

Abstract
The phosphine-catalyzed addition of 2,3-butadienoates to aldehydes has been extended to the formation of disubstituted dihydro-2-pyrones. The requisite shift in equilibrium of the intermediate zwitterionic β-phosphonium dienolates toward the s-cis intermediate was accomplished through the use of a Brønsted acid additive, which disrupts the favorable Coulombic interaction present in the s-trans intermediate. The detailed nature of the synergistic interactions involving the Brønsted acid additives and phosphine involved in the formation of s-cis β-phosphonium dienolates was analyzed through a series of DFT calculations. Unlike previously reported annulations of aldehydes with allenoates, where trialkylphosphines are optimal catalysts, in this study triphenylphosphine was also found for the first time to be a suitable catalyst for the synthesis of dihydropyrones. This method provides a one-step route toward functionalized dihydropyrones from simple, stable starting materials. In addition, new reaction pathways of phosphine-catalyzed allene annulations are unveiled, with the formation of dihydropyrones being the first example of dual activation in this sphere.
Keywords: Phosphine, Catalysis, Lewis Base, Brønsted Acid, Dihydropyrone, Aldehyde, Allenoate
1. Introduction
Dual activation is a powerful technology in the development of novel reaction pathways.1 In particular, the combined use of compatible acidic and basic reagents to activate multiple reactive centers in reactants can have a powerful synergistic effect on rate and reaction modality; additionally, enantioselective processes are possible with a single chiral activator.2 We became interested in applying this concept of dual activation to the phosphine-catalyzed annulations of 2,3-butadienoates. Although numerous catalytic asymmetric Morita–Baylis–Hillman (MBH) processes have been reported employing chiral Brønsted acids that are compatible with nucleophilic Lewis bases,3,4 the literature is devoid of examples of dual activation for the mechanistically similar nucleophilic phosphine-catalyzed allene annulations. Indeed, the use of an additive to manipulate the reactivity in phosphine-catalyzed annulations of allenes has as of yet been explored only for the synthesis of dihydropyrones, as we describe in further detail below.
There is much evidence supporting dual activation in the nucleophile-catalyzed MBH reaction.3 In addition to using a Lewis basic nucleophile as a catalyst, the presence of certain hydrogen bond donors4 can dramatically increase the rate of reaction, due in part to the increase in concentration of the zwitterionic intermediate.5 The addition of an alcohol or water has also been demonstrated in several cases to substantially increase the reaction rate,6 which is often attributed to added assistance during the proton transfer steps.7 Chiral Lewis and Brønsted acidic components possessing free hydrogen bond donors have been applied to activate the electrophilic partner, thereby increasing the reaction rate and inducing facial selectivity.8 Bifunctional catalysis,9 a particularly promising type of dual activation, has also led to many extremely rewarding discoveries in the MBH reaction. Such catalysts, which commonly comprise a nucleophilic reactive site and a hydrogen bond donor to activate the electrophilic partner,10 increase the rate of the reaction and its scope, frequently inducing high levels of enantiomeric excess in the products. The power of dual activation by means of nucleophilic catalysis in conjunction with an additional component is well documented.1, 2
The scope of phosphine-catalyzed annulations of 2,3-butadienoates with unsaturated electrophiles has been greatly expanded over the last decade or so.11 In addition to the reported reactions of electron-poor alkenes and imines, we have previously demonstrated that aldehydes are viable electrophiles for the selective formation of dioxane and pyrone oxacycles (Scheme 1).12 The use of small phosphines, such as trimethylphosphine, leads to the formation of dioxane products (2), whereas bulky phosphines form pyrone products (3) exclusively. It is believed that the key mechanistic difference lies in the equilibrium between the two isomeric β-phosphonium dienolates formed upon addition of the tertiary phosphine catalyst to the butadienoate; we suspected that an alternative means of influencing this equilibrium, other than changing the phosphine’s size, would enable the formation of dihydropyrone oxacycles (4).
Scheme 1.
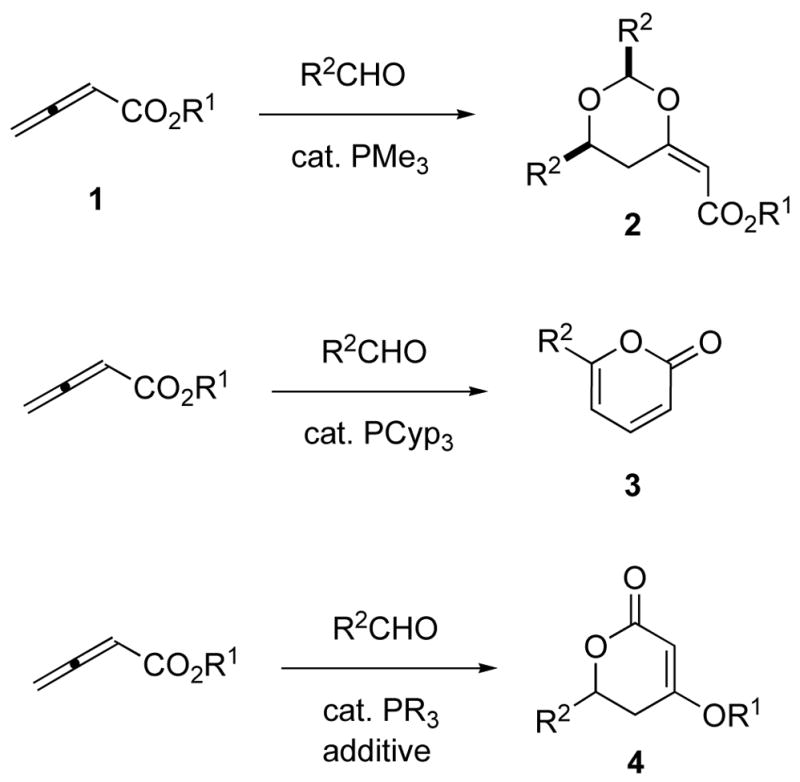
Phosphine-catalyzed annulations of 2,3-butadienoates with aldehydes as electrophiles
2. Results and Discussion
In the first step of the nucleophilic phosphine catalysis of allenes, the addition of the phosphine catalyst to the 2,3-butadienoate 1 results in an equilibrium mixture of dienolates 5 and 5′ (Scheme 2).12c if the phosphine is small, such as trimethylphosphine, then the intermediate dienolate 5′ having s-trans geometry is dominant because of Coulombic attraction between the phosphonium unit and the delocalized negative charge of the enolate alkoxide moiety. The addition of an aldehyde to the s-trans intermediate results in the formation of the β-phosphonium enoate 6′ having Z geometry, which incorporates a second equivalent of aldehyde and undergoes 6-exo-trig cyclization onto the enoate to release the catalyst and form the dioxane 2.12a In an alternative reaction pathway, if the phosphine catalyst is bulky, for instance triisopropyl-, tricyclohexyl-, or tricyclopentylphosphine, steric hindrance around the phosphine unit appears to disrupt the favorable Coulombic interaction and shift the equilibrium toward the dienolate 5 having s-cis geometry. Upon addition of the aldehyde to the γ-carbon atom,12b the β-phosphonium enoate 6 having E geometry is formed. The spatial proximity between the alkoxide and enoate units in intermediate 6 allows lactonization, resulting in the formation of 7. The concomitantly generated alkoxide R1O− facilitates deprotonation of the β-phosphonium lactone 7; the resulting zwitterion proceeds to form the pyrone 3 and regenerate the phosphine catalyst. We suspect that conjugate addition of the alkoxide followed by facile β-elimination of the phosphine, resulting in dihydropyrone 4, is blocked by the bulky phosphine required for the formation of 5. An alternative method for stabilizing the s-cis β-phosphonium dienolate 5 would potentially allow a smaller phosphine to act as a catalyst, with subsequent formation of disubstituted dihydropyrones 4. Given that the electrostatic attraction between the phosphonium cation and the alkoxide anion facilitates the formation of the s-trans β-phosphonium dienolate 5′, we envisioned the possibility of using hydrogen bond donors to stabilize the alkoxide moiety and induce the desired s-cis dienolate 5. It occurred to us that addition of an alcohol R1OH would be especially attractive because the competing reaction pathway to the pyrone 3 would be discouraged.
Scheme 2.
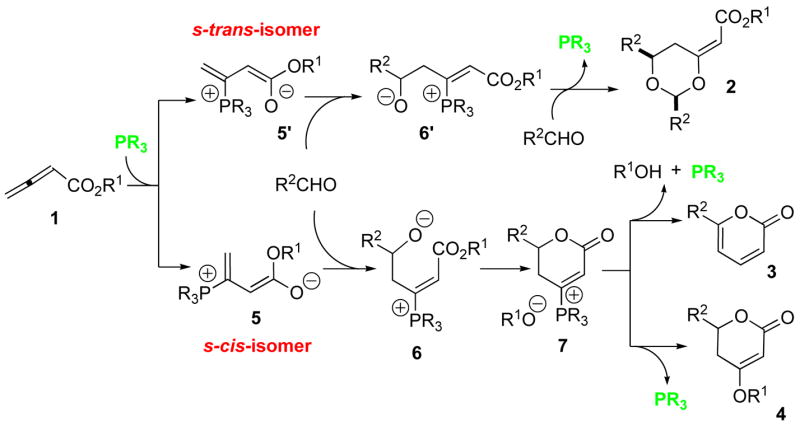
Formation of dioxanes 2, 2-pyrones 3, and dihydro-2-pyrones 4
To evaluate the viability of this mechanistic proposal, we performed a series of quantum chemical calculations rooted within the formalism of the generalized gradient approximation (GGA)-hybrid Kohn–Sham density functional theory (KS-DFT) using Becke’s three-parameter exchange functional (B3)13 with inclusion of Lee, Yang, and Parr’s electron correlation method (LYP)14 and Pople’s 6-3lG(d) basis set.15 The reported energies represent computed values from single-point implicit conductor-like polarizable continuum model (CPCM) solvation calculations (benzene ε = 2.25) performed on B3LYP/6-31G(d)-(CPCM)//B3LYP/6-31G(d)-optimized geometries.16 All quantum chemical calculations were undertaken using the Gaussian suite of programs G0317 and Gaussview 3.0.18 To accomplish this goal, we conducted a systematic survey of the potential energy surface (PES) within the vicinity corresponding to the transition state for P+—C bond formation during the phosphine-to-allenoate addition in the presence of MeOH (Scheme 3). From this survey, we located two nearly energetically equivalent lowest-energy first-order saddle points TSa (ΔΔ G‡solv = 0 kcal/mol) and TSb (ΔΔG‡solv = 0.7 kcal/mol). The defining metrics of these transition state structures included P+—C bond forming distances of 2.24 Å and 2.20 Å, respectively. In addition, present within each structure was a hydrogen bond (H-bond) contact between the MeOH hydroxyl group and the Oδ−-carbonyl oxygen atom of the reacting allenoate, measuring 1.82 Å for TSa and 1.85 Å for TSb, which functionally impart degrees of stability to these transition states. Upon subsequent optimization of TSa and TSb to their respective local minima 5 and 5a, corresponding to the immediate products of P+—C bond formation, a most revealing mechanistic feature was unveiled: In the presence of Brønsted-acidic methanol, the intermediate products formed were the minima 5 and 5a possessing s-cisoid geometries. These findings represent a dramatic departure from our previous in silico mechanistic studies addressing nucleophilic phosphine-triggered zwitterionic enolate formation in the absence of Brønsted-acidic alcoholic additives (see 5′ in Scheme 3).19 In our prior studies we established that the s-trans enolate 5′ was the direct and most stable product formed from the transition state TSc of the phosphine-to-allenoate addition. Accordingly, based upon our new calculations—in what certainly reflects a cause-and-effect relationship—the addition of a Brønsted acid, in this case methanol, leads to a Boltzmann distribution toward the s-cis zwitterions 5 and 5a from favoring the s-trans zwitterion 5′ in nonpolar media. These two minima, 5 and 5a, vary structurally solely in terms of the relative orientation of their hydrogen-bond-coordinated methanol units.20
Scheme 3.
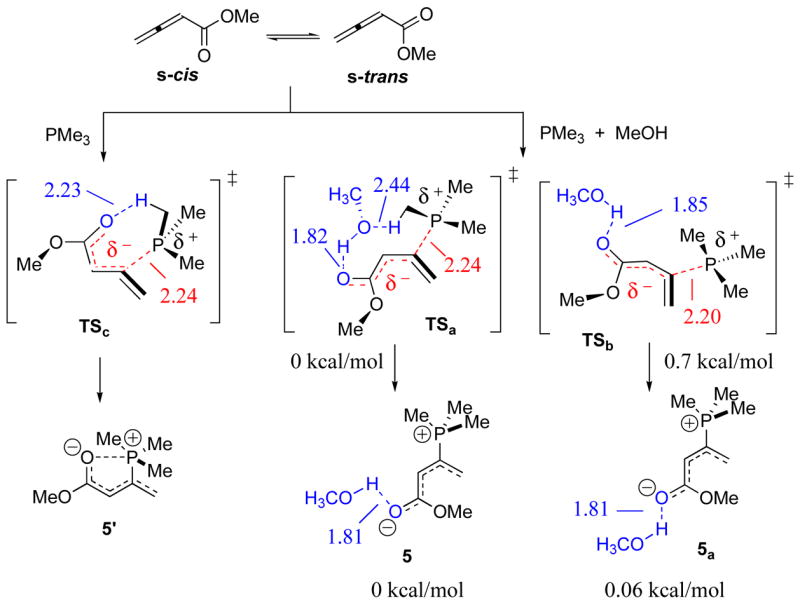
Structures of the transition states and the resulting phosphonium dienolate zwitterions in the presence and absence of methanol
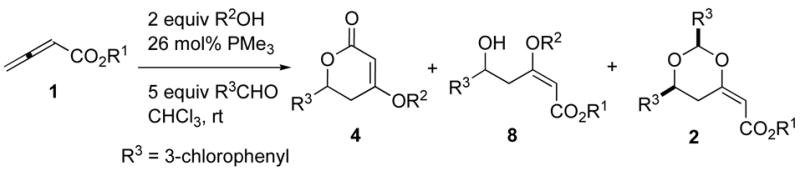
Having verified, through DFT calculations, that addition of a hydrogen bond donor could induce formation of the s-cis isomer 5 by disrupting the putative Coulombic attraction stabilizing 5′, we acted upon this mechanistic insight experimentally. Promisingly, initial experiments using isopropanol and ethanol (Table 1, entries 1 and 2) revealed a small induction of the desired dihydropyrones, but the majority of the product was the dioxane, presumably because the phosphonium dienolate equilibrium favored the undesired s-trans isomer 5′. The use of methanol provided more encouraging results, with an increase in total mass recovery to 78% and the major product being the dihydropyrone (Table 1, entry 3). The second major product was the three-component-coupling adduct 8, formed from the aldehyde, allenoate, and alcohol, which formed presumably through regeneration of the phosphine catalyst via Michael addition of the alkoxide prior to lactonization of the intermediate 6 (see Scheme 2).21 We suspected that a decrease in the nucleophilicity of the additive would suppress this interruptive Michael addition. Accordingly, the slightly more acidic benzyl alcohol provided exclusively the dihydropyrone product 4, although the mass recovery was unexpectedly low (Table 1, entry 4). This observation led us to explore more-acidic alcohols. We expected that the commercially available halogenated ethanol derivatives, which exhibit decreased nucleophilicity and, simultaneously, decreased pKa, would preferentially induce formation of the desired dihydropyrone (Table 1, entries 5–12). Indeed, a decrease in the yield of the non-cyclic product 8 occurred when we used the less-nucleophilic 2-fluoroethanol and 2-chloroethanol additives (Table 1, entries 5 and 6), with an overall increase in the mass recovery relative to that obtained using benzyl alcohol. Further decreases in pKa were detrimental: we observed no apparent consumption of the starting butadienoate when the additive was 2-bromoethanol, 2,2-dichlorothanol, 2,2,2-trichloroethanol, or 2,2,2-trifluoroethanol (Table 1, entries 7–10). The use of mismatched alkyl ester and additive alkoxides resulted in mixtures of products through transesterification and the potential addition of two Michael donors; subsequently, we found that the application of matched 2-chloroethyl- and 2-fluoroethyl-2,3-butadienoates resulted in single dihydropyrone products with similar efficiencies (Table 1, entries 11 and 12). In addition to alcohols, we screened an assortment of other Brønsted acidic additives having values of pKa within the desirable range (Table 1, entries 13–22); disappointingly, only N,N′-dimethylurea and water produced the dihydropyrone product, but with diminished yields and mass recoveries. Due to their promising mix of pKa and nucleophilicity, we subjected the 2-chloroethanol and 2-fluoroethanol additives to further evaluation. The structural connectivity of dihydropyrone product was established unequivocally through X-ray crystallography of 6-(4-bromophenyl)-4-(2-fluoroethyl)-5,6-dihydro-2H-pyran-2-one (Figure 1).22
Table 1.
Screening of various hydrogen-bond-donating additives
 | ||||
|---|---|---|---|---|
| entry | R1 | R2or additive | pKa of R2OH in H2O (DMSO)a | yieldb (%;4:8c:2) |
| 1 | iPr | iPr | 16.5 (30.3) | 54 (16:0:84) |
| 2 | Et | Et | 16.0 (29.8) | 70 (23:21:55) |
| 3 | Me | Me | 15.5 (29.0) | 78 (54:39:7) |
| 4 | Bn | Bn | 15.2, 15.4 | 36 (100:0:0) |
| 5 | Me | CH2CH2Cl | 14.3 | 55 (90:10:0)d |
| 6 | Me | CH2CH2F | 13.9, 14.2 | 49 (85:15:0)d |
| 7 | iPr | CH2CH2Br | 13.8 | 0 |
| 8 | Me | CH2CHCl2 | 12.9 | trace |
| 9 | Me | CH2CCl3 | 11.8, 12.2 | 0 |
| 10 | Me | CH2CF3 | 11.4, 12.4 (23.5) | 0 |
| 11 | CH2CH2 Cl | CH2CH2Cl | 14.3 | 41 (81:19:0) |
| 12 | CH2CH 2F | CH2CH2F | 13.9, 14.2 | 51 (89:11:0) |
| 13 | Me | H2O | 15.7 (31.4) | 31 (46:4:50)e |
| 14 | Me |
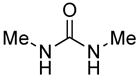
|
14.6 (26.9)f | 11 (100:0:0)e |
| 15 | Me |
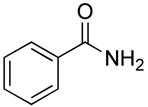
|
14.5 (23.3) | 0 |
| 16 | CH2CH2Cl |
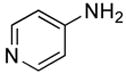
|
(26.5) | 0 |
| 17 | CH2CH2F |
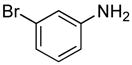
|
(28.4) | 0 |
| 18 | CH2CH2F |
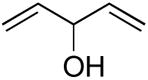
|
13.7 | 0 |
| 19 | CH2CH2F |
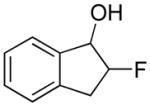
|
13.0 | 0 |
| 20 | CH2CH2F |
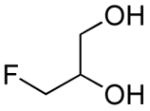
|
13.1 | 0 |
| 21 | CH2CH2F |
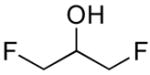
|
12.7 | 0 |
| 22 | iPr | ethylene glycol | 14.77 | decomposition |
Data collected from Ref. 23; if two sources report slightly different pKa values in water, both values are provided.23
Isolated yields.
The alkene stereochemistry was exclusively E (NOESY NMR spectroscopy).
Both methoxide and the 2-haloethoxide were incorporated as β-substituents (OR2) in 4 and 8.
Methoxide incorporated as the β-substituent.
Value of pKa for urea in DMSO.
Figure 1.
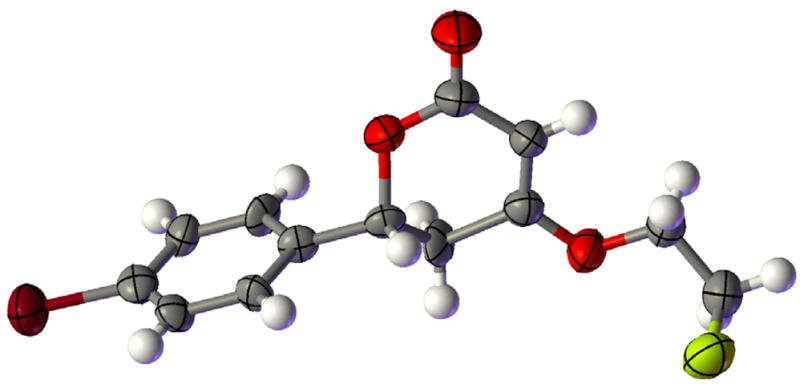
X-ray crystallographic representation of 6-(4-bromophenyl)-4-(2-fluoroethyl)-5,6-dihydro-2H-pyran-2-one 4i.
Next, we further optimized the reaction parameters for the 2-chloroethyl and 2-fluoroethyl butadienoates (Table 2). Diminished yields of the desired dihydropyrone were observed when using tributylphosphine and triethylphosphine as catalysts (Table 2, entries 1 and 2), perhaps due to their increased steric bulk, relative to that of trimethylphosphine, being detrimental to the requisite Michael addition of the alcohol. Trimethylphosphine was the optimal catalyst for the coupling of 3-chlorobenzaldehyde with 2-chloroethyl allenoate (Table 2, entry 3). Unexpectedly, triphenylphosphine also induced dihydropyrone formation, albeit with diminished yield (Table 2, entry 4). This result is notable because it is the first example of triphenylphosphine facilitating the coupling between a butadienoate and an aldehyde. Additionally, in this case we observed no non-cyclic product, presumably because a decrease in rate of the Michael addition/elimination step allowed lactonization to compete effectively with catalyst regeneration. From a solvent screening evaluation, the optimal solvent was dichloromethane, which combined an increase in efficiency with a higher yield of cyclized product (Table 2, cf. entry 5 with entries 6–11 for other common organic solvents). We believe that the favorable outcome with nonpolar solvents is the result of more-efficient association of the alcohol additives with the zwitterionic intermediates. In dichloromethane, the combination of 2-fluoroethyl allenoate and 2-fluoroethanol furnished a higher yield of the dihydropyrone as well as better ratio with respect to the non-cyclic product (Table 2, entry 12). Interestingly, the yield increased further when using triphenylphosphine as the catalyst; more importantly, no non-cyclic product was formed (57%; Table 2, entry 13).
Table 2.
Screening of solvents and phosphine catalysts
 | ||||
|---|---|---|---|---|
| entry | R | solvent | phosphinea | yieldb (%; 4:8) |
| 1 | CH2CH2Cl | CHCl3 | P(n-Bu)3 | 0 |
| 2 | CH2CH2Cl | CHCl3 | PEt3 | 15 (100:0) |
| 3 | CH2CH2Cl | CHCl3 | PMe3 | 41 (81:19) |
| 4 | CH2CH2Cl | CHCl3 | PPh3 | 18 (100:0) |
| 5 | CH2CH2Cl | CH2Cl2 | PMe3 | 48 (88:13) |
| 6 | CH2CH2Cl | DCE | PMe3 | 31 (86:14) |
| 7 | CH2CH2Cl | benzene | PMe3 | 29 (86:14) |
| 8 | CH2CH2Cl | toluene | PMe3 | 28 (84:16) |
| 9 | CH2CH2Cl | Et2O | PMe3 | 12 (100:0) |
| 10 | CH2CH2Cl | THF | PMe3 | 8 (100:0) |
| 11 | CH2CH2Cl | DMF | PMe3 | 0 |
| 12 | CH2CH2F | CH2Cl2 | PMe3 | 51 (89:11) |
| 13 | CH2CH2F | CH2Cl2 | PPh3 | 57 (100:0) |
Trialkylphosphine: 26 mol%; PPh3: 20 mol%.
Isolated yields.
The alkene stereochemistry was exclusively E (NOESY NMR spectroscopy).
With the choice of catalyst, solvent, and Brønsted acid additive optimized, we proceeded to probe the scope of the formation of dihydropyrones (Table 3). In the hope of reducing the degree of undesirable allenoate oligomerization as a side reaction limiting the mass recovery, the 0.25 M butadienoate solution was added slowly. Benzaldehydes presenting a variety of electron-withdrawing substituents provided their dihydropyrones in good yields (Table 3, entries 1–5). Interestingly, trimethylphosphine proved to be a more effective catalyst for some substrates, such as the trifluoromethyl-substituted benzaldehydes (Table 3, entries 6 and 7). As demonstrated in previous allene/aldehyde annulations,12 electron-rich aromatic aldehydes exhibited low efficiencies (entries 8–11). A heteroaromatic substrate, 3-pyridine carboxaldehyde, was also compatible with the optimized reaction conditions, providing the corresponding substituted pyridine in 68% yield (entry 12). Disubstituted dihydropyrones were obtained rapidly in a single step from simple, bench-stable starting materials in moderate to good yields.
Table 3.
Syntheses of various 6-aryl-4-(2-fluoroethoxy)-5,6-dihydro-2-pyronesa
 | ||||
|---|---|---|---|---|
| entry | Ar | phosphineb | product | yield (%)c |
| 1 | 4-NCC6H4 | PPh3 | 4a | 81 |
| 2 | 3-NCC6H4 | PPh3 | 4b | 72 |
| 3 | 3-O2NC6H4 | PPh3 | 4c | 77 |
| 4 | 3-ClC6H4 | PPh3 | 4d | 61 |
| 5 | 4-AcC6H4 | PPh3 | 4e | 58 |
| 6 | 4-F3CC6H4 | PMe3 | 4f | 58 |
| 7 | 2-F3CC6H4 | PMe3 | 4g | 55 |
| 8 | C6H5 | PMe3 | 4h | 30 |
| 9 | 4-BrC6H4 | PMe3 | 4i | 40 |
| 10 | 3-MeC6H4 | PMe3 | 4j | 21 |
| 11 | 3-MeOC6H4 | PMe3 | 4k | 39 |
| 12 | 3-pyridyl | PMe3 | 4l | 68 |
Dilute allenoate addition; see the Experimental section for a detailed procedure.
PPh3: 20 mol%; PMe3: 25 mol%.
Isolated yield.
3. Conclusion
We have uncovered a new mode for the phosphine-catalyzed reaction of 2,3-butadienoates with aromatic aldehydes, namely the formation of disubstituted dihydropyrones, through the addition of Brønsted-acidic additives, which presumably interact with the zwitterionic β-phosphonium dienolate 5, shifting the equilibrium toward its s-cis isomer. Previously, such a shift had required the use of an extremely bulky phosphine catalyst, which inhibited Michael addition of an alkoxide, resulting instead in the formation of pyrone products. Using an additive, in addition to a nucleophilic phosphine catalyst, to induce an alternate reaction pathway is an example of dual activation of the starting material substrates. Adjustment of the pKa and nucleophilicity of the additives was necessary to prevent formation of non-cyclized products, which formed presumably through premature catalyst regeneration; 2-fluoroethyl butadienoate and 2-fluoroethanol were the optimal substrate and additive. Additionally, the use of triphenylphosphine as the catalyst led to complete inhibition of the non-cyclized product, possibly due to a reduction in rate of Michael addition of the alkoxide so that complete lactonization could occur. The dual activation present in the mechanism of this reaction pathway suggests many avenues for chiral induction: e.g., bifunctional catalysts featuring both hydrogen-bond-donating and nucleophilic reaction sites, asymmetric phosphine catalysts, or chiral Brønsted-acidic additives. In addition, increasing the allenoate’s complexity through either α- or γ-substitution would increase the complexity of the resultant dihydropyrones.
4. Experimental
4.1. General
All reactions were performed under an argon atmosphere with dry solvents and anhydrous conditions, unless otherwise noted. Tetrahydrofuran (THF) and diethyl ether were distilled over sodium/benzophenone ketyl; dichloromethane, benzene, and toluene were freshly distilled from CaH2. All other anhydrous solvents were packaged in Sure/Seal™ bottles and were used as received from Aldrich; chloroform was further distilled from calcium chloride immediately prior to use. All the aldehydes were commercially available and purchased from Aldrich or Acros Organics. The liquid aldehydes were washed sequentially with saturated sodium bicarbonate and saturated sodium chloride and then distilled prior to use. Solid aldehydes were dissolved in dichloromethane, washed with saturated sodium bicarbonate, evaporated to dryness, and then stored under vacuum over phosphorus pentoxide. All the phosphines were commercially available and purchased from Aldrich, Strem Chemicals, Inc., Organometallics, Inc., or Maybridge Chemicals. All other reagents were used as received from commercial sources, with the alcohols stored over 4Å molecular sieves. Reactions were monitored through thin layer chromatography (TLC) on 0.25-mm Silicycle silica gel plates (TLG-R10011B-323), visualizing with UV light or staining with permanganate or anisaldehyde. Flash column chromatography was performed using Silicycle Silia-P gel (50-μm particle size, R12030B) and compressed air. IR spectra were recorded on a Perkin–Elmer Paragon1000 FT-IR spectrometer. NMR spectra were obtained on a Bruker Avance-500 instrument and calibrated using residual non-deuterated chloroform as an internal reference (7.26 ppm for 1H NMR; 77.00 ppm for 13C NMR). Data for 1H NMR spectra are reported as follows: chemical shift (δppm), multiplicity, coupling constant (Hz), and integration. Data for 13C NMR spectra are reported in terms of chemical shift and multiplicities, with coupling constants (Hz) in the case of JCF coupling. The following abbreviations are used to explain the multiplicities: s, singlet; d, doublet; t, triplet; q, quartet; sept, septet; m, multiplet; br, broad; and app, apparent. High-resolution electrospray ionization/time of flight (ESI-TOF) mass spectrometry data were collected on a QStar Elite Hybrid LC/MS/MS system, with caffeine and MARFA peptide used as internal calibration standards.
4.2. General Procedure for the Synthesis of Dihydropyrones
Dry solvent (10 mL) was added to a flame-dried, 15-mL round-bottom flask under an argon atmosphere, followed by the neat aldehyde (5.0 equiv), the alcohol additive (2.0 equiv), and the phosphine (20 mol% for PPh3; 26 mol% for PMe3); in the case of a solid phosphine, additive, or aldehyde, the compound was added prior to the solvent and the argon was replenished. Using a 250-μL micro syringe, the allenoate (1.0 mmol) was added dropwise over a period of 30 min. An orange-red solution was obtained; the mixture was stirred at room temperature until TLC (eluant: 20% EtOAc/hexanes; permanganate stain) revealed consumption of the allenoate. The reaction mixture was concentrated and the residue purified through flash column chromatography (SiO2; 33–50% EtOAc/hexanes) to give the products.
4.2.1. 4-Benzyloxy-6-(3-chlorophenyl)-5,6-dihydro-2H-pyran-2-one
36% yield; thick oil: IR (neat) νmax 1700, 1622, 1558, 1540, 1506, 1472, 1456, 1224 cm−1; 1H NMR (500 MHz, CDCl3) δ7.44–7.28 (m, 9H), 5.44 (dd, J = 12.2, 3.8 Hz, 1H), 5.36 (d, J = 1.5 Hz, 1H), 4.99 (d, J = 2.5 Hz, 2H), 2.84 (ddd, J = 17.0, 12.0, 1.5 Hz, 1H), 2.68 (dd, J = 17.0, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ170.9, 166.2, 140.1, 134.6, 134.1, 129.9, 128.8, 128.7, 128.7, 127.8, 126.1, 123.9, 91.6, 76.2, 71.1, 35.0; MS (ESI-TOF) calcd for C18H16O3Cl+ [M + H]+ 315.0788, found 315.0782.
4.3. Optimized Experimental Procedure for the Synthesis of Dihydropyrones
Dry solvent (8 mL) was added to a flame-dried, 15-mL round-bottom flask under an argon atmosphere, followed by the neat aldehyde (5.0 equiv), the alcohol additive (2.0 equiv), and the phosphine (20 mol% for PPh3; 26 mol% for PMe3); in the case of a solid phosphine, additive, or aldehyde, the compound was added prior to the solvent and then the argon was replenished. A solution of the allenoate (1 mmol) in dry solvent (4 mL) was added over 30 min using a syringe pump. An orange-red solution was obtained; the mixture was stirred at room temperature until TLC (eluant: 20% EtOAc/hexanes; permanganate stain) revealed consumption of the allenoate. The reaction typically took 1.5 h for PMe3 and 6 h for PPh3. The reaction mixture was concentrated and the residue purified through flash column chromatography (SiO2; 33–50% EtOAc/hexanes) to give the products. Spectral data for the new dihydropyrones listed in Table 3 are provided below.
4.3.1. 6-(4-Cyanophenyl)-4-(2-fluoroethoxy)-5,6-dihydro-2H-pyran-2-one (4a)
80.8% yield; white solid: IR (neat) νmax 1712, 1626, 1391, 1228, 1076, 918, 835, 583 cm−1; 1H NMR (500 MHz, CDCl3) δ7.71 (dd, J = 6.8, 1.7 Hz, 2H), 7.55 (d, J = 8.5 Hz, 2H), 5.50 (dd, J = 11.8, 4.2 Hz, 1H), 5.26 (d, J = 1.5 Hz, 1H), 4.73 (dt, J = 47.5, 4.0 Hz, 2H), 4.25–4.10 (m, 2H), 2.81 (ddd, J = 17.0, 12.0, 1.5 Hz, 1H), 2.71 (dd, J = 17.3, 4.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ170.6, 165.6, 143.1, 132.5, 126.4, 118.2, 112.6, 91.5, 80.4 (JCF = 172 Hz), 75.9, 68.0 (JCF = 20 Hz), 34.7; MS (ESI-TOF) calcd for C14H13O3NF [M + H]+ 262.0879, found 262.0873.
4.3.2. 6-(3-Cyanophenyl)-4-(2-fluoroethoxy)-5,6-dihydro-2H-pyran-2-one (4b)
72.3% yield; white solid: IR (neat) νmax 1710, 1621, 1392, 1294, 1235, 1191, 1072, 1021 cm−1; 1H NMR (500 MHz, CDCl3) δ7.74 (s, 1H), 7.68 (t, J = 6.5 Hz, 2H), 7.54 (t, J = 8.0 Hz, 1H), 5.48 (dd, J = 12.0, 4.0 Hz, 1H), 5.27 (d, J = 1.5 Hz, 1H), 4.74 (dt, J = 47.5, 4.0 Hz, 2H), 4.26–4.11 (m, 2H), 2.83 (ddd, J = 17.0, 12.0, 1.5 Hz, 1H), 2.71 (dd, J = 17.5, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ170.6, 165.5, 139.6, 132.2, 130.1, 129.6, 129.4, 118.1, 113.0, 91.5, 80.43 (JCF = 172 Hz), 75.7, 68.0 (JCF = 20 Hz), 34.7; MS (ESI-TOF) calcd for C14H13O3NF [M + H]+ 262.0879, found 262.0880.
4.3.3. 4-(2-Fluoroethoxy)-6-(3-nitrophenyl)-5,6-dihydro-2H-pyran-2-one (4c)
77.0% yield; white solid: IR (neat) νmax 1715, 1625, 1532, 1353, 1228, 1074, 816, 740 cm−1; 1H NMR (500 MHz, CDCl3) δ 8.31 (t, J = 1.5 Hz, 1H), 8.24 (d, J = 8.0 Hz, 1H), 7.80 (t, J = 8.0 Hz, 1H), 7.62 (t, J = 8.0 Hz, 1H), 5.56 (dd, J = 12.0, 4.0 Hz, 1H), 5.29 (d, J = 1.5 Hz, 1H), 4.74 (dt, J = 47.0, 4.0 Hz, 2H), 4.27–4.12 (m, 2H), 2.88 (ddd, J = 17.3, 12.3, 1.5 Hz, 1H), 2.76 (dd, J = 17.3, 4.3 Hz, 1H); 13 170.6, 165.5, 148.3, C NMR (125 MHz, CDCl3) δ 140.1, 131.9, 129.9, 123.6, 120.9, 91.5, 80.5 (JCF = 172 Hz), 75.7, 68.1 (JCF = 20 Hz), 34.7; MS (ESI-TOF) calcd for C13H13O5NF+ [M + H]+ 282.0778, found 282.0772.
4.3.4. 6-(4-Acetylphenyl)-4-(2-fluoroethoxy)-5,6-dihydro-2H-pyran-2-one (4e)
57.6% yield; yellow oil: IR (neat) νmax 1711, 1681, 1625, 1360, 1270, 1227, 1074, 603 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.99 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 5.51 (dd, J = 12.0, 4.0 Hz, 1H), 5.26 (d, J = 1.5 Hz, 1H), 4.73 (dt, J = 47.0, 4.0 Hz, 2H), 4.25–4.10 (m, 2H), 2.84 (ddd, J = 17.0, 12.0, 1.5 Hz, 1H), 2.71 (dd, J = 17.0, 4.0, 1H), 2.61 (s, 3H); 13 C NMR (125 MHz, CDCl3) δ 197.4, 170.8, 165.9, 143.0, 137.2, 128.7, 125.9, 91.5, 80.5 (JCF = 171 Hz), 76.4, 67.9 (JCF = 20 Hz), 34.8, 26.6; MS (ESI-TOF) calcd for C15H16O4F+ [M + H]+ 279.1033, found 279.1027.
4.3.5. 4-(2-Fluoroethoxy)-6-(4-trifluoromethylphenyl)-5,6-dihydro-2H-pyran-2-one (4f)
57.8% yield; white solid: IR (neat) νmax 1713, 1626, 1327, 1228, 1168, 1125, 1068, 1018 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 8.5 Hz, 2H), 7.55 (d, J = 8.0 Hz, 2H), 5.52 (dd, J = 12.0 Hz, 4.0 Hz, 1H), 5.27 (d, J = 1.5 Hz, 1H), 4.74 (dt, J = 47.5, 4.0 Hz, 2H), 4.26–4.10 (m, 2H), 2.84 (ddd, J = 17.0, 12.0, 1.5 Hz, 1H), 2.72 (dd, J = 17.0, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 170.8, 165.8, 141.9, 130.8 (q, JCF = 32 Hz), 126.1, 125.7 (q, JCF = 3.8 Hz), 123.8 (q, JCF = 270.4 Hz), 91.5, 80.5 (JCF = 172 Hz), 76.2, 68.0 (JCF = 20 Hz), 34.8; MS (ESI-TOF) calcd for C14H13O3F4 [M + H]+ 305.0801, found 305.0806.
4.3.6. 4-(2-Fluoroethoxy)-6-(2-trifluoromethylphenyl)-5,6-dihydro-2H-pyran-2-one (4g)
54.9% yield; white solid: IR (neat) νmax 1722, 1712, 1626, 1315, 1229, 1165, 1118, 1072 cm−1; 1H NMR (500 MHz, CDCl3) δ7.85 (d, J = 7.5 Hz, 1H), 7.69–7.64 (m, 2H), 7.48 (t, J = 7.7 Hz, 1H), 5.82 (dd, J = 12.5, 4.0 Hz, 1H), 5.27 (d, J = 2.0 Hz, 1H), 4.80–4.68 (dm, J = 47.0 Hz, 2H), 4.26–4.21 (m, 2H), 2.79 (ddd, J = 17.5, 12.5, 1.5 Hz, 1H), 2.65 (dd, J = 17.5, 3.8 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ171.0, 166.1, 136.9, 132.4, 128.7, 128.2, 126.9 (q, JCF = 30 Hz), 125.6 (q, JCF = 6 Hz), 124.0 (q, JCF = 272 Hz), 91.2, 80.5 (d, JCF = 171 Hz), 73.4, 68.0 (d, JCF = 20 Hz), 35.7; MS (ESI-TOF) calcd for C14H13O3F4 [M + H]+ 305.0801, found 305.0802.
4.3.7. 4-(2-Fluoroethoxy)-6-phenyl-5,6-dihydro-2H-pyran-2-one (4h)
30.4% yield; white solid: IR (neat) νmax 1726, 1711, 1632, 1223, 1068, 917, 702, 577 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.43–7.34 (m, 5H), 5.45 (dd, J = 12.0, 4.0 Hz, 1H), 5.25 (d, J = 1.5 Hz, 1H), 4.73 (dt, J = 47.5, 4.0 Hz, 2H), 4.24–4.12 (m, 2H), 2.88 (ddd, J = 17.3, 12.3, 1.5 Hz, 1H), 2.67 (dd, J = 17.0, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 171.2, 166.4, 138.0, 128.6, 128.6, 125.9, 91.4, 80.57 (JCF = 171 Hz), 77.1, 67.8 (JCF = 20 Hz), 34.8; MS (ESI-TOF) calcd for C13H14O3F [M + H]+ 237.0927, found 237.0927.
4.3.8. 6-(4-Bromophenyl)-4-(2-fluoroethoxy)-5,6-dihydro-2H-pyran-2-one (4i)
40.1% yield; white solid: IR (neat) νmax 1704, 1630, 1389, 1347, 1224, 1059, 914, 828, 443 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.53 (d, J = 8.5 Hz, 2H), 7.30 (d, J = 8.5 Hz, 2H), 5.41 (dd, J = 12.0, 4.0 Hz, 1H), 5.24 (d, J = 1.5 Hz, 1H), 4.73 (dt, J = 47.0, 4.0 Hz, 2H), 4.24–4.09 (m, 2H), 2.82 (ddd, J = 17.5, 12.0, 1.5 Hz, 1H), 2.66 (dd, J = 17.5, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 171.0, 166.1, 137.1, 131.9, 127.6, 122.7, 91.5, 80.6 (d, JCF = 171 Hz), 76.4, 68.0 (d, JCF = 20 Hz), 34.5; MS (ESI-TOF) calcd for C13H13O3FBr+ [M + H]+ 315.0032, found 315.0026.
4.3.9. 4-(2-Fluoroethoxy)-6-(3-tolyl)-5,6-dihydro-2H-pyran-2-one (4j)
21.3% yield; white solid: IR (neat) νmax 1711, 1625, 1391, 1291, 1225, 1072, 924, 885 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.30–7.25 (m, 2H), 7.18 (t, J = 9.0 Hz, 2H), 5.42 (dd, J = 12.0, 4.0 Hz, 1H), 5.24 (d, J = 2.0 Hz, 1H), 4.74 (dt, J = 47.5, 4.0 Hz, 2H), 4.24–4.09 (m, 2H), 2.87 (ddd, J = 17.0, 12.0, 1.5 Hz, 1H), 2.66 (dd. J = 17.0, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 171.2, 166.5, 138.4, 137.9, 129.3, 128.5, 126.6, 122.9, 91.4, 80.5 (JCF = 171 Hz), 77.1, 67.8 (JCF = 20 Hz), 34.9, 21.3; MS (ESI-TOF) calcd for C14H16O3F [M + H]+ 251.1083, found 251.1083.
4.3.10. 6-(3-Anisyl)-4-(2-fluoroethoxy)-5,6-dihydro-2H-pyran-2-one (4k)
39.1% yield; white solid: IR (neat) νmax 1710, 1624, 1458, 1392, 1226, 1045, 924, 884 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 8.3 Hz, 1H), 6.98–6.96 (m, 2H), 6.90 (dd, J = 8.5, 2.0 Hz, 1H), 5.43 (dd, J = 12.0, 4.0 Hz, 1H), 5.24 (d, J = 1.5 Hz, 1H), 4.73 (dt, J = 47.0, 4.0 Hz, 2H), 4.24–4.10 (m, 2H), 3.83 (s, 3H), 2.87 (ddd, J = 17.3, 12.3, 1.5 Hz, 1H), 2.67 (dd, J = 17.0, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 171.1, 166.4, 159.8, 139.5, 129.7, 118.0, 114.2, 111.3, 91.4, 80.5 (JCF = 171 Hz), 76.9, 67.8 (JCF = 20.4 Hz), 55.2, 34.9; MS (ESI-TOF) calcd for C14H16O4F+ [M + H]+ 267.1033, found 267.1023.
4.3.11. 4-(2-Fluoroethoxy)-6-(3-pyridyl)-5,6-dihydro-2H-pyran-2-one (4l)
68.4% yield; yellow oil: IR (neat) νmax 1710, 1622, 1392, 1228, 1076, 1023, 916, 820 cm−1; 1H NMR (500 MHz, CDCl3) δ8.66 (d, J = 2.0 Hz, 1H), 8.63 (dd, J = 4.5, 1.5 Hz, 1H), 7.83 (dt, J = 8.0, 1.8 Hz, 1H), 7.38 (dd, J = 8.0, 4.5 Hz, 1H), 5.55 (dd, 12.5, 4.0 Hz, 1H), 5.31 (d, J = 1.5 Hz, 1H), 4.74 (dt, J = 47.0, 4.0 Hz, 2H), 4.26–4.11 (m, 2H), 2.89 (ddd, J = 17.0, 12.0, 1.5 Hz, 1H), 2.71 (dd, 17.5, 4.0 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ170.8, 165.8, 150.0, 147.4, 133.8, 133.7, 123.6, 91.5, 80.4 (JCF = 171.5 Hz), 74.8, 68.0 (JCF = 20 Hz), 34.5; MS (ESI-TOF) calcd for C12H13O3NF [M + H]+ 238.0879, found 238.0868.
Acknowledgments
This study was supported by the NIH (R01GM071779 and P41GM081282). T.D. thanks the Research Corporation (Cottrell College Science Award), the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI), and Merck for financial support. We thank Dr. Saeed Khan for performing the X-ray crystallographic analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References and Notes
- 1.For a review of dual activation, see: Ma JA, Cahard D. Angew Chem, Int Ed. 2004;43:4566. doi: 10.1002/anie.200300635.
- 2.For a review on additives and cocatalysts, see: Vogl EM, Gröger H, Shibasaki M. Angew Chem, Int Ed. 1999;38:1570. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1570::AID-ANIE1570>3.0.CO;2-Y.For examples, see: Nishiwaki N, Knudsen KR, Gothelf KV, Jørgensen KA. Angew Chem, Int Ed. 2001;40:2992. doi: 10.1002/1521-3773(20010817)40:16<2992::AID-ANIE2992>3.0.CO;2-3.Itoh K, Kanemasa S. J Am Chem Soc. 2002;124:13394. doi: 10.1021/ja027313+.
- 3.For reviews on asymmetric Morita–Baylis–Hillman reactions, see: Masson G, Housseman C, Zhu J. Angew Chem, Int Ed. 2007;46:4614. doi: 10.1002/anie.200604366.Langer P. Angew Chem, Int Ed. 2000;39:3049. doi: 10.1002/1521-3773(20000901)39:17<3049::aid-anie3049>3.0.co;2-5.
- 4.Morita–Baylis–Hillman reactions using Lewis bases and external Brønsted acids; for phenol/binol, see: Yamada YMA, Ikegami S. Tetrahedron Lett. 2000;41:2165.McDougal NT, Schaus SE. J Am Chem Soc. 2003;125:12094. doi: 10.1021/ja037705w.McDougal NT, Trevellini WL, Rodgen SA, Kliman LT, Schaus SE. Adv Synth Catal. 2004;346:1231.Rodgen SA, Schaus SE. Angew Chem, Int Ed. 2006;45:4929. doi: 10.1002/anie.200601076.Shi M, Liu YH. Org Biomol Chem. 2006;4:1468. doi: 10.1039/b600854b.For proline, see: Shi M, Jiang JK, Li CQ. Tetrahedron Lett. 2002;43:127.Imbriglio JE, Vasbinder MM, Miller SJ. Org Lett. 2003;5:3741. doi: 10.1021/ol035466b.Chen SH, Hong BC, Su CF, Sarshar S. Tetrahedron Lett. 2005;46:8899.Vasbinder MM, Imbriglio JE, Miller SJ. Tetrahedron. 2006;62:11450.. For (thio)urea, see: Maher DJ, Connon SJ. Tetrahedron Lett. 2004;45:1301.Sohtome Y, Tanatani A, Hashimoto Y, Nagasawa K. Tetrahedron Lett. 2004;45:5589.Raheem IT, Jacobsen EN. Adv Synth Catal. 2005;347:1701.Berkessel A, Roland K, Neudörfl JM. Org Lett. 2006;8:4195. doi: 10.1021/ol061298m.For chiral ionic liquid, see: Pégot B, Vo-Thanh G, Gori D, Loupy A. Tetrahedron Lett. 2004;45:6425.Gausepohl R, Buskens P, Kleinen J, Bruckmann A, Lehmann CW, Klankermayer J, Leitner W. Angew Chem, Int Ed. 2006;45:3689. doi: 10.1002/anie.200600327.
- 5.(a) Aggarwal VK, Mereu A, Tarver GJ, McCague R. J Org Chem. 1998;63:7183. doi: 10.1021/jo980421n. [DOI] [PubMed] [Google Scholar]; (b) Aggarwal VK, Dean DK, Mereu A, Williams R. J Org Chem. 2002;67:510. doi: 10.1021/jo016073y. [DOI] [PubMed] [Google Scholar]
- 6.(a) Augé J, Lubin N, Lubineau A. Tetrahedron Lett. 1994;35:7947. [Google Scholar]; (b) Jenner G. High Pressure Res. 1999;16:243. [Google Scholar]; (c) Yu C, Liu B, Hu L. J Org Chem. 2001;66:5413. doi: 10.1021/jo015628m. [DOI] [PubMed] [Google Scholar]; (d) Cai J, Zhou Z, Zhao G, Tang C. Org Lett. 2002;4:4723. doi: 10.1021/ol027197f. [DOI] [PubMed] [Google Scholar]; (e) Luo S, Zhang B, He J, Janczuk A, Wang PG, Cheng JP. Tetrahedron Lett. 2002;43:7369. [Google Scholar]; (f) Aggarwal VK, Emme I, Fulford SY. J Org Chem. 2003;68:692. doi: 10.1021/jo026671s. [DOI] [PubMed] [Google Scholar]; (g) Park KS, Kim J, Choo H, Chong Y. Synlett. 2007;3:395. [Google Scholar]
- 7.(a) Robiette R, Aggarwal VK, Harvey JN. J Am Chem Soc. 2007;129:15513. doi: 10.1021/ja0717865. and references therein. [DOI] [PubMed] [Google Scholar]; (b) Xia Y, Liang Y, Chen Y, Wang M, Jiao L, Huang F, Liu S, Li Y, Yu ZX. J Am Chem Soc. 2007;129:3470. doi: 10.1021/ja068215h. [DOI] [PubMed] [Google Scholar]; (c) Mercier E, Fonovic B, Henry C, Kwon O, Dudding T. Tetrahedron Lett. 2007;48:3617. [Google Scholar]
- 8.(a) Aggarwal VK, Tarver GJ, McCague R. Chem Commun. 1996:2713. [Google Scholar]; (b) Yang KS, Lee WD, Pan JF, Chen K. J Org Chem. 2003;68:915. doi: 10.1021/jo026318m. [DOI] [PubMed] [Google Scholar]; (c) Matsui K, Takizawa S, Sasai H. Tetrahedron Lett. 2005;46:1943. [Google Scholar]; (d) Mocquet CM, Warriner SL. Synlett. 2004;2:356. [Google Scholar]
- 9.For a review on bifunctional catalysis, see: Shibasaki M, Sasai H, Arai T. Angew Chem, Int Ed. 1997;36:1236.For examples, see: Hiemstra H, Wynberg H. J Am Chem Soc. 1981;103:417.Trost BM, Yeh VSC. Angew Chem, Int Ed. 2002;41:861. doi: 10.1002/1521-3773(20020301)41:5<861::aid-anie861>3.0.co;2-v.Buskens P, Klankermayer J, Leitner W. J Am Chem Soc. 2005;127:16762. doi: 10.1021/ja0550024.
- 10.(a) Oishi T, Oguri H, Hirama M. Tetrahedron: Asymmetry. 1995;6:1241. [Google Scholar]; (b) Markó IE, Giles PR, Hindley NJ. Tetrahedron. 1997;53:1015. [Google Scholar]; (c) Barrett AGM, Cook AS, Kamimura A. Chem Commun. 1998:2533. [Google Scholar]; (d) Iwabuchi Y, Nakatani M, Yokoyama N, Hatakeyama S. J Am Chem Soc. 1999;121:10219. [Google Scholar]; (e) Shi M, Xu YM. Angew Chem, Int Ed. 2002;41:4507. doi: 10.1002/1521-3773(20021202)41:23<4507::AID-ANIE4507>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]; (f) Shi M, Jiang JK. Tetrahedron: Asymmetry. 2002;13:1941. [Google Scholar]; (g) Kawahara S, Nakano A, Esumi T, Iwabuchi Y, Hatakeyama S. Org Lett. 2003;5:3103. doi: 10.1021/ol035102j. [DOI] [PubMed] [Google Scholar]; (h) Balan D, Adolfsson H. Tetrahedron Lett. 2003;44:2521. [Google Scholar]; (i) Shi M, Chen LH. Chem Commun. 2003:1310. doi: 10.1039/b301863f. [DOI] [PubMed] [Google Scholar]; (j) Krishna PR, Kannan V, Reddy PVN. Adv Synth Catal. 2004;346:603. [Google Scholar]; (k) Matsui K, Takizawa S, Sasai H. J Am Chem Soc. 2005;127:3680. doi: 10.1021/ja0500254. [DOI] [PubMed] [Google Scholar]; (l) Shi M, Chen LH, Li CQ. J Am Chem Soc. 2005;127:3790. doi: 10.1021/ja0447255. [DOI] [PubMed] [Google Scholar]; (m) Wang J, Li H, Yu X, Zu L, Wang W. Org Lett. 2005;7:4293. doi: 10.1021/ol051822+. [DOI] [PubMed] [Google Scholar]; (n) Shi M, Xu YM, Shi YL. Chem Eur J. 2005;11:1794. doi: 10.1002/chem.200400872. [DOI] [PubMed] [Google Scholar]; (o) Shi M, Li CQ. Tetrahedron: Asymmetry. 2005;16:1385. [Google Scholar]; (p) Nakano A, Kawahara S, Akamatsu S, Morokuma K, Nakatani M, Iwabuchi Y, Takahashi K, Ishihara J, Hatakeyama S. Tetrahedron. 2006;62:381. [Google Scholar]; (q) Matsui K, Tanaka K, Horii A, Takizawa S, Sasai H. Tetrahedron: Asymmetry. 2006;17:578. [Google Scholar]; (r) Nakano A, Takahashi K, Ishihara J, Hatakeyama S. Org Lett. 2006;8:5357. doi: 10.1021/ol0622561. [DOI] [PubMed] [Google Scholar]
- 11.(a) Zhang C, Lu X. J Org Chem. 1995;60:2906. [Google Scholar]; (b) Zhu G, Chen Z, Jiang Q, Xiao D, Cao P, Zhang X. J Am Chem Soc. 1997;119:3836. [Google Scholar]; (c) Xu Z, Lu X. Tetrahedron Lett. 1997;38:3461. [Google Scholar]; (d) Xu Z, Lu X. J Org Chem. 1998;63:5031. [Google Scholar]; (e) Xu Z, Lu X. Tetrahedron Lett. 1999;40:549. [Google Scholar]; (f) Kumar K, Kapur A, Ishar MPS. Org Lett. 2000;2:787. doi: 10.1021/ol000007l. [DOI] [PubMed] [Google Scholar]; (g) Kumar K, Kapoor R, Kapur A, Ishar MPS. Org Lett. 2000;2:2023. doi: 10.1021/ol0000713. [DOI] [PubMed] [Google Scholar]; (h) Ung AT, Schafer K, Lindsay KB, Pyne SG, Amornraksa K, Wouters R, Van der Linden I, Biesmans I, Lesage ASJ, Skelton BW, White AH. J Org Chem. 2002;67:227. doi: 10.1021/jo010864i. [DOI] [PubMed] [Google Scholar]; (i) Liu B, Davis R, Joshi B, Reynolds DW. J Org Chem. 2002;67:4595. doi: 10.1021/jo016154u. [DOI] [PubMed] [Google Scholar]; (j) Du Y, Lu X, Yu Y. J Org Chem. 2002;67:8901. doi: 10.1021/jo026111t. [DOI] [PubMed] [Google Scholar]; (k) Lu C, Lu X. Org Lett. 2002;4:4677. doi: 10.1021/ol0270733. [DOI] [PubMed] [Google Scholar]; (l) Wang JC, Krische MJ. Angew Chem, Int Ed. 2003;42:5855. doi: 10.1002/anie.200352218. [DOI] [PubMed] [Google Scholar]; (m) Zhu XF, Lan J, Kwon O. J Am Chem Soc. 2003;125:4716. doi: 10.1021/ja0344009. [DOI] [PubMed] [Google Scholar]; (n) Du Y, Lu X. J Org Chem. 2003;68:6463. doi: 10.1021/jo034281f. [DOI] [PubMed] [Google Scholar]; (o) Kuroda H, Tomita I, Endo T. Org Lett. 2003;5:129. doi: 10.1021/ol020198n. [DOI] [PubMed] [Google Scholar]; (p) Jung CK, Wang JC, Krische MJ. J Am Chem Soc. 2004;126:4118. doi: 10.1021/ja049377l. [DOI] [PubMed] [Google Scholar]; (q) Pham TQ, Pyne SG, Skelton BW, White AH. J Org Chem. 2005;70:6369. doi: 10.1021/jo050827h. [DOI] [PubMed] [Google Scholar]; (r) Wurz RP, Fu GC. J Am Chem Soc. 2005;127:12234. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]; (s) Zhao GL, Shi M. Org Biomol Chem. 2005;3:3686. doi: 10.1039/b510572b. [DOI] [PubMed] [Google Scholar]; (t) Zhu XF, Henry CE, Kwon O. Tetrahedron. 2005;61:6276. [Google Scholar]; (u) Tran YS, Kwon O. Org Lett. 2005;7:4289. doi: 10.1021/ol051799s. [DOI] [PubMed] [Google Scholar]; (v) Wilson JE, Fu GC. Angew Chem, Int Ed. 2006;45:1426. doi: 10.1002/anie.200503312. [DOI] [PubMed] [Google Scholar]; (w) Nair V, Biju AT, Mohanan K, Suresh E. Org Lett. 2006;8:2213. doi: 10.1021/ol0604623. [DOI] [PubMed] [Google Scholar]; (x) Lu X, Lu Z, Zhang X. Tetrahedron. 2006;62:457. [Google Scholar]; (y) Jean L, Marinetti A. Tetrahedron Lett. 2006;47:2141. [Google Scholar]; (z) Virieux D, Guillouzic AF, Cristau HJ. Tetrahedron. 2006;62:3710. [Google Scholar]; (aa) Scherer A, Gladysz JA. Tetrahedron Lett. 2006;47:6335. [Google Scholar]; (bb) Castellano S, Fiji HDG, Kinderman SS, Watanabe M, de Leon P, Tamanoi F, Kwon O. J Am Chem Soc. 2007;129:5843. doi: 10.1021/ja070274n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (cc) Wallace DJ, Sidda RL, Reamer RA. J Org Chem. 2007;72:1051. doi: 10.1021/jo062170l. [DOI] [PubMed] [Google Scholar]; (dd) Henry CE, Kwon O. Org Lett. 2007;9:3069. doi: 10.1021/ol071181d. [DOI] [PubMed] [Google Scholar]; (ee) Gabillet S, Lecercle’ D, Loreau O, Carboni M, De’zard S, Gomis JM, Taran F. Synthesis. 2007:515. doi: 10.1021/ol701563e. [DOI] [PubMed] [Google Scholar]; (ff) Gabillet S, Lecercle’ D, Loreau O, Carboni M, De’zard S, Gomis JM, Taran F. Org Lett. 2007;9:3925. doi: 10.1021/ol701563e. [DOI] [PubMed] [Google Scholar]; (gg) Zhu XF, Henry CE, Kwon O. J Am Chem Soc. 2007;129:6722. doi: 10.1021/ja071990s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (hh) Cowen BJ, Miller SJ. J Am Chem Soc. 2007;129:10988. doi: 10.1021/ja0734243. [DOI] [PubMed] [Google Scholar]; (ii) Tran YS, Kwon O. J Am Chem Soc. 2007;129:12632. doi: 10.1021/ja0752181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (jj) Sriramurthy V, Barcan GA, Kwon O. J Am Chem Soc. 2007;129:12928. doi: 10.1021/ja073754n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (kk) Fang YQ, Jacobsen EN. J Am Chem Soc. 2008 doi: 10.1021/ja801344w. ASAP. [DOI] [PubMed] [Google Scholar]
- 12.(a) Zhu XF, Henry CE, Wang J, Dudding T, Kwon O. Org Lett. 2005;7:1387. doi: 10.1021/ol050203y. [DOI] [PubMed] [Google Scholar]; (b) Zhu XF, Schaffner AP, Li RC, Kwon O. Org Lett. 2005;7:2977. doi: 10.1021/ol050946j. [DOI] [PubMed] [Google Scholar]; (c) Dudding T, Kwon O, Mercier E. Org Lett. 2006;8:3643. doi: 10.1021/ol061095y. [DOI] [PubMed] [Google Scholar]; (d) Creech GS, Kwon O. Org Lett. 2008;10:429. doi: 10.1021/ol702462w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; (b) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623. [Google Scholar]
- 14.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 15.(a) Hehre WJ, Ditchfield R, Pople JA. J Chem Phys. 1972;56:2257. [Google Scholar]; (b) Hariharan PC, Pople JA. Theoret Chim Acta. 1973;28:213. [Google Scholar]; (c) Hariharan PC, Pople JA. Mol Phys. 1974;27:209. [Google Scholar]; (d) Francl MM, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA. J Chem Phys. 1982;77:3654. [Google Scholar]; (e) Rassolov VA, Pople JA, Ratner MA, Windus TL. J Chem Phys. 1998;109:1223. [Google Scholar]
- 16.(a) Barone V, Cossi M. J Phys Chem A. 1998;102:1995. [Google Scholar]; (b) Cossi M, Rega N, Scalmani G, Barone V. J Comp Chem. 2003;24:669. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 17.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian, Inc.; Wallingford CT: 2004. Gaussian 03, Revision C.02. [Google Scholar]
- 18.Dennington R, II, Keith T, Millam J, Eppinnett K, Hovell WL, Gilliland R. Semichem, Inc.; Shawnee Mission, KS: 2003. GaussView, Version 3.09. [Google Scholar]
- 19.It is noted that an energetically less favorable twisted-dienolate minimum, originating from MeOH-mediated hydrogen bond stabilization, possessing a conjugated π-system perturbed from what is an optimal syn-periplanar geometry was obtained from this PES-study. The reaction barrier for aldehyde addition to this intermediate was, however, calculated to be more endergonic than the aldehyde addition modes computed to date for 5 and 5a; consequently, it does not contribute to a productive pathway.
- 20.We are currently investigating the subsequent aldehyde addition to elucidate the complete mechanism of dihydropyrone formation.
- 21.The non-cyclized product, once isolated, did not undergo equilibration or cyclization when subjected to the reaction conditions.
- 22.Crystallographic data (excluding structure factors) for compound 4i have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 675758. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-(0)1223-336033; e-mail: deposit@ccdc.cam.ac.uk).
- 23.(a) Ripin DH, Evans DA. Table of pKa values compiled for Chem 206. accessed online at http://daecr1.harvard.edu/pdf/evans_p-Ka_table.pdf.; (b) Williams R, Jencks WP, Westheimer FH. Table of pKa values. accessed online at www.webqc.org/pkaconstants.php.; (c) Dalby KN, Kirby AJ, Hollfelder F. J Chem Soc, Perkin Trans 2. 1993:1269. [Google Scholar]; (d) Calculated using Advanced Chemistry Development (ACD/Labs) Software (v. 8.14) for Solaris (1994–2007 ACD/Labs); error reported as ±0.10.; (e) Menger FM, Ladika M. J Am Chem Soc. 1987;109:3145. [Google Scholar]; (f) Nakagawa Y, Uehara K, Mizuno N. Inorg Chem. 2005;44:9068. doi: 10.1021/ic051163x. [DOI] [PubMed] [Google Scholar]; (g) Krogsgaard-Larsen P, Liljefors T, Madsen U, editors. Textbook of Drug Design and Discovery. 3. CRC Press; New York: 2002. p. 432. [Google Scholar]