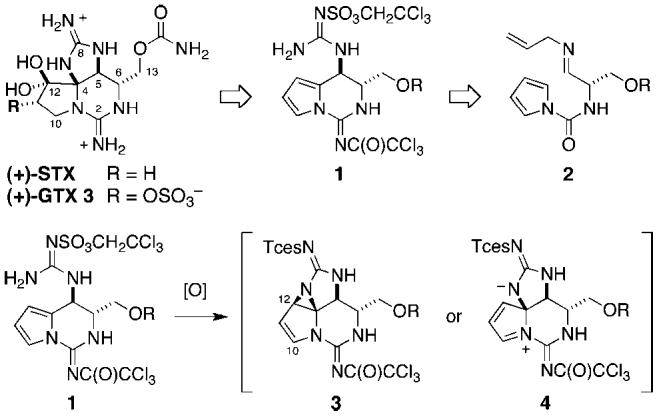
Pyrrole oxidation highlights synthetic approach to GTX 3.
The five-membered cyclic guanidine in GTX 3 became the focal point of our synthetic analysis following our recent disclosure of an oxidative method for 2-aminoimidazoline formation.8 This transformation is thought to proceed through the intermediacy of a Rh-bound guanidine nitrene, a reactive species capable of modifying both C-H and π-bonds. For the purpose of crafting GTX 3, amination of a pyrrole nucleus by the guanidine nitrenoid presented a novel application of this technology (Figure 1). Such a reaction could occur through either a strained aziridine 3 or dipolar species 4, attack of which by a nucleophile at either C10 or C12 would generate the desired tricyclic core.9 This regiochemical issue notwithstanding, such a strategy simplifies the GTX problem to a rather unassuming bicyclic intermediate 1. Pursuant to this approach, a route to bis-guanidine 1 was formulated that would exploit an intramolecular addition of a pyrrole to an activated imine. Although limited in precedent, this type of Pictet-Spengler reaction could be quickly evaluated, as the necessary precursor 2 is easily accessed from serine.
The synthesis of GTX 3 commences with a three-step sequence that transforms L-serine methyl ester to aldehyde 5 (Scheme 1).10 Condensation of this aldehyde with allylamine is followed by treatment with BF3 · OEt2, which effects the desired ring closure to furnish the trans-substituted urea 6 with >20:1 diastereoselectivity.11 Assuming the C5/C6 stereochemistry (GTX numbering) in this product is established under kinetic control, a conformation that minimizes allylic strain between the substituents on C6 and N7 could account for the observed sense of induction. Forwarding 6 to the requisite amination precursor 7 was efficiently achieved through a sequence of four transformations; of note is the development of a single step process for sequential allyl deprotection and isothiourea formation (cf., step e, 6→7, Tces = SO3CH2CCl3).
Scheme 1a.

a Conditions: (a) pyrrole-1-carboxylic acid, DCC, Et3N, CH2Cl2, 65%; (b) tBuPh2SiCl, imidazole, DMF, 97%; (c) iBu2AlH, CH2Cl2, -90 °C; (d) allylamine, BF3•OEt2, CH2Cl2, 56% (2 steps, >20:1 trans/cis); (e) Pd(PPh3)4, 1,3-dimethylbarbituric acid, CH2Cl2; then Na2CO3, TcesN=(SMe)Cl, 94%; (f) EtOSO2CF3, 2,4,6-tri-tert-butylpyrimidine, CH2Cl2, 47 °C, 78%; (g) NH3, NH4OAc, MeOH, 60 °C, 82%; (h) CCl3C(O)Cl, iPr2NEt, CH2Cl2, -20 °C, 87%; (i) 5 mol% Rh2(esp)2, PhI(OAc)2, MgO, CH2Cl2, 42 °C, 61%; (j) Et3SiH, BF3 • OEt2, CH2Cl2, 81%; (k) nBu4NF, THF; (l) Cl3CC(O)NCO, CH2Cl2, -20 °C; then MeOH, 76% (2 steps); (m) 2 mol% OsO4, NMO, THF/H2O, 81%; (n) PhC(O)CN, DMAP, CH2Cl2/MeCN, -78 °C, 67%; (o) Dess-Martin periodinane, CH2Cl2, 79%; (p) H2, Pd/C, CF3CO2H, MeOH; then NH3, MeOH, 83%; (q) DMF•SO3, 2,6-di-tert-butyl-4-methylpyridine, NMP, 71%.
Successful application of the Rh-catalyzed amination reaction with guanidine 7 assembles the tricyclic frame of GTX 3 in a singular, defining event. The reaction is chemoselective, as C-H insertion into the proximal C6 center does not appear to compete with pyrrole modification. Acetic acid, produced as a byproduct in this transformation, adds regio- and stereoselectively to the putative aziridine, affording N,O-acetal 8 as the only product based on 1HNMR analysis of the reaction mixture.12,13 In spite of the fact that acetate attack occurs exclusively at C10 instead of C12, the isolated tricycle 8 is suitably disposed for completion of the GTX 3 synthesis.
The stability of 8 toward handling and purification proved somewhat capricious, thus prompting a decision to reduce the presumably labile N,O-acetal unit. This transformation is smoothly performed with Et3SiH and BF3 · OEt2, giving the C11-C12 alkene in 81% yield. None of the transposed olefin product is detected under these conditions. Installation of the 1° carbamate is then made possible using C13CC(O)NCO.14 Intermediate 9 contains all of the necessary carbon centers found in the natural product.
Alternative approaches for transforming alkene 9 to the corresponding α-ketol have been examined. Regioselective ketohydroxylation would provide the most expeditious route to the desired target; such conditions have not yet been identified.4f By contrast, olefin dihydroxylation using 2 mol % OsO4 and N-methylmorpholine-N-oxide is quite effective and affords diol 10 as a single stereoisomer. Analysis of molecular models indicates that the β-face of the alkene in 9 is more exposed, consistent with the observed selectivity. Protection of the C11-OH is accomplished under highly optimized conditions that employ benzoyl cyanide and DMAP. Other, more standard acylating agents (e.g., PhC(O)Cl) in combination with 3° amine or pyridine bases produce inseparable mixtures of isomeric, benzoylated materials. While it is possible to install alternative blocking groups such as tBuMe2Si- at C11, their larger steric volume prevents subsequent oxidation of the C12 alcohol. With 11, ketone formation at C12 is enabled using Dess-Martin periodinane.15
Removal of all three protecting groups in 12 through a single operation affords 11 β-hydroxysaxitoxin, which is isolated as the bis-C3F7CO-2 salt.4f Analytical data for this material (1H NMR, HRMS) match those found in the literature.6a,b,16
To complete the synthesis of GTX 3, selective sulfation of the C11 alcohol is achieved using DMF · SO3 in combination with 2,6-di-tert-butyl-4-methylpyridine as an acid scavenger. The pure toxin is obtained as the C3F7CO-2 adduct following reversed-phase HPLC. This material matches the reported physical properties of natural GTX 3 in all respects and blocks electrical transmission in heterologously expressed NaV1.4 ion channels with an IC50 value of ~20 nM (reported IC50 = 13.2-33.5 nM).17 Upon standing GTX 3 in aqueous solution at pH = 8, epimerization at C11 occurs to give GTX 2, also in agreement with published observations (Figure 2).6a,16a
Figure 2.
Epimerization at C11 occurs upon standing.
The completed synthesis of GTX 3 offers a unique strategic approach for preparing this family of guanidinium toxins and related structures, while underscoring the power of Rh-catalyzed amination for heterocycle assembly.18 Access to unnatural forms of the toxin should be possible following this work.
Supplementary Material
Acknowledgment
J.V.M. is grateful to the National Science Foundation for a graduate fellowship. This work has been supported by a grant from the NIH and by generous gifts from Pfizer, Amgen, Boehringer-Ingelheim, and GlaxoSmithKline.
Footnotes
Supporting Information Available: Analytical data for selected compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Botana LM, editor. Seafood and Freshwater Toxins: Pharmacology, Physiology, and Detection. Marcel Dekker; New York: 2000. [Google Scholar]
- (2).For leading reviews, see: Llewellyn LE. Nat. Prod. Rep. 2006;23:200–222. doi: 10.1039/b501296c.. Hall S, Strichartz G, Moczydlowski E, Ravindran A, Reichardt PB. ACS Symp. Series. 1990;418:29–65.
- (3).(a) Kao CY, Levinson SR, editors. Tetrodotoxin, Saxitoxin, and the Molecular Biology of the Sodium Channel. Vol. 479. New York Academy of Sciences; New York: 1986. [PubMed] [Google Scholar]; (b) Tikhonov DB, Zhorov BS. Biophys. J. 2005;88:184–197. doi: 10.1529/biophysj.104.048173. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tanino H, Nakata T, Kaneko T, Kishi Y. J. Am. Chem. Soc. 1977;99:2818–2819. doi: 10.1021/ja00450a079.. Kishi Y. Heterocycles. 1980;14:1477–1495.. Jacobi PA, Martinelli MJ, Polanc S. J. Am. Chem. Soc. 1984;106:5594–5598.. Martinelli MJ, Brownstein AD, Jacobi PA, Polanc S. Croat. Chem. Acta. 1986;59:267–295.. Fleming JJ, Du Bois J. J. Am. Chem. Soc. 2006;128:3926–3927. doi: 10.1021/ja0608545.. Fleming JJ, McReynolds MD, Du Bois J. J. Am. Chem. Soc. 2007;129:9964–9975. doi: 10.1021/ja071501o.. Following their earlier report, Kishi et al. have described an asymmetric synthesis of (-)-decarbamoylsaxitoxin; see: Hong CY, Kishi Y. J. Am. Chem. Soc. 1992;114:7001–7006.
- (5).Iwamoto O, Koshino H, Hashizume D, Nagasawa K. Angew. Chem., Int. Ed. 2007;46:8625–8628. doi: 10.1002/anie.200703326. [DOI] [PubMed] [Google Scholar]
- (6).(a) Shimizu Y, Buckley LJ, Alam M, Oshima Y, Fallon WE, Kasai H, Miura I, Gullo VP, Nakanishi K. J. Am. Chem. Soc. 1976;98:5414–5416. doi: 10.1021/ja00433a072. [DOI] [PubMed] [Google Scholar]; (b) Boyer GL, Schantz EJ, Schnoes HK. J. Chem. Soc., Chem. Commun. 1978:889–890. [Google Scholar]; (c) Onodera H, Satake M, Oshima Y, Yasumoto T, Carmichael WW. Nat. Toxins. 1997;5:146–151. doi: 10.1002/1522-7189(1997)5:4<146::AID-NT4>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- (7).An approach to GTX 2 and 3 has been described, see: Hannick SM, Kishi Y. J. Org. Chem. 1983;48:3833–3835.
- (8).Kim M, Mulcahy JV, Espino CG, Du Bois J. Org. Lett. 2006;8:1073–1076. doi: 10.1021/ol052920y. [DOI] [PubMed] [Google Scholar]
- (9).Analogous oxidation reactions with indole derivatives give evidence for a zwitterionic intermediate; see: Padwa A, Flick AC, Leverett CA, Stengel T. J. Org. Chem. 2004;69:6377–6386. doi: 10.1021/jo048990k.
- (10).Boger DL, Patel M. J. Org. Chem. 1987;52:2319–2323. [Google Scholar]
- (11).An X-ray crystal structure of a modified form of this intermediate confirms the trans stereochemical assignment
- (12).The instability of this material on SiO2 is likely responsible for the reduced isolated yields
- (13).The choice of solvent has a rather substantial influence on the performance of this step, CH2Cl2 being the only medium in which complete consumption of the starting guanidine 7 is observed
- (14).Kocovsky P. Tetrahedron Lett. 1986;27:5521–5524. [Google Scholar]
- (15).Hexavalent chromium, TEMPO, and DMSO-based oxidation protocols universally consumed starting material without generating ketone 12
- (16).(a) Wichmann CF, Boyer GL, Divan CL, Schantz EJ, Schnoes HK. Tetrahedron Lett. 1981;22:1941–1944. [Google Scholar]; (b) Shimizu Y, Kobayashi M, Genenah A, Oshima Y. Tetrahedron. 1984;40:539–544. [Google Scholar]
- (17).(a) Choudhary G, Shang L, Li X, Dudley SC., Jr. Biophys. J. 2002;83:912–919. doi: 10.1016/S0006-3495(02)75217-X. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kao CY, Kao PN, James-Kracke MR, Koehn FE, Wichmann CF, Schnoes HK. Toxicon. 1985;23:647–655. doi: 10.1016/0041-0101(85)90369-1. [DOI] [PubMed] [Google Scholar]
- (18).An attempt to utilize guanidine C-H insertion for assembling bromopyrrole-derived natural products has been described; see: Wang S, Romo D. Angew. Chem., Int. Ed. 2008;47:1284–1286. doi: 10.1002/anie.200703998.. For a recent review of this family of molecules, see:KÖck M, Grube A, Seiple IB, Baran PS. Angew. Chem., Int. Ed. 2007;46:6586–6594. doi: 10.1002/anie.200701798.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.