

Abstract
Colorectal cancer (CRC) is a major cause of morbidity and mortality from cancers in the United States. Recent studies have revealed the paradigm in which sequential genetic changes (mutations) result in the progression from normal colonic tissues to frank carcinoma. In particular, the study of hereditary colorectal cancer and polyposis syndromes such as familial adenomatous polyposis and hereditary nonpolyposis colon cancer has contributed enormously to the understanding of the pathogenesis of CRC. Here we describe some of the common genetic pathways in CRC and the mechanisms of action for some of the key genes involved in the formation of CRC. The understanding of the genetic pathways and functions in CRC may lead to the development of novel therapeutic approaches for treating this deadly disease.
Introduction
Colorectal cancer (CRC) is a leading cause of cancer mortality in the Western World. In the United States, CRC is the third most commonly diagnosed cancer in men and women and the second leading cause of cancer-related mortality [1]. Although the mortality associated with CRC has been steadily declining over the past few decades, partly due to earlier detection by screening, CRC remains to be a major public health concern.
The pathogenesis of CRC is complex and includes hereditary and environmental factors. Recent research has shed significant light on the molecular mechanisms leading to CRC formation. Among all cases of CRC, the majorities (~ 75%) are sporadic in origin and the remainders are familial or related to inflammatory bowel diseases [2]. Of the familial cases, approximately one third has a clearly defined genetic basis [2]. Two well-defined hereditary CRC syndromes in particular, familial adenomatous polyposis (FAP) and hereditary nonpolyposis colon cancer (HNCC), have aided in determining the genetic basis underlying most CRC, including sporadic ones. Here we will review the genetic paradigm of CRC, the molecular genetics of hereditary polyposis syndromes, and the molecular mechanisms by which some of the CRC-causing genes function.
The Genetic Paradigm of Colorectal Cancer
Fifteen years ago, Fearon and Vogelstein [3] proposed a genetic model to explain the stepwise formation of CRC from normal colonic tissues. This model states that 1) CRC is the result of changes (mutations) of genes with important functions in regulating cell proliferation or repair of DNA damages, 2) mutations in more than one gene are required, and 3) the sequence of mutations is important in determining the eventual formation of CRC. The model is illustrated in Figure 1, which also incorporated information from more recent studies. The genes involved in the genetic paradigm leading to CRC can be broadly divided into two classes: tumor suppressor genes (TSGs) and oncogenes. TSGs encode proteins that either inhibit cell proliferation or promote cell death (apoptosis). TSGs are often inactivated in CRC. In contrast, oncogenes are activated versions of proto-oncogenes, which are often involved promoting cell proliferation or development. Once activated, oncogenes can lead to accelerated cell growth and contribute to tumor formation. Two independent pathways that lead to CRC are depicted in Figure 1. One is initiated by the mutational inactivation of the TSG adenomatous polyposis coli (APC), which is responsible for FAP and approximately 85% of sporadic CRC [4]. A subset of CRC in this pathway is initiated upon the mutational activation of (β-catenin, the activity of which is normally regulated by APC [5]. The second pathway is through inactivation of a family of TSGs involved in DNA mismatch repair (MMR) including human mutS homolog 2 (MSH2), mutation of human mutL homolog 1 (MLH1), and PMS2. MMR mutation is found in approximately 15% of sporadic CRC and is responsible for HNPCC [4]. Mutational inactivation of additional TSGs, such as DPC4/SMAD4 and p53, and mutational activation of oncogenes, such as COX-2 and K-RAS, are also present in later stages of CRC formation (Fig. 1). Recent studies have also implicated several additional TSGs or oncogenes not included in Figure 1 but that are involved in both hereditary polyposis syndromes and sporadic colorectal cancer. Readers are referred to a recent review on the functions of these genes [6••].
Figure 1.
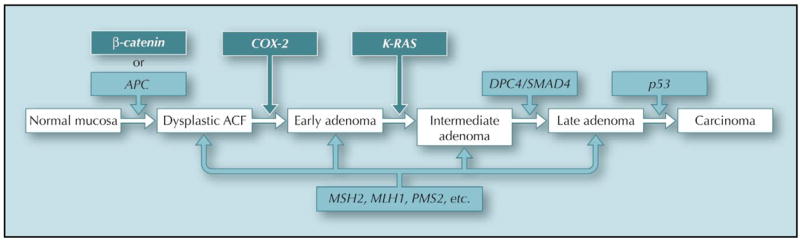
. The genetic paradigm of colorectal cancer. The genetic changes that accompany the stepwise transformation of normal colonic mucosal tissues to carcinoma are depicted in the model. Both mutational inactivation of tumor suppressor genes (light shaded) and activation of oncogenes (dark shaded) are involved. The sequence of gene mutations in the APC pathway is shown on the top and those in the mismatch repair pathway is shown in the bottom. ACF—aberrant crypt foci; APC— adenomatous polyposis coli; COX-2—cyclooxygenase-2.
Hereditary Colorectal Cancer and Polyposis Syndromes
Approximately one third of all familial cases of CRC have a definitive genetic etiology [2]. These genetic predispositions to CRC are usually characterized by the formation of precursor lesions in the form of either adenoma or hamartoma [6••]. An adenoma and a hamartoma differ from each other by their cells of origin: adenoma is derived from cells of the colonic epithelium and hamartoma from cells of the stroma. However, both lesions pose an increased risk for CRC. The majorities of genes involved in hereditary CRC syndromes are TSGs, and mutations in these genes are transmitted in the germ-line. There are two major types of familial predisposition to adenoma formation: FAP and HNPCC with mutation in the APC and MMR genes as the primary cause, respectively. Familial predisposition to hamartoma formation is also called juvenile polyposis syndrome (JPS) and contains many sub-types. Table 1 is a summary of the various hereditary colorectal cancer and polyposis syndromes. With regard to the mechanisms of action of the respective TSGs responsible for the various hereditary colorectal cancer syndromes, it has been likened that APC is a “gatekeeper” because of the huge number of polyps formed when the gene is mutated [7]. In contrast, the MMR genes are dubbed “caretaker” because of the significantly increased mutation rate when mismatch repairs becomes defective [7]. Lastly, the genes responsible for JPS are christened “landscaper” because their mutations lead to an abnormal stromal environment [7]. The clinical pictures for some of the hereditary colorectal cancer syndromes and the functions of the genes involved in these syndromes are provided below.
Familial adenomatous polyposis
FAP is an autosomal dominant disease characterized by the appearance of hundreds to thousands of adenomatous polyps throughout the gastrointestinal tract, but primarily in the colon, early in the life of affected patients [8]. If left untreated, patients invariably develop colorectal cancer in early adulthood. FAP accounts for approximately 1% of all CRC cases in the United States. FAP results from germline mutations in the gene encoding APC [9–11]. There are several variants of FAP. Gardner’s syndrome is characterized by the presence of extracolonic manifestations, such as osteomas, epidermoid cysts, and a characteristic retinal lesion called the congenital hypertrophy of the retinal pigment epithelium (CHRPE). Turcot’s syndrome is defined as typical FAP and brain tumors including medulloblastoma and glioblastoma, the latter often associated with MMR mutations. Attenuated form of FAP (AAPC) is characterized by fewer than 100 colonic polyps and a later stage of onset of CRC. It is often associated with mutations in either the extreme 5′ or 3′ terminus of the APC transcript [12]. Prophylactic colectomy is the treatment of choice in FAP once colonic polyposis becomes manifested.
The APC I1307K mutation
Most of the APC mutations found in hereditary and sporadic CRC are nonsense mutations—ie, they lead to the formation of a truncated protein. The APC I1307K mutation, in contrast, is a missense mutation in nucleotide position 3920 of the APC coding region, causing a T to A transverion and consequently the substitution of a lysine (K) residue for an isoleucine (I) residue in amino acid position 1307 of the APC protein [13]. Although the amino acid change by itself does not alter the function of the protein, the nucleotide change predisposes to a short hypermutable region of the APC gene. This increases the likelihood of somatic mutation in the gene and confers a twofold increased risk for CRC in carriers of the I1307K mutation, which is relatively prevalent in Ashkenazi Jews [13]. The identification of the cancer susceptibility APC I1307K allele underscores a new paradigm for cancer genetics, with a shift in emphasis from the rare, high-risk alleles that have served as a model for cancer genetics earlier to the common, low-risk alleles that are now being identified in specific populations [14].
MYH-associated polyposis
Germ-line mutations in the human mutY homolog (MYH) gene are found in a small number of patients with classical or attenuated form of adenomatous polyposis but without identifiable APC gene mutation [15•]. The MYH gene functions in base excision repair (BER) and acts in an autosomal recessive manner, with bi-allelic mutations necessary for expression of the phenotype, now called MYH-associate polyposis (MAP). Somatic mutations of the APC gene can be found in tumors from patients with bi-allelic missense or nonsense mutations [15•]. It has been estimated that MAP contributes to approximately 1% of unselected CRC [16].
Hereditary nonpolyposis colorectal cancer
HNPCC is the most common form of inherited CRC syndromes and comprises about 5% of all CRC cases. The minimal criterion of HNPCC is that colorectal carcinoma is diagnosed and histologically verified in at least three first-degree relatives belonging to two or more successive generations. Moreover, the age of onset should be less than 50 years in at least one patient. In addition to the colon (most often the right side), organs commonly affected with cancer include the endometrium, ovary, stomach, biliary and pancreatic system, and urinary tract [17]. HNPCC is an autosomal dominant disorder caused primarily by mutations in no fewer than five MMR genes (Table 1), with those in MLH1 and MSH2 accounting for a majority (90%) of the cases [18]. The MMR genes identify and repair minor errors that occur during DNA replication. The absence of this critical function leads to the accumulation of errors in short, repeated DNA sequences called microsatellites. Tumors from patients with HNPCC exhibit microsatellite instability (MSI). A consequence of mutations in the MMR genes is the inactivation of downstream genes due to MSI. Some of the target genes exhibit important function in regulating cell proliferation. Examples include the type II receptor for transforming growth factor β (TGF-β RII) [19], the receptor for insulin-like growth factor II (IGFIIR) [20], and BAX that is involved in the control of apoptosis [21]. Inactivation of these genes leads to a derangement in cell proliferation and subsequent tumor formation.
Table I.
Summary of the hereditary colorectal cancer and polyposis syndromes
| Syndromes | Genes | Inheritance | Penetrance | Risk of CRC | Polyp burden | Polyp histology |
|---|---|---|---|---|---|---|
| Adenomatous polyposis syndromes | ||||||
| Familial adenomatous polyposis (FAP) | APC | AD | High | 1 | 100-1000s | Adenoma |
| Attenuated FAP | APC | AD | High | >90% | < 100 | Adenoma |
| I1307K mutation | APC | AD | Low | ~ 10% | Few | Adenoma |
| Hereditary nonpolyposis colorectal cancer |
hMLH1
hMSH2 hMSH6 hPMS1 hPMS2 |
AD | High | 0.8 | Few | Adenoma |
| Muir-Torre |
hMLH1
HMSH2 |
AD | High | 0.8 | Few | Adenoma |
| MYH-associated polyposis | MYH | AR | High | 1 | < 100 | Adenoma |
| Blooms syndrome | BLM | AR | Low | 0.1 | Few | Adenoma |
| Hamartomatous (juvenile) polyposis syndromes | ||||||
| Peutz-Jeghers syndrome | LKB1/STK11 | AD | High | 0.4 | Few | Hamartoma |
| Juvenile polyposis coli |
SMAD4
BMPRIA PTEN |
AD | High | 10%–40% | Few | Hamartoma |
| Cowden syndrome | PTEN | AD | High | No change | Few | Hamartoma |
| Bannayan-Riley-Ruvalcaba syndrome | PTEN | AD | High | 10%–40% | Few | Hamartoma |
| Others | ||||||
| Turcot’s syndrome (FAR variant) |
APC
hMLHI hMSH2 |
AD | High | 1 | 100s-1000s | Adenoma |
| Hereditary mixed polyposis syndrome | Unknown | AD | Unknown | 0.3 | Few | Mixed |
AD—autosomal dominant; APC—adenomatous polyposis coli; AR—autosomal recessive; CRC—colorectal cancer.
The hamartomatous polyposis syndromes
Hamartoma refers to the focal excessive growth of cells of stromal origin. Although the cellular elements are mature, they do not reproduce the normal architecture in the surrounding tissue. In the intestinal tract, several discrete familial syndromes characterized by multiple hamartomatous or juvenile polyps have been described. They include Peutz-Jeghers syndrome (PJS), juvenile polyposis coli or JPS, and related syndromes such as Cowden syndrome and Bannayan-Riley-Ruval-caba syndrome.
Peutz-Jeghers syndrome
PJS is an autosomal dominant disorder characterized by melanocytic macules of the lips, buccal mucosa, and digits, multiple gastrointestinal hamartomatous polyps, and an increased risk of various neoplasms, including those of the gastrointestinal tract, breast, ovary, and testicles. Germ-line mutations of the gene encoding LKB1/STK11, a serine-threonine kinase, are found in more than half of the patients with PJS [22,23]. However, not all families with PJS are linked to this locus, suggesting that additional genes are involved in its pathogenesis.
Juvenile polyposis syndrome
JPS is defined by any one of the following criteria: 1) 10 or more colonic juvenile polyps, 2) juvenile polyps throughout the gastrointestinal tract, or 3) any number of juvenile polyps in a person with a family history of JPS. The risk of malignant transformation of a juvenile polyp is between 10% and 40%. Three genes have been linked to JPS: SMAD4 and BMPR1A that are involved in the TGF-β signaling [24], and PTEN, a tumor suppressor gene with phosphatase activity [25]. Mutations in PTEN have also been seen in Cowden and Bannayan-Riley-Ruvalcaba syndromes [26].
The Functions of a Select Number of Genes Involved in CRC
Adenomatous polyposis coli
The APC gene is located on chromosome 5q21 and encodes a large polypeptide of 2843 amino acids with a predicted molecular weight of greater than 300,000 kDa [27]. It is present in a variety of epithelial tissues, often in cells that are postmitotic [28]. Studies indicate that APC participates in the regulation of a myriad of cellular functions including proliferation, differentiation, apoptosis, adhesion, migration, and chromosomal segregation [29••,30]. It accomplishes these multiple tasks by exerting two major but very different kinds of activities. In one, APC serves as a crucial member of the Wnt/β-catenin signaling pathway, which is an important determinant of cell proliferation, differentiation and apoptosis. The other depends on APC’s ability to regulate cytoskeletal proteins including F-actin and microtubules, thus allowing it to regulate adhesion, migration and mitosis. Each of these two functions is briefly summarized below.
The Wnt/β-catenin signaling pathway is also called the canonical Wnt pathway and is essential in for controlling intestinal epithelial cell proliferation [31,32]. A model for Wnt signaling is depicted in Figure 2. Wnt, a secreted glycoprotein, interacts with two cell surface receptors, Frizzled (Fz) and low-density lipoprotein receptor-related protein (LRP) [33–35]. In the absence of Wnt, β-catenin is sequestered in the cytoplasm in a “destruction complex” that includes APC, axin, and glycogen synthase kinase-3β, and is subjected to ubiquitin-mediated degradation [31,32]. Upon binding to its cell surface receptors, Wnt releases β-catenin from the complex, which can also be a result of APC mutation (Fig. 2, right panel). Free β-catenin is shuttled into the nucleus, where it frees T-cell factor (TCF) from their represser, CtBP and Groucho, and converts TCF from a represser into an activator of gene transcription [36]. Among the target genes stimulated by β-catenin/TCF are c-Myc and cyclin D, both essential for the progression of the cell cycle during proliferation [37,38]. The binding between APC and β-catenin is crucial for APC to function as a tumor suppressor as the mutations in APC found in CRC are often those that abolish its ability to down-regulate β-catenin level [39]. The significance of Wnt/β-catenin pathway in the pathogenesis of CRC is further demonstrated by the observations that in the absence of APC mutation, CRC sometimes contains inactivating mutation of axin [40,41] or activating mutation of β-catenin [5].
Figure 2.
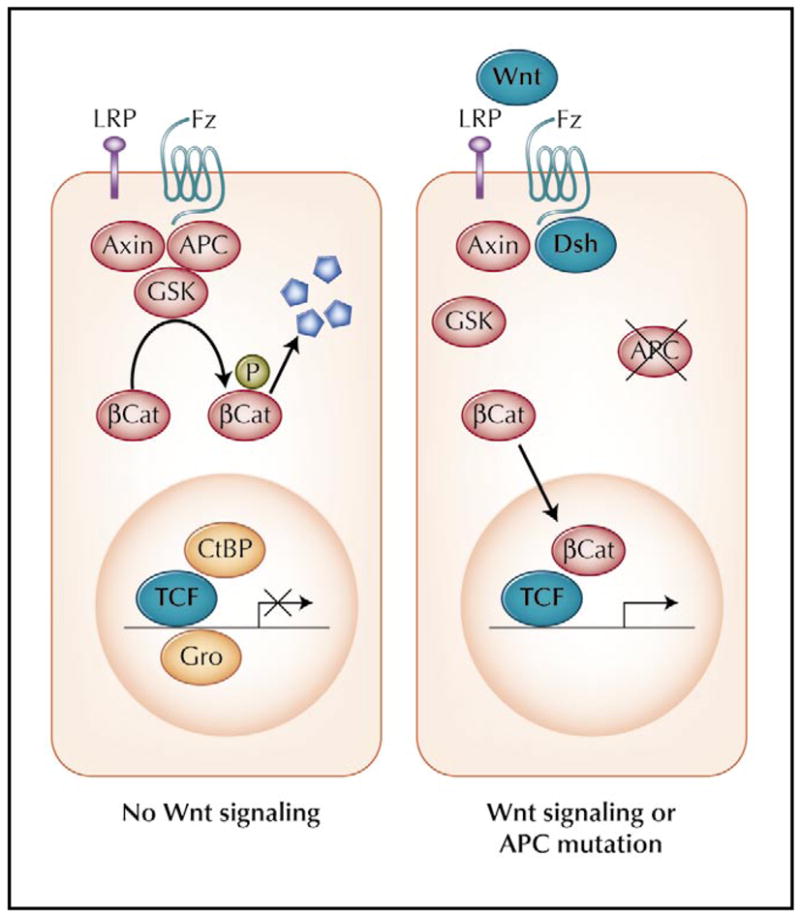
The Wnt/β-catenin signaling pathway. Readers are referred to the text for a detailed description of the pathway. βCat—β-catenin; CtBP—C-terminal binding protein; Dsh—disheveled; Fz—frizzled; Gro—Groucho; GSK—glycogen synthase kinase 3β; LRP—low-density lipoprotein receptor-related protein; TCF—T-cell factor.
Studies indicate that the carboxyl terminal portion of APC binds cytoskeletal proteins including microtubules [42], a microtubule-binding protein EB1 [43], and PDZ domain-containing proteins, through which APC binds F-actin [44]. As a microtubule-associated protein, APC contributes to mitotic spindle formation and function. In early mitosis, APC localizes to the outer kinetochores, consistent with its accumulation at microtubule plus ends [45]. In addition, APC can be detected in centrosomes and mitotic spindles [46]. Cells lacking APC are prone to defects in chromosomal segregation [45]. Importantly, APC mutations have been correlated with chromosomal abnormalities in early colorectal adenomas [47]. These results suggest that loss of APC from normal colonic tissue may lead to chromosomal instability, which may be a contributing factor to tumorigenesis.
p53
The tumor suppressor p53 is mutated in approximately 50% of all human cancers worldwide [48]. Although thought to have oncogenic activity after it was initially identified, p53 was subsequently shown to be a tumor suppressor that is mutated in colorectal cancer [49]. p53 is a transcription factor that normally inhibits cell growth and stimulates cell death when induced by cellular stress [50]. The most common mechanism to disrupt the p53 pathway is through a missense mutation that inactivates its ability to bind specifically to its cognate recognition sequence. However, there are several other ways to achieve the same effect, including amplification of the gene encoding MDM2, a ubiquitin ligase that binds and degrades p53 [51], inactivation of p14/p19ARF that binds and inactivates MDM2 [52], and infection with DNA tumor viruses whose products (such as the E6 protein of the human papilloma-virus) binds and inactivates p53 [53].
p53 has a myriad of functions. It has been shown to be involved in cell cycle regulation, apoptosis, development, differentiation, homologous recombination, DNA excision repair, senescence, and chromosomal segregation [53]. The amount and activity of p53, which is mostly latent in the absence of stress, is increased in response to a variety of signals, such as DNA damage, nucleoside depletion, hypoxia and presence of oncogenes [54]. The regulation of p53 activity by the various stimuli is highly complex and involves multiple proteins such as ataxia telengiectasia mutated (ATM), cell cycle checkpoint kinase (CHK), Nijmegen breakage syndrome (NBS), and DNA-dependent protein kinase (DNAPK) [54]. Phosphorylation of p53 by some of these upstream mediators is crucial for its activation. In addition, several other forms of modification including acetylation, methylation, ubiquitination, and sumoylation have been shown to be important in modulating p53’s activity [55•].
Once activated, p53 initiates a transcription program that reflects the nature of the stress. The genes involved in this network initiate one of several programs including cell cycle arrest at the G1/S and G2/M boundaries, apoptosis and senescence, among others [55•]. An important player in the p53-mediated G1 arrest is the cyclin-dependent kinase inhibitor p21WAF1/CIP1 [56] The G2 arrest is achieved by the ability of p53 to inhibit expression of cyclin B and CDC2 [57], which is essential for the G2/M phase transition, and to stimulate expression of 14-3-3δ [58], which sequesters CDC25 in the cytoplasm rendering it incapable of activating cyclin B/CDC2. Another important event controlled by p53 is apoptosis. p53 activates a large number of genes that contribute to apoptosis [55•]. Several p53-regulated genes (BAX, NOXA, PUMA) enhance secretion of cytochrome c from the mitochondria into the cytoplasm, which leads to activation of caspases and subsequent apoptosis [55•]. This is the intrinsic apoptotic pathway initiated by various stress signals that activate p53. In addition to the intrinsic pathway, p53 also regulates several genes (Fas ligands, killer Dr receptor) that are involved in the extrinsic apoptotic pathway. In contrast to the well studied mechanisms by which p53 mediates cell cycle arrest and apoptosis, little information is available on how p53 causes cell senescence. Moreover, the decision as to just which of these three pathways, cell cycle arrest, apoptosis or senescence, is chosen for the fate of a cell is also not well understood.
K-RAS
The RAS family comprises three isoforms (H-RAS, K-RAS, and N-RAS) with high degrees of sequence identity. All RAS proteins cycle between an active GTP-bound conformation and an inactive GDP-bound conformation [59]. The RAS signaling cascade constitutes a major pathway controlling cell proliferation, and RAS proteins are able to integrate extracellular signals from diverse receptor types. Signal transduction downstream of RAS is accompanied by several effectors. The classical pathway consists of an evolutionarily conserved module of three sequentially activated kinases: RAF, mitogen-activated protein kinase kinase (MAPKK or MEK), and mitogen-activated protein kinase (MAPK). Another effector for RAS is phosphoinositide-3-kinase (PI3K), which regulates the AKT/PKB pathway involved in promotion of cell survival [60]. The significance of RAS in regulating cellular proliferation is illustrated by the presence of activating mutations of K-RAS in approximately one third of all human cancers [61], including up to 50% of colorectal cancer [62]. Moreover, 20% of colorectal tumors with wild type K-RAS bear activating mutations in B-RAF [63]. The complementarity of mutations between K-RAS and B-RAF implies a central role for the RAS-RAF-MAPK pathway in the pathogenesis of colorectal cancer. This conclusion is supported by the observation that targeted expression of oncogenic K-RAS in the intestinal epithelium can cause spontaneous tumorigenesis in mice [64,65]
Conclusions
Immense progress has been made over the past two decades in understanding the molecular genetics of CRC. This has been made possible in part from the study of hereditary conditions that predispose to CRC formation. The biochemical mechanisms by which many of the genes involved in CRC function have also been elucidated. These advances are likely to help develop targeted diagnostic or therapeutic approaches to the management and prevention of CRC.
Acknowledgments
This work was in part support by grants from the National Institutes of Health (DK52230 and CA84197). Vincent W. Yang is the recipient of a Georgian Cancer Coalition Distinguished Cancer Clinician Scientist Award.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.American Cancer Society. Cancer Facts & Figures 2005. [Accessed January 31, 2006]; http://www.cancer.org/downloads/STTCAFF2005f4PWSecured.pdf.
- 2.Hisamuddin IM, Yang VW. Genetics of colorectal cancer. Med Gen Med. 2004;6:13–19. [PMC free article] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 5.Sparks AB, Morin PJ, Vogelstein B, et al. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- 6••.de la Chapelle A. Genetic predisposition to colorectal cancer. Nat Rev Cancer. 2004;4:769–780. doi: 10.1038/nrc1453. This excellent review article discussed the various genetic mechanisms that predispose to colorectal cancer, including those with low-penetrance. [DOI] [PubMed] [Google Scholar]
- 7.Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280:1036–1037. doi: 10.1126/science.280.5366.1036. [DOI] [PubMed] [Google Scholar]
- 8.Cruz-Correa M, Giardiello FM. Familial adenomatous polyposis. Gastrointest Endosc. 2003;58:885–894. doi: 10.1016/s0016-5107(03)02336-8. [DOI] [PubMed] [Google Scholar]
- 9.Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–665. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 10.Nishisho I, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 11.Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 12.Soravia C, Berk T, Madlensky L, et al. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet. 1998;62:1290–1301. doi: 10.1086/301883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laken SJ, Petersen GM, Gruber SB, et al. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC. Nat Genet. 1997;17:79–83. doi: 10.1038/ng0997-79. [DOI] [PubMed] [Google Scholar]
- 14.Gruber SB, Petersen GM, Kinzler KW, et al. Cancer, crash sites, and the new genetics of neoplasia. Gastroenterology. 1999;116:210–212. doi: 10.1016/s0016-5085(99)70246-5. [DOI] [PubMed] [Google Scholar]
- 15•.Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791–799. doi: 10.1056/NEJMoa025283. This article described the identification of biallelic mutation of MYH as the cause of classic adenomatous polyposis syndrome in the absence of any APC mutations. [DOI] [PubMed] [Google Scholar]
- 16.Halford SE, Rowan AJ, Lipton L, et al. Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol 2003. 162:1545–1548. doi: 10.1016/S0002-9440(10)64288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mecklin JP, Jarvinen HJ. Tumor spectrum in cancer family syndrome (hereditary nonpolyposis colorectal cancer) Cancer. 1991;68:1109–1112. doi: 10.1002/1097-0142(19910901)68:5<1109::aid-cncr2820680535>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 18.Papadopoulos N, Lindblom A. Molecular basis of HNPCC: mutations of MMR genes. Hum Mutat. 1997;10:89–99. doi: 10.1002/(SICI)1098-1004(1997)10:2<89::AID-HUMU1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 19.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 20.Souza RF, Appel R, Yin J, et al. Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet. 1996;14:255–257. doi: 10.1038/ng1196-255. [DOI] [PubMed] [Google Scholar]
- 21.Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 22.Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391:184–187. doi: 10.1038/34432. [DOI] [PubMed] [Google Scholar]
- 23.Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet. 1998;18:38–43. doi: 10.1038/ng0198-38. [DOI] [PubMed] [Google Scholar]
- 24.Sayed MG, Ahmed AF, Ringold JR, et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol. 2002;9:901–906. doi: 10.1007/BF02557528. [DOI] [PubMed] [Google Scholar]
- 25.Olschwang S, Serova-Sinilnikova OM, Lenoir GM, et al. PTEN germ-line mutations in juvenile polyposis coli. Nat Genet. 1998;18:12–14. doi: 10.1038/ng0198-12. [DOI] [PubMed] [Google Scholar]
- 26.Marsh DJ, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461–1472. doi: 10.1093/hmg/8.8.1461. [DOI] [PubMed] [Google Scholar]
- 27.Yang VW. APC as a checkpoint gene: the beginning or the end? Gastroenterology. 2002;123:935–939. doi: 10.1053/gast.2002.35773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Midgley CA, White S, Howitt R, et al. APC expression in normal human tissues. J Pathol. 1997;181:426–433. doi: 10.1002/(SICI)1096-9896(199704)181:4<426::AID-PATH768>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 29••.Nathke IS. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu Rev Cell Dev Biol. 2004;20:337–366. doi: 10.1146/annurev.cellbio.20.012103.094541. This paper gave an excellent review of the biochemical and cell biologic mechanisms of action of the tumor suppressor APC. [DOI] [PubMed] [Google Scholar]
- 30.van Es JH, Giles RH, Clevers HC. The many faces of the tumor suppressor gene APC. Exp Cell Res. 2001;264:126–134. doi: 10.1006/excr.2000.5142. [DOI] [PubMed] [Google Scholar]
- 31.Reya T, Clevers H. Wnt signaling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 32.Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 2005;19:877–890. doi: 10.1101/gad.1295405. [DOI] [PubMed] [Google Scholar]
- 33.Bhanot P, Brink M, Samos CH, et al. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- 34.Tamai K, Semenov M, Kato Y, et al. LDL-receptor-related proteins in Wnt signal transduction. Nature. 2000;407:530–535. doi: 10.1038/35035117. [DOI] [PubMed] [Google Scholar]
- 35.Pinson KI, Brennan J, Monkley S, et al. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 36.Behrens J, von Kries JP, Kuhl M, et al. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 37.He TC, Sparks AB, Rago C, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 38.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 39.Miyoshi Y, Nagase H, Ando H, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 40.Jin LH, Shao QJ, Luo W, et al. Detection of point mutations of the Axinl gene in colorectal cancers. Int J Cancer. 2003;107:696–699. doi: 10.1002/ijc.11435. [DOI] [PubMed] [Google Scholar]
- 41.Webster MT, Rozycka M, Sara E, et al. Sequence variants of the axin gene in breast, colon, and other cancers: an analysis of mutations that interfere with GSK3 binding. Genes Chromosomes Cancer. 2000;28:443–453. [PubMed] [Google Scholar]
- 42.Smith KJ, Levy DB, Maupin P, et al. Wild-type but not mutant APC associates with the microtubule cytoskeleton. Cancer Res. 1994;54:3672–3675. [PubMed] [Google Scholar]
- 43.Su LK, Burrell M, Hill DE, et al. APC binds to the novel protein EB1. Cancer Res. 1995;55:2972–2977. [PubMed] [Google Scholar]
- 44.Matsumine A, Ogai A, Senda T, et al. Binding of APC to the human homolog of the Drosophila discs large tumor suppressor protein. Science. 1996;272:1020–1023. doi: 10.1126/science.272.5264.1020. [DOI] [PubMed] [Google Scholar]
- 45.Fodde R, Kuipers J, Rosenberg C, et al. Mutations in the APC tumor suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 46.Kaplan KB, Burds AA, Swedlow JR, et al. A role for the adenomatous polyposis coli protein in chromosome segregation. Nat Cell Biol. 2001;3:429–432. doi: 10.1038/35070123. [DOI] [PubMed] [Google Scholar]
- 47.Leslie A, Stewart A, Baty DU, et al. Chromosomal changes in colorectal adenomas: relationship to gene mutations and potential for clinical utility. Genes Chromosomes Cancer. 2005;45:126–135. doi: 10.1002/gcc.20271. [DOI] [PubMed] [Google Scholar]
- 48.Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 49.Baker SJ, Fearon ER, Nigro JM, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 50.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 51.Momand J, Zambetti GP, Olson DC, et al. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 52.Honda R, Yasuda H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. Embo J. 1999;18:22–27. doi: 10.1093/emboj/18.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wsierska-Gadek J, Horky M. How the nucleolar sequestration of p53 protein or its interplayers contributes to its (re)-activation. Ann N YAcad Sci. 2003;1010:266–272. doi: 10.1196/annals.1299.046. [DOI] [PubMed] [Google Scholar]
- 54.Agarwal ML, Taylor WR, Chernov MV, et al. The p53 network. /Biol Chem. 1998;273:1–4. doi: 10.1074/jbc.273.1.1. [DOI] [PubMed] [Google Scholar]
- 55•.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. This article reviewed the various positive and negative mechanisms of regulation of the tumor suppressor p53’s activity. [DOI] [PubMed] [Google Scholar]
- 56.el-Deiry WS, Tokino T, Velculescu VEL, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 57.Taylor WR, Stark GR. Regulation of the G2/M transition byp53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 58.Hermeking H, Lengauer C, Polyak K, et al. 14-3-3 sigma is a p53-regulated inhibitor of G2/M progression. Mol Cell. 1997;1:3–11. doi: 10.1016/s1097-2765(00)80002-7. [DOI] [PubMed] [Google Scholar]
- 59.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 60.Shields JM, Pruitt K, McFall A, et al. Understanding Ras: it ain’t over ‘til it’s over. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 61.Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 62.Andreyev HJ, Norman AR, Cunningham D, et al. Kirsten ras mutations in patients with colorectal cancer: the “RASCAL II” study. Br J Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rajagopalan H, Bardelli A, Lengauer C, et al. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 64.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 65.Janssen KP, el-Marjou F, Pinto D, et al. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi: 10.1053/gast.2002.34786. [DOI] [PubMed] [Google Scholar]