

Abstract
Several widely used antibiotics such as β-lactams, glycopeptides, and lipoglycopeptides exhibit their activity by interfering with peptidoglycan biosynthesis. Ever-increasing resistance to these and other agents has placed a renewed emphasis on the need to understand the reactions in this bio-synthetic pathway at the molecular level. While efficient access to many of the biosynthetic enzymes has been gained, the isolation of their natural substrates has proven difficult. Chemical synthesis has provided valuable tools to circumvent this problem and has allowed convenient access to several key intermediates and analogs thereof. Recent advances in the synthesis of the late-stage intermediates, including the Park nucleotide, lipid I, lipid II, fragments of the bacterial cell wall, along with other biochemical probes are reviewed. A brief discussion on the use of these substrates in study of this important biosynthetic pathway is also included.
Keywords: Peptidoglycan, Bacterial cell wall, Antibiotics, Glycopeptide
Introduction
Bacterial peptidoglycan consists of a network of β-[1,4]-linked carbohydrate polymers that are cross-linked by pendant peptide side chains.[1,2] The biosynthetic pathway is believed to occur in three distinct stages (Scheme 1). The first stage takes place within the bacterial cytoplasm resulting in the conversion of UDP-N-acetylglucosamine (UDP-GlcNAc) into UDP-N-acetyl muramic acid pentapeptide (Park nucleotide).[2] The initial step in this pathway is facilitated by the MurA enzyme and involves incorporation of an enolpyruvyl group onto the C(3) hydroxyl group of the UDP-GlcNAc precursor. Subsequent reduction mediated by MurB, an NADPH-dependent enolpyruvyl reductase, provides UDP-N-acetylmuramic acid (UDP-MurNAc, 2) that contains the lactate handle that serves as the anchor for incorporation of the pentapeptide (or stem peptide) side chain. The stem peptide side chain is then incorporated through a series of reactions mediated by ATP-dependent amino acid ligases (MurC-F). The ligases facilitate sequential incorporation of 3-Ala, 3-Glu, 3-Lys,[3] and 3-Ala-3-Ala to provide UDP-N-acetylmuramic acid pentapeptide, also known as the Park nucleotide 3.[4]
Scheme 1.

The bacterial cell wall biosynthesis pathway.
The second stage of the peptidoglycan biosynthetic cascade takes place at the cytoplasmic surface of the bacterial cell membrane. The first reaction is a pyrophosphate exchange reaction wherein the UDP-MurNAc-pentapeptide precursor is coupled to a membrane-anchored C55 lipid carrier to provide undecaprenylpyrophosphoryl-MurNAc-pentapeptide, also known as lipid I 4. In a second reaction, MurG catalyzes the transfer of GlcNAc from a UDP-GlcNAc precursor to the C(4) hydroxyl group of the lipid-linked MurNAc-pentapeptide. The product of this reaction, lipid II 5, is the final monomeric intermediate utilized in the peptidoglycan biosynthetic cascade.[5]
In the third and final stage of peptidoglycan biosynthesis, lipid II is translocated to the extracellular surface of the bacterial cell membrane where the membrane-anchored disaccharyl pentapeptide is polymerized into β-[1,4]-linked glycan strands through the action of the membrane-bound bacterial transglycosylases. The undecaprenyl carrier lipid released during the glycosylation reaction is subsequently translocated across the cytoplasmic membrane to be recycled. In reactions mediated by the bacterial transpeptidases, the glycan polymers are then cross-linked through amide bond formation between the terminal amino group of the lysine residue, or the amino terminus of an attached peptide chain (vide supra), and the penultimate 3-Ala residue of a neighboring glycan strand. The amide bond is established with concomitant loss of the terminal 3-Ala residue. Mature peptidoglycan is a single macromolecule, which may contain multiple layers of cross-linked glycan strands, and precisely defines the size and shape of the bacterial cell.
Chemotherapeutic agents that inhibit bacterial cell wall (peptidoglycan) biosynthesis have dominated treatment regimens for bacterial infections in both hospital and outpatient settings for over fifty years.[6] Glycopeptide antibiotics (e.g., vancomycin) and β-lactams, two of the most widely utilized classes of antibiotics, derive their antibiotic activity through inhibition of the enzyme-mediated transglycosylation and/or transpeptidation reactions that occur on the extracellular surface of the bacterial cell membrane.[6,7] The rapid emergence of antimicrobial resistance has now begun to erode the once dependable clinical efficacy of these chemotherapeutic agents and has become an increasingly significant threat to public health.
In response to the rapid increase in bacterial resistance, a renewed emphasis has been placed on the study of the bacterial enzymes within the peptidoglycan biosynthesis pathway. Structural and mechanistic information gained from these studies could make possible the design of chemotherapeutic agents with novel mechanisms of action. Advances in protein biochemistry and molecular biology have now provided efficient access to the enzymes utilized for peptidoglycan biosynthesis; however, isolation of their respective substrates from natural sources remains a significant problem. Access to substantial quantities of the bio-synthetic precursors is necessary to enable detailed study of peptidoglycan biosynthesis and resistance mechanisms that have evolved through use of cell wall active agents.
Isolation of the biosynthetic intermediates from bacterial sources poses some significant challenges. First, each of the intermediates is present in very low copy numbers. For example, in E. coli, the copy numbers for lipid I and lipid II are estimated to be approximately 700 and 1000–2000 molecules per cell, respectively.[8] Second, separation of miniscule amounts of the cell wall precursors from the comparatively large amounts of cellular lipid and membrane components is technically challenging and further compromises the ability to isolate sufficient quantities of the natural substrates to enable detailed mechanistic studies.
Over the last decade, efforts have leveraged organic synthesis as a vehicle to procure these valuable biosynthetic intermediates. Here we discuss the recent advances in this area that have targeted the late-stage intermediates; namely, the Park nucleotide, lipid I, and lipid II. This review will focus on methods, both chemical and chemoenzymatic, for the preparation of these intermediates. Substrate analogues of the lipid intermediates, while having demonstrated their utility in the development of assay systems to identify inhibitors, and for kinetic characterization of the biosynthetic enzymes, will not be discussed in significant detail. These have already been the subject of an excellent review.[9] We will also present a brief discussion on how the cell wall bio-synthetic intermediates, and cell wall fragments, can be used to study peptidoglycan biosynthesis along with the structure of peptidoglycan itself.
Synthesis of Park Nucleotide
The total synthesis of Park nucleotide[10] demonstrated the power and utility of chemical synthesis as a means to procure advanced intermediates utilized in the peptidoglycan biosynthetic pathway and provided the foundation for subsequent synthetic approaches directed toward lipid I and lipid II. The synthetic strategy relied upon a Khorana–Moffatt coupling[11] protocol for introduction of the glycosyl nucleoside diphosphate linkage (Scheme 2). This process couples glycosyl phosphates (e.g., 6) with nucleoside 5′-phosphoromorpholidates (e.g., 7) and is typically performed, after global deprotection, as a final step in glycosyl nucleoside diphosphate syntheses. These reactions are often limited by the insolubility of unmasked glycosyl phosphates in suitable organic solvents. Because the pentapeptide chain present in the muramyl coupling partner was likely to exacerbate this problem, the coupling reaction was performed on a protected form of peptidomuramyl phosphate 6.
Scheme 2.

Park nucleotide retrosynthetic analysis.
Protecting groups were chosen that allowed base-mediated global deprotection in a single step in order to preserve the acid sensitive anomeric pyrophosphate moiety. Peptidylmuramyl phosphate 6 was prepared by peptide coupling of the protected pentapeptide 9 with an activated ester deriving from the protected muramic acid derivative 8.
The synthesis began with conversion of the benzyl N-acetyl-4,6-benzylidenemuramic acid 10, available in three steps from N-acetylglucosamine, to the corresponding phenylsulfonylethyl ester. Acid-mediated cleavage of the benzylidene acetal followed by acetylation provided the di-acetate 11.
A phosphitylation/oxidation protocol was utilized for introduction of the anomeric phosphate.[12] This choice was made based on the premise that methods that employ electrophilic carbohydrate components with participating groups at C(2) generally favor formation of 1,2-trans-linked (β-anomer) glycosyl phosphates. The anomeric hydroxyl group was unmasked by hydrogenolytic cleavage of the benzyl ether protective group. Treatment of the intermediate lactol with dibenzyl N,N-diethylphosphoramidite in the presence of 1,2,4-triazole afforded the anomeric phosphite 12 as the major component of a 2.5:1 (α/β) anomeric mixture. The labile anomeric phosphites were oxidized to their respective phosphates and purified by silica gel chromatography (Scheme 3).
Scheme 3.

Chemical synthesis UDP-N-acetylmuramyl pentapeptide (Park nucleotide).
Under these conditions, the α-anomer 8 was obtained exclusively in 42 % overall yield from 11; the β-anomer (1,2-trans) was not isolable likely due to rapid oxazoline formation involving the acetamido group at C(2).
The carboxyl group of 8 was unmasked through DBU-mediated β-elimination. The pentapeptide side chain 9 was assembled from commercially available Cbz-3-Ala-3-Ala-(OMe) using standard Boc protection and EDC coupling procedures. Coupling of peptide 9 with the muramyl carboxyl fragment provided the muramyl pentapeptide 13 in 70 % overall yield from 8. Hydrogenolytic debenzylation of 13 in the presence of cyclohexylamine provided the corresponding cyclohexylammonium salt, a salt chosen to enhance solubility of the phosphate intermediate in organic solvents. Khorana–Moffatt coupling of the cyclohexyl-ammonium phosphate salt with uridine 5′-phosphoromor-pholidate under anhydrous conditions in DMF afforded the protected UDP-N-acetylmuramyl pentapeptide. Global de-protection with aqueous sodium hydroxide followed by reverse phase HPLC purification provided pure UDP-N-acetylmuramyl pentapeptide in 32 % overall yield from 13.
Wong and co-workers have disclosed a chemoenzymatic route to the Park nucleotide (Scheme 4).[13] In this synthesis, UDP-GluNAc was transformed into UDP-N-acetylmuramic acid upon treatment with MurA and MurB. Stereoselective reduction of the intermediate UDP-GlcNAc enolpyruvate (mediated by MurB) was facilitated through in situ generation of NADPH from NADP using glucose and a thermostable glucose dehydrogenase. A depsipentapeptide analog 17, which served as a vancomycin resistant cell wall precursor, was also efficiently accessed using depsipeptide 16 in the peptide-coupling step.
Scheme 4.

Chemoenzymatic synthesis of Park nucleotide and UDP-N-acetylmuramyl depsipentapeptide.
Although the chemoenzymatic strategy is generally efficient and obviates the need of lengthy protecting group manipulations, it suffers from some inherent limitations. First, isolation and purification of enzymes on preparative scale is necessary. This can be difficult to achieve especially with the downstream, membrane associated enzymes. Second, enzyme-mediated synthesis may not be suitable for analog preparation due to substrate specificity of the enzymes. Thus, chemical synthesis offers the most versatile and reliable route for large-scale preparation of all the advanced cell wall intermediates and their analogs.
Synthesis of Lipid I
Compared to the Park nucleotide, lipid I is a much more challenging synthetic target due to presence of the unsaturated undecaprenyl side chain. The lipid side chain is linked to the carbohydrate backbone by an anomeric diphosphate. The anomeric diphosphate linkage is extremely acid sensitive because it is both allylic and glycosidic. Thus, any synthetic transformations that take place after introduction of the lipid diphosphate linkage cannot utilize acidic reaction conditions. This consideration also mandated that base-cleavable protective groups must be utilized for the carbohydrate core and the peptide side chain if a late-stage global deprotection was desired. In addition, the hydrophobic nature of the lipid side chain raised the possibility that solubility issues or micelle formation might interfere with synthetic transformations subsequent to its introduction. Taken together, these considerations clearly suggested a strategy that involved a late-stage introduction of the lipid diphosphate while minimizing the number of synthetic transformations required after its introduction.
Two groups have independently reported a chemical synthesis of lipid I and both were qualitatively similar to the synthetic route developed by Hitchcock for synthesis of the Park nucleotide.[10] The first synthesis was reported by Walker within the context of an effort directed toward the identification of an optimal substrate for a bacterial transglycosylase assay.[14] The synthetic lipid I served as a substrate for conversion into lipid II through incubation with purified MurG isolated from E. coli. VanNieuwenhze and co-workers subsequently reported a total synthesis of lipid I.[15] The synthetic routes used by each group were qualitatively very similar; the latter of these two synthetic efforts will be discussed in the paragraphs that follow.
One of the shortcomings of the Park nucleotide synthesis was the lack of anomeric selectivity for the phosphitylation/ oxidation sequence utilized to install the anomeric phosphate. For example, α-lactol 18 (the product deriving from hydrogenolysis of the benzyl ether 11, Scheme 3) displayed only a modest preference (2.5:1) for the desired α-phosphate (Scheme 5). In related work, directed toward the synthesis of lipid I analogues for use in the development of an assay to measure MurG inhibition, Walker reported a similar phosphitylation/oxidation sequence for a related substrate that provided the α-anomer as the exclusive product in good chemical yield (Scheme 5).[16]
Scheme 5.
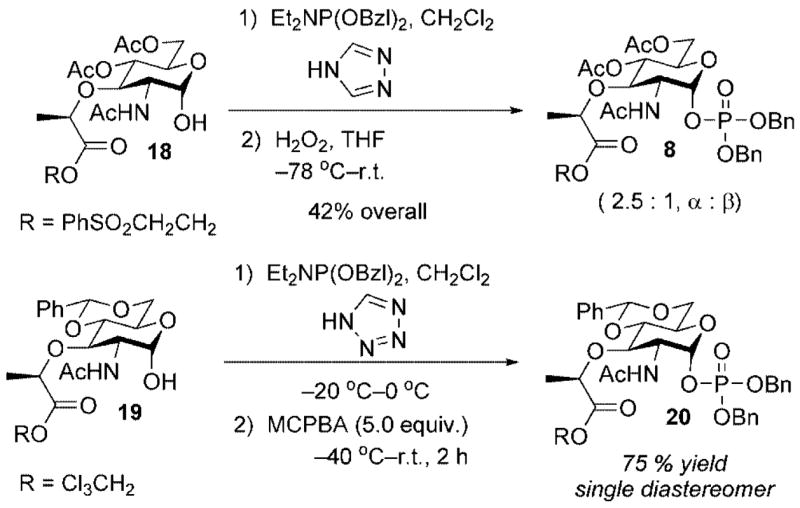
Comparison of phosphitylation/oxidation protocols.
Although different protective group schemes were used in each example, we felt that the enhanced α-selectivity observed by the Walker group could be due to the enhanced acidity of 1H-tetrazole (pKa = 4.9) relative to 1,2,4-triazole (pKa = 10.0). The greater acidity of 1H-tetrazole would provide a greater equilibrium concentration of the activated phosphoramidite reagent such that capture by the α-lactol occurs at a much faster rate than anomerization and capture by the more nucleophilic β-lactol. Improving the anomeric selectivity for this reaction was important in terms of providing a sufficient throughput of material to complete the lipid I and lipid II synthetic efforts (vide infra).
Thus, as depicted in Scheme 6, exposure of α-lactol 18 to dibenzyl-N,N-diethylphosphoramidite and 1H-tetrazole in dichloromethane followed by oxidation of the intermediate anomeric phosphite with 30 % hydrogen peroxide provided the target α-phosphate 8 in 86 % yield. The lactyl handle required for incorporation of the peptide side chain was cleanly unmasked upon exposure of 8 to DBU and provided carboxylic acid 21 in 95 % yield. Activation of 21 (EDCI, NHS, DMF) followed by addition of pentapeptide 9 and iPr2NEt provided differentially protected muramyl pentapeptide 13 in 75 % yield.
Scheme 6.

Synthesis of lipid I.
Having achieved efficient access to a differentially protected version of phosphomuramyl pentapeptide 13, the stage was now set for incorporation of the lipid diphosphate and completion of the lipid I total synthesis. A number of methods, both chemical and enzymatic, were available for preparation of glycosyl diphosphates. Application of the Khorana–Moffat protocol,[11] mentioned earlier within the context of the Park nucleotide synthesis, would have required independent synthesis of a phosphoromorpholidate deriving from the lipid or carbohydrate fragment. Given this limitation, and the potential instability of such activated glyosidic or allylic phosphate intermediates, we chose to investigate in situ activation protocols for installation of the prenyl-linked diphosphate.
Several procedures have been developed for the construction of lipid-linked glycosyl diphosphates. For example, phosphoric anhydrides,[17] phosphoryl dichlorides,[18] and phosphoroimidazolidates[19,20] have been used as electrophiles for the addition of nucleophilic phosphate salts. The phosphoroimidazolidate protocol[20] was selected because, of the three methods highlighted above, it was the only method that proceeded through initial activation of the carbohydrate component. This allowed the use of commercially available undecaprenyl monophosphate directly in a coupling reaction with the activated carbohydrate component and allowed facile introduction of the chemically sensitive diphosphate linkage under relatively mild reaction conditions.
Thus, cleavage of the benzyl ether protective groups from the anomeric phosphodiester was easily achieved by hydrogenolysis; the intermediate anomeric phosphate was isolated as its monopyridyl salt 22 and used directly without purification. Addition of 1,1′-carbonyldiimidazole (CDI) to a solution of 22 in a solution of THF/DMF (4:1) provided the intermediate phosphoroimidazolidate. After careful quenching of excess CDI by slow addition of methanol, a solution of undecaprenyl monophosphate diammonium salt and 1H-tetrazole was added by syringe. The progress of the reaction was conveniently monitored by mass spectrometry until complete disappearance of activated phosphoroimidazolidate intermediate was observed. This crude reaction mixture was concentrated in vacuo, dissolved in a dioxane/ water (1:1) solvent mixture, and treated with 1 3 NaOH. After stirring at room temperature for two hours, the mixture was filtered and purified by reverse-phase HPLC. Lyophilization of the pure fractions provided pure lipid I in 40 % overall yield from muramyl pentapeptide 13.
Synthesis of Lipid II
Lipid II is the ultimate monomeric intermediate utilized in bacterial cell wall biosynthesis and has been the target of synthetic efforts by three research groups. Walker and co-workers have published a chemoenzymatic approach to lipid II. In this route, lipid I was prepared by chemical synthesis and was then converted to lipid II, and various analogues containing modified lipid side chains, using purified MurG from E. coli (vide supra).[14] A series of lipid II analogues (25a–25e) was prepared in this manner. Unfortunately, even though it is the natural substrate for MurG, lipid I (4) itself reacted slowly and provided the lowest amount of product – in this case, lipid II (5) – when compared to the other precursors containing modified lipid side chains (Scheme 7). Thus, this approach was not optimal for preparative scale synthesis of endogenous lipid II.
Scheme 7.

Enzymatic conversion of lipid I to lipid II.
Two chemical syntheses of lipid II have been reported; one by VanNieuwenhze and co-workers[21] and a second by Schwartz and co-workers.[22] Both share a qualitatively identical synthetic pathway and differ from one another with respect to the protecting group scheme that was employed.[23]
The retrosynthetic analysis from the VanNieuwenhze synthesis of lipid II is presented below and will illustrate the general strategy used by both groups (Scheme 8).[24] The analysis further shows that although the strategic bond constructions closely parallel those used for the lipid I total synthesis, the additional carbohydrate residue, N-acetylglucosamine (NAG), and its β-[1,4] linkage to the adjoining N-acetylmuramic acid (NAM) residue added further complexity to the overall synthetic strategy.
Scheme 8.

Retrosynthetic analysis for lipid II.
Starting from the lipid II target 5, and in analogy to the lipid I synthesis presented above, the lipid diphosphate was incorporated at a late stage of the synthesis. This minimized the number of subsequent synthetic transformations that this chemically sensitive linkage needed to withstand and diminished any solubility-related issues that might arise upon incorporation of the lipid side chain. In addition, this left only a global deprotection as a final step. Thus, the first disconnection revealed disaccharyl pentapeptide 26 and commercially available undecaprenyl monophosphate 27. Base-cleavable protective groups were used for all peripheral functionality in 26 in order to insure the acid-sensitive lipid diphosphate linkage would survive the subsequent global deprotection step.
A second retrosynthetic disconnection revealed disaccharyl monopeptide 28 and tetrapeptide 29. The tetrapeptide fragment could be prepared using standard peptide chemistry. Disaccharyl monopeptide 29 employed a triply orthogonal protective group scheme in order to insure selective unmasking of the C-terminal carboxyl group for peptide coupling. This disconnection, however, brought with it the risk for epimerization of the α-stereocenter of the 3-Ala residue upon carboxyl activation for peptide coupling. Our original synthetic plan called for introduction of the entire pentapeptide side chain in a single operation. The rationale for this change in strategy will be discussed below.
The anomeric phosphate in 28 would be introduced by a two-step sequence involving unmasking of the anomeric hydroxyl group (protected as its benzyl ether) followed by application of the identical phosphitylation/oxidation sequence utilized for lipid I. It is again important to note here that the protective group scheme needed to be triply orthogonal in order to allow selective unmasking of the anomeric hydroxyl and lactate carboxyl groups in the presence of other protected functionality. In turn, disaccharide 28 could be obtained from a Königs–Knorr glycosidation reaction between glycosyl donor 30 and acceptor 31. A carbamate protective group was chosen for the C(2) amino substituent of glycosyl donor 30 based upon its ability to participate in neighboring group stabilization of the oxonium ion intermediate and thus favoring selective production of the desired β-glycosidic linkage.
Glycosyl donor 30 was prepared on three steps from glucosamine[25] while acceptor 31 was easily prepared in three steps from a commercially available muramic acid derivative 32 (Scheme 9).[26]
Scheme 9.

Synthesis of glycosyl acceptor 31.[26]
The identification of 31 as the optimal glycosyl acceptor was based upon the premise that an ether protective group for the C(6) hydroxyl would result in more efficient nucleophilic capture of the donor by the adjacent C(4) hydroxyl during the Königs–Knorr glycosylation reaction when compared to donors possessing acyl protective groups at C(6). This, in turn, required a protecting group interconversion (benzyl to acetate), subsequent to the glycosylation reaction, in order to achieve global deprotection in a single operation in the final stages of the synthesis.
A second consideration was also involved in the selection of 31 as the glycosyl acceptor for the Königs–Knorr reaction used for preparation of disaccharide 28. This consideration arose from the need to execute a regioselective reductive opening of benzylidene acetal 34, or a derivative, in order to selectively unmask the C(4) hydroxy group of the acceptor for capture of the glycosyl donor. Preliminary investigation of the reductive ring-opening, using conditions previously reported by DeNinno[28] for regioselective ring opening of 4,6-O-benzylidene acetals resulted in isolation of the lactone 35 as the major reaction product (Scheme 10). The simplest solution to this problem was to install the first amino acid residue present in the stem peptide side chain. While this very effectively suppressed lactonization during the reductive ring opening, this modification came with the inherent risk of epimerization at the 3-Ala α-stereocenter during the carboxyl activation necessary for introduction of the remainder of the pentapeptide side chain.[27]
Scheme 10.
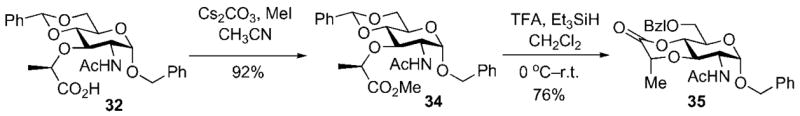
Reductive lactonization of muramic acid derivative 32.
When the glycosyl donor 30 (Pg2 = Troc) and the acceptor 31 (R2 = CH2CH2SO2Ph, R3 = Bzl) were combined under rigorously anhydrous Königs–Knorr conditions (Ag-OTf/CH2Cl2), the anticipated β-linked disaccharide was isolated in 74 % yield (Scheme 11). The choice of the Troc protective group was driven by two major considerations: First, we needed a participating group that would favor production of the desired β-[1,4] linkage during the glycosylation reaction without the risk of competitive oxazoline formation. Second, previous work by Wong and co-workers had demonstrated that Troc-protected 2-amino-2-deoxyglucopyranose donors showed a forty-fold rate enhancement in their glycosylation reactions when compared to phthaloyl-protected 2-amino-2-deoxyglucopyranose donors.[29]
Scheme 11.

Preparation of NAG-NAM disaccharide 37.
At this point it was necessary to change the protective group scheme believed to be necessary for maximizing the efficiency of the of glycosylation reaction with a scheme optimized for late-stage global deprotection. This was conveniently achieved in a one-pot reaction that accomplished two protective group exchange reactions. Glycosylation product 36 was dissolved in acetic anhydride/acetic acid. Exposure of this solution to anhydrous zinc chloride cleanly removed the C(6) benzyl ether, revealing a free hydroxyl group, that was rapidly acylated in situ. Zinc dust was then added to the same reaction mixture to facilitate reductive cleavage of the Troc group from the glucosamine nitrogen; the free amino group was also acylated under the reaction conditions. Disaccharide 37 was obtained in gram quantity in 67 % overall yield after purification by silica gel chromatography.
Access to an ample supply of an orthogonally protected NAG-NAM disaccharide subsequently enabled our final approach to lipid II (Scheme 12). The synthetic operations are virtually identical to those utilized for the synthesis of lipid I. The lipid II end game began with liberation of the anomeric hydroxy group through hydrogenolysis of the corresponding benzyl ether 37. The alcohol 38 was obtained in 94 % yield and exposed to the two-step phosphitylation/ oxidation procedure described above. Anomeric phosphate 28 was obtained in 78 % overall yield from 38. DBU-initiated β-elimination of phenyl vinyl sulfone provided an intermediate carboxylate salt that was activated for peptide coupling by conversion to the corresponding NHS ester. Exposure of the activated ester intermediate to tetrapeptide 29 – prepared by standard amino acid coupling chemistries – provided the disaccharyl pentapeptide 39 in 46 % overall yield for the three step deprotection/activation/coupling sequence. Reductive cleavage of the phosphodiester protective groups, followed by exposure of the intermediate phosphate to pyridine, provided the monopyridyl salt 26 in 94 % yield. Finally, activation of the anomeric phosphate as its phosphoroimidazolidate, coupling with undecaprenyl monophosphate 27, and a final global deprotection provided lipid II 5 as a white solid after reverse phase HPLC purification.
Scheme 12.

Completion of the lipid II synthesis.
Peptidoglycan Fragments
Recently, there has been a great deal of activity directed toward study of the final stages of bacterial cell wall biosynthesis. For example, Mobashery and co-workers have devised versatile synthetic routes to cell wall fragments of varied architecture in order to study the balance between D,D-transpeptidase and D,D-peptidase activity of a soluble version of a penicillin binding protein[30] and to develop a model for the three-dimensional structure of bacterial cell wall peptidoglycan.[31]
With respect to the former goal, the Mobashery group developed efficient synthetic routes toward peptidoglycan fragments 40 and 41 (Scheme 13). Each is an analog of lipid I (proceeding from 42), and lipid II (proceeding from 46), lacking the full undecaprenyl diphosphate side chain. The synthetic route to 40 also demonstrated the feasibility for late-stage introduction of the lactate handle onto the protected NAG-NAM disaccharide; a modification that avoided the possibility of unwanted side reactions [e.g., intramolecular lactonization (Scheme 10), and racemization at the lactate stereocenter during peptide coupling].[30]
Scheme 13.

Synthesis of cell wall fragments used to characterize penicillin binding proteins.
The Mobashery group has also prepared a bacterial cell wall fragment 47 that is comprised of a NAG-NAM-NAG-NAM tetrasaccharide with two pendant stem peptides. Their synthesis relied on use of the tetrasaccharide 48 containing orthogonal protective groups (e.g., benzyl vs. acetate) to differentiate the C(3) hydroxyl groups in the emerging NAG and NAM carbohydrate subunits (Scheme 14). The C(3) acetate groups in 48 were cleaved by treatment with NaOMe and the lactyl ethers were installed by alkylation of the free hydroxyl groups in the presence of excess (S)-2-chloropropionic acid. The pentapeptide 45 was then coupled with the free carboxylic acids through generation of a p-nitrophenol ester intermediate. Finally, cleavage of the benzylidene acetal and subsequent global deprotection afforded the target cell wall fragment 47.[31]
Scheme 14.

Synthesis of cell wall fragment 47.

The preceding sections highlight the impact that chemical synthesis has had on the preparation of intermediates utilized in the peptidoglycan biosynthesis pathway. This capability, when coupled with access to the biosynthetic enzymes, and/or compounds that in some way disrupt the bio-synthetic pathway, has provided new avenues for the study and development of novel agents for the treatment of bacterial infections. In the following section, we will highlight some of the recent applications of these tools toward the study of novel antibacterial agents and peptidoglycan biosynthesis.
Utility of Bacterial Cell Wall Intermediates
Although agents have been identified that inhibit many of the steps in the peptidoglycan biosynthetic pathway,[32] most antibiotic agents that inhibit bacterial PG biosynthesis exert their activity at the later stages of this biochemical pathway.[33] Therefore, availability of the advanced intermediates involved in the process has greatly facilitated studies on such antibiotics.[34] These studies have been mainly directed toward identification of the precise cellular targets and delineating the binding mode between the antibiotic and its target substrate at the molecular level. Recent work on mechanism of action of ramoplanin A2 serves as an illustration.[35]

Initial reports by Somner and Reynolds suggested that ramoplanin likely inhibits a membrane-associated step in bacterial cell wall biosynthesis, although it did not block the formation of lipid I.[36] Thus, they proposed that ramoplanin inhibited the glycosyltranferase reaction catalyzed by MurG through binding and sequestering the lipid I intermediate. This hypothesis gained broad acceptance over the next decade.
The cellular location of MurG was unknown at the time of the Somner and Reynolds work. Moreover, because there was no reliable source of lipid I at the time, there was no means to directly monitor MurG activity nor was there a straightforward method to assess ramoplanin binding to lipid I. Later, with the natural substrate(s) available by synthetic routes, Walker and co-workers carried out systematic kinetic studies of the reactions mediated by the MurG and transglycosylase (E. coli PBP1b) enzymes. They unambiguously showed that ramoplanin inhibits transglycosylation by binding to lipid II (in 2:1, ramoplanin: lipid II stoichiometry).[37] Additional data suggested that MurG inhibition by ramoplanin may involve direct binding to MurG rather than binding to lipid I.[38] Furthermore, the Walker group also showed that ramoplanin inhibited MurG at concentrations at least 10-fold higher than the MIC values. On the other hand, the kinetics for translycosylase inhibition indicated that ramoplanin bound tightly to lipid II (at 10–7 3), which was in accordance with the MIC values.[37] These experiments strongly suggested that the transglycosylasylation reaction was the preferred target of ramoplanin, not the MurG-mediated glycosyltransferase reaction as originally proposed by Somner and Reynolds.[36]
Research efforts in our laboratories have been focused on chemical synthesis and biological studies on peptide antibiotics that active against vancomycin- and methicillin-resistant Gram-positive strains. We are currently investigating three peptides, katanosin B 51, plusbacin A3 52, and mersacidin 53 with the ultimate goal of developing novel peptide antibiotics with enhanced antibacterial activity. All three peptides are believed to exert their activity through sequestration of the lipid intermediates (i.e., lipid I and lipid II); however, competition experiments have suggested their respective binding sites are different than that used by vancomycin.[39–41] Their precise mechanism of action remains to be unequivocally established although the available evidence suggests that each may inhibit the transglycosylation reaction. It may also be possible that each has a secondary mode of action; namely, inhibition of MurG through sequestration of lipid I. Plusbacin A3 and katanosin B are potent against vancomycin-resistant enterococci as well as methicillin-resistant Staphylococcus aureus (MIC = 0.39–3.13 μg/ mL).[41] We are interested in determining the substrate specificities, and the correlation between the binding affinity for the lipid intermediate(s) and antibacterial activities of these peptides and their synthetic analogs. Also, the knowledge of the three-dimensional structure of the antibiotics bound with their lipid substrates, in a lipid-like environment will be extremely useful. To achieve these goals, access to the late stage lipid intermediates is of paramount importance.
Mersacidin is active against several Gram-positive bacteria including streptococci, bacilli, and methicillin-resistant Staphylococcus aureus.[42,43] Its mode of action is believed to involve complex formation with the lipid II intermediate and inhibition of the transglycosylation reaction.[39,40] The mersacidin-lipid II complex appears to be selective for lipid II because none of the lipid II biosynthetic precursors were able to antagonize the activity of the antibiotic. Because several of these precursors possess peptide side chains terminating in 3-Ala-3-Ala, it seems likely that complex formation does not involve the vancomycin-binding site. Binding of mersacidin to growing cells and isolated membranes was not inhibited by vancomycin, thus implicating simultaneous adsorption at two distinct binding sites. In addition, mersacidin was found to have activity against vancomycin-resistant Enterococcus faecium, a strain that synthesizes PG precursors terminating in 3-Ala-3-Lac.[39]

Studies with mersacidin have demonstrated that it has a preference for complexation with lipid II as compared to other biosynthetic intermediates. Bonvin has recently published a report on NMR study of mersacidin and lipid II interaction in dodecylphosphocholine (DCP) micelles, which were used to mimic the membrane environment. This study showed that the compact, globular structure of mersacidin opens up at a hinge region (Ala-12 and Abu-13) upon complexation with lipid II.[44] This structural change directly affects the exposure of charged groups suggesting that electrostatic interactions might dictate the binding mechanism. The structure of mersacidin is highly sensitive to the surrounding environment. Therefore, such structural data when obtained in a lipid bilayer environment, which better mimics the cell surface, might provide more relevant information.
Synthetic derivatives of the late-stage biosynthetic intermediates have also found several other applications in the study of the mechanics of bacterial cell wall biosynthesis, assay development for the identification of inhibitors of this process, and for bacterial cell wall engineering. For example, Mobashery has reported the synthesis of cell wall fragments 40 and 41 (Scheme 13) as probe substrates to assess the balance between D,D-transpeptidase activity (i.e., the stem-peptide cross linking reaction) and D,D-peptidase activity.[30] The latter reaction results in the cleavage of the C-terminal 3-Ala residue and serves as a control to modulate the extent of cross-linking by the D,D-transpeptidases. Both 40 and 41 were shown to be substrates for cloned and purified PBP5 from E. coli and enabled determination of the kinetic parameters for the reactions mediated by this enzyme. In addition, Mobashery has developed a model for the three-dimensional structure of cell wall fragment 47[31] wherein the carbohydrate backbone of peptidoglycan adopts a right-handed helix of defined periodicity – three NAG-NAM repeats per helix turn. This information has provided the foundation for a proposed model for the three-dimensional structure of bacterial cell wall peptidoglycan.[45]
In addition to the elegant work elucidating the likely mode of action of ramoplanin A2, the Walker group has made several important contributions to the study of the peptidoglycan biosynthetic pathway; specifically, the MurG and transglycosylation reactions, that have made use of modified peptidoglycan precursors. Lipid I derivative 54 was prepared for use as a substrate in the development of a MurG assay system.[16,46] The citronellyl lipid side chain enhances the solubility of 54, relative to lipid I itself, and improves stability by removing the chemically sensitive allylic double bond. The substrate was prepared by a synthetic route that was qualitatively similar to that illustrated for lipid I (vide supra); the major difference being the presence of a citronellyl diphosphate linkage in place of the natural undecaprenyl diphosphate linkage.[16,46] Compound 54 was incubated with UDP-(14C)-GlcNAc and MurG. Reaction progress was monitored through measuring the amount of radiolabeled lipid II product. Initial attempts to use 54 in this assay system uncovered problems with separation of the radiolabeled product from excess UDP-(14C)-GlcNAc. This issue was conveniently addressed by conversion of 54 into the biotin-tagged derivative 56 thus allowing the radiolabeled product to be easily separated from other radioactive components in the reaction mixture through use of an avidinderivatized resin.[16,46]
The Walker group has also reported a detailed study of the relationship between lipid chain length, along with geometry of the allylic double bond, and substrate reactivity in transglycosylase assays employing lipid II derivatives (Scheme 7). Interestingly, lipid II analogs bearing lipid chains much shorter than the natural C55 substrate, or analogs having a trans geometry at the double bond proximal to the diphosphate linkage (e.g., 25b,e) did not react, while an analog containing a lipid side chain with seven prenyl units (25d) was an excellent substrate (Scheme 15).[9,14]
Scheme 15.

Synthesis of a biotin-tagged lipid I derivative for MurG assay development.
Schwartz and co-workers (vide supra) have used lipid II for the development of an assay system for observation of the glycosyl transfer and transpeptidation reactions catalyzed by PBP1b of E. coli (Scheme 16).[22] Progress of the glycosyl transfer reaction was readily monitored by incubation of lipid II with recombinant PBP1b and removal of aliquots of the reaction mixture at specified time points. After adjusting the pH of the reaction mixture (pH ≥9), fluorescamine 57 was added to the reaction mixture. Unreacted lipid II formed the fluorescent condensation product 58, whose presence was readily monitored by HPLC. Significantly, the transpeptidation reaction could also be monitored by the same reaction system through visualization of the fluorescent adduct 59 formed through condensation of the by-product of the transpeptidation reaction, the terminal 3-Ala residue of the stem peptide, with fluorescamine. This, in principle, would allow simultaneous monitoring of the transglycosylation and transpeptidation processes from a single reaction mixture.[22] The Schwartz group has also developed a continuous fluorescence assay system that utilizes a dansylated lipid II reporter substrate in order to determine the effect of pH on the glycosyl transfer reaction.[47]
Scheme 16.

Lipid II labeling for monitoring transglycosylation reaction.
Finally, Nishimura has used derivatives of the Park nucleotide to provide functional handles to derivatize the surface of bacterial cell walls (Scheme 17).[48–50] This method relies upon the display of ketone functional handles on cell surfaces that are presented for capture by external nucleophiles. Compound 61 was prepared by a simple carbodiimide-mediated coupling reaction between the ε-amino group if the lysine residue in the Park nucleotide (UDP-MurNAc-pentapeptide) and 4-oxopentanoic acid 60.
Scheme 17.

Display of fluorescent and epitope tags on bacterial cell surface.
Bacterial cells were incubated with 61 that resulted in its incorporation into the cell wall. After collection of the bacteria by centrifugation, the bacteria were resuspended in buffer and treated with a fluorescent dye (e.g., Alexa Fluor® 388 hydrazide 62) or oligomannose derivative 63. Fluorescent labeling of the cell wall was easily confirmed by fluorescence microscopy;[48–50] display of oligomannose on the surface of bacterial cell walls was demonstrated by an increase in adhesion of the sugar-displaying bacteria onto concancavalin A films as measured by surface plasmon resonance.[50] Chemical engineering of the bacterial cell surface may allow for a variety of epitopes to be displayed on the bacterial cell surface that could find several applications, such as in the development of novel drugs and vaccines or in the modulation of bacterial pathogenicity.
In summary, the intent of this review was to highlight the significant contributions that organic synthesis has made to the study of bacterial cell wall biosynthesis. This survey was not intended to be an exhaustive review of this field, but rather to illustrate how organic chemistry has contributed to this important field of research. Organic synthesis has provided powerful solutions to the problems of substrate availability along with biochemical tools to enable more detailed study of the peptidoglycan biosynthetic pathway. This, in turn, has provided the foundation to conceptually novel approaches (e.g., epitope tagging) for treatment of infections due to pathogenic bacteria. With the continued increase in bacterial resistance, increasing emphasis will be placed upon the quest for new antibacterial agents with novel mechanisms of action. As the examples presented in this review illustrate, organic synthesis can have a significant role in support of this quest.
Biographies

Radha S. Narayan received her BS in chemistry from University of Pune, India, in 1995, and the MS in chemistry from the Indian Institute of Technology, Bombay, in 1997. The same year, she was awarded a Research Fellowship from the Council of Scientific and Industrial Research, India, to conduct research at the National Chemical Laboratory, Pune. She then moved to Michigan State University and obtained her PhD in 2004 working with Prof. Babak Borhan. In 2004, she joined the laboratory of Prof. Michael S. VanNieuwenhze at the University of California, San Diego, as a postdoctoral researcher, where she worked on the total synthesis of the peptide antibiotic mersacidin. Dr. Narayan is currently working at the Center for Chemical Methodology and Library Development at Boston University.

Michael S. VanNieuwenhze received his BA from Kalamazoo College. He received his PhD degree from Indiana University in 1992 under the guidance of W. R. Roush and subsequently worked as a postdoctoral fellow with K. B. Sharpless at the Scripps Research Institute. In 1994 he accepted a position in discovery chemistry research at Eli Lilly and Company, Indianapolis. In 2002, he joined the faculty at the University of California, San Diego. His research interests include: the synthesis and mechanistic study of peptide antibiotics, bacterial cell wall biosynthesis, and cellular processes involving lipid metabolites.
References
- 1.Bugg TDH, Walsh CT. Nat Prod Rep. 1992;9:199. doi: 10.1039/np9920900199. [DOI] [PubMed] [Google Scholar]
- 2.Höltje JV. Microbiol Mol Biol Rev. 1998;62:181. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.In Gram negative bacteria, and some Gram positives, meso-diaminolimelic acid (meso-DAP) is incorporated in place of 3-Lys (see refs 1,2).
- 4.Park JT. J Biol Chem. 1952;194:877. [PubMed] [Google Scholar]
- 5.In some Gram-positive bacteria 1–5 amino acid residues may be attached to the ε-amino group of the lysine (or meso-DAP) residue. These are typically glycine although organisms incorporating 3-serine, 3-threonine, and other amino acids are known (see ref.[2]).
- 6.Gale EF, Cundliffe E, Reynolds PE, Richmond MH, Waring MJ. The Molecular Basis of Antibiotic Action. Vol. 2. Wiley-Interscience; New York: 1981. [Google Scholar]
- 7.Chu DTW, Plattner JJ, Katz L. J Med Chem. 1996;39:3853. doi: 10.1021/jm960294s. [DOI] [PubMed] [Google Scholar]
- 8.van Heijenoort Y, Gomez M, Derrien M, van Heijenoort J. J Bacteriol. 1992;174:3549. doi: 10.1128/jb.174.11.3549-3557.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazar K, Walker S. Curr Opin Chem Biol. 2002;6:786. doi: 10.1016/s1367-5931(02)00355-1. [DOI] [PubMed] [Google Scholar]
- 10.Hitchcock SA, Eid CN, Aikins JA, Zia-Ebrahimi M, Blaszczak LC. J Am Chem Soc. 1998;120:1916. [Google Scholar]
- 11.Roseman S, Distler JJ, Moffatt JG, Khorana HG. J Am Chem Soc. 1961;83:659. [Google Scholar]
- 12.Sim MM, Kondo H, Wong CH. J Am Chem Soc. 1993;115:2260. [Google Scholar]
- 13.Liu H, Sadamoto R, Sears PS, Wong CH. J Am Chem Soc. 2001;123:9916. doi: 10.1021/ja011708w. [DOI] [PubMed] [Google Scholar]
- 14.Ye XY, Lo MC, Brunner L, Walker D, Kahne D, Walker S. J Am Chem Soc. 2001;123:3155. doi: 10.1021/ja010028q. [DOI] [PubMed] [Google Scholar]
- 15.VanNieuwenhze MS, Mauldin SC, Zia-Ebrahimi M, Aikins JA, Blaszczak LC. J Am Chem Soc. 2001;123:6983. doi: 10.1021/ja016082o. [DOI] [PubMed] [Google Scholar]
- 16.Men H, Park P, Ge M, Walker S. J Am Chem Soc. 1998;120:2484. [Google Scholar]
- 17.Warren CD, Jeanloz RW. Biochemistry. 1972;11:2565. doi: 10.1021/bi00764a002. [DOI] [PubMed] [Google Scholar]
- 18.Imperiali B, Zimmerman JW. Tetrahedron Lett. 1990;31:6485. [Google Scholar]
- 19.Danilov LL, Maltsev SD, Shibaev VN, Kochetkov NK. Carbohydr Res. 1981;88:203. [Google Scholar]
- 20.Fang X, Gibbs BS, Coward JK. Bioorg Med Chem Lett. 1995;5:2701. [Google Scholar]
- 21.VanNieuwenhze MS, Mauldin SC, Zia-Ebrahimi M, Winger BE, Hornback WJ, Saha SL, Aikins JA, Blaszczak LC. J Am Chem Soc. 2002;124:3656. doi: 10.1021/ja017386d. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz B, Markwalder JA, Wang Y. J Am Chem Soc. 2001;123:11638. doi: 10.1021/ja0166848. [DOI] [PubMed] [Google Scholar]
- 23.The syntheses also differ with respect to the identity of the second amino acid residue in the stem peptide. In the Schwartz synthesis, this residue is a γ-3-Glu, while in the Van-Nieuwenhze synthesis it is γ-3-Gln.
- 24.The protecting group schemes illustrated in Scheme 8 reflect those utilized in both of the reported synthetic routes to lipid II.
- 25.Imoto M, Yoshimura H, Shimoto T, Sakaguchi N, Kusumoto S, Shiba T. Bull Chem Soc Jpn. 1987;60:2205. [Google Scholar]
- 26.Jeanloz RW, Walker E, Sinäy P. Carbohydr Res. 1968;6:184. doi: 10.1016/s0008-6215(00)85075-2. [DOI] [PubMed] [Google Scholar]
- 27.Saha SL, VanNieuwenhze MS, Hornback WJ, Aikins JA, Blaszczak LC. Org Lett. 2001;3:3575. doi: 10.1021/ol016692t. [DOI] [PubMed] [Google Scholar]
- 28.DeNinno MP, Etienne JB, Duplantier KC. Tetrahedron Lett. 1995;36:669. [Google Scholar]
- 29.Zhang Z, Ollman IR, Ye XY, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734. [Google Scholar]
- 30.Hesek D, Suvorov M, Morio K-i, Lee M, Brown S, Vakulenko SB, Mobashery S. J Org Chem. 2004;69:778. doi: 10.1021/jo035397e. [DOI] [PubMed] [Google Scholar]
- 31.Hesek D, Lee M, Morio K, Mobashery S. J Org Chem. 2004;69:2137. doi: 10.1021/jo035583k. [DOI] [PubMed] [Google Scholar]
- 32.Silver LL. Curr Opin Microbiol. 2003;6:431. doi: 10.1016/j.mib.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Silver LL. Biochem Pharmacol. 2006;71:996. doi: 10.1016/j.bcp.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 34.Breukink E, de Kruiff B. Nat Rev Drug Discovery. 2006;5:321. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 35.Walker S, Chen L, Hu Y, Rew Y, Shin D, Boger DL. Chem Rev. 2005;105:449. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- 36.Somner EA, Reynolds PE. Antimicrob Agents Chemother. 1990;34:413. doi: 10.1128/aac.34.3.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu Y, Helm JS, Chen L, Ye XY, Walker S. J Am Chem Soc. 2003;125:8736. doi: 10.1021/ja035217i. [DOI] [PubMed] [Google Scholar]
- 38.Helm JS, Chen L, Walker S. J Am Chem Soc. 2002;124:13790. doi: 10.1021/ja021097n. [DOI] [PubMed] [Google Scholar]
- 39.Brotz H, Bierbaum G, Leopold K, Reynolds PE, Sahl HG. Antimicrob Agents Chemother. 1998;42:154. doi: 10.1128/aac.42.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brotz H, Bierbaum G, Markus A, Molitor E, Sahl HG. Antimicrob Agents Chemother. 1995;39:714. doi: 10.1128/AAC.39.3.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maki H, Miura K, Yamano Y. Antimicrob Agents Chemother. 2001;45:1823. doi: 10.1128/AAC.45.6.1823-1827.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chatterjee S, Chatterjee DK, Jani RH, Blumbach J, Ganguli BN, Klesel N, Limbert M, Seibert G. J Antibiot. 1992;45:839. doi: 10.7164/antibiotics.45.839. [DOI] [PubMed] [Google Scholar]
- 43.Limbert MD, Isert D, Klesel N, Markus A, Seibert G, Chatterjee S, Chatterjee DK, Jani RH, Ganguli BN. In: Nisin and Novel Lantibiotics. Jung G, Sahl H-G, editors. ES-COM; Leiden: 1991. p. 448. [Google Scholar]
- 44.Hsu STD, Breukink E, Bierbaum G, Sahl HG, de Kruiff B, Kaptein R, van Nuland NAJ, Bonvin AMJJ. J Biol Chem. 2003;278:13110. doi: 10.1074/jbc.M211144200. [DOI] [PubMed] [Google Scholar]
- 45.Meroueh SO, Bencze KZ, Hesek D, Lee M, Fisher JF, Stemmler TL, Mobashery S. Proc Natl Acad Sci USA. 2006;103:4404. doi: 10.1073/pnas.0510182103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ha S, Chang E, Lo MC, Men H, Park P, Ge M, Walker S. J Am Chem Soc. 1999;121:8415. [Google Scholar]
- 47.Schwartz B, Markwalder JA, Seitz SP, Wang Y, Stein RL. Biochemistry. 2002;41:12552. doi: 10.1021/bi026205x. [DOI] [PubMed] [Google Scholar]
- 48.Sadamoto R, Niikura K, Monde K, Nishimura SI. Methods Enzymol. 2003;362:273. doi: 10.1016/s0076-6879(03)01019-x. [DOI] [PubMed] [Google Scholar]
- 49.Sadamoto R, Niikura K, Sears PS, Liu H, Wong CH, Suksomcheep A, Tomita F, Monde K, Nishimura SI. J Am Chem Soc. 2002;124:9018. doi: 10.1021/ja026133x. [DOI] [PubMed] [Google Scholar]
- 50.Sadamoto R, Niikura K, Ueda T, Monde K, Fukuhara N, Nishimura SI. J Am Chem Soc. 2004;126:3755. doi: 10.1021/ja039391i. [DOI] [PubMed] [Google Scholar]