

Abstract
On the one hand, owing to its electronegativity, relatively small size, and notable leaving group ability from anionic intermediates, fluorine offers unique opportunities for mechanism-based enzyme inhibitor design. On the other, the “bio-orthogonal” and NMR-active 19-fluorine nucleus allows the bioorganic chemist to follow the mechanistic fate of fluorinated substrate analogues or inhibitors as they are enzymatically processed. This article takes an overview of the field, highlighting key developments along these lines. It begins by highlighting new screening methodologies for drug discovery that involve appropriate tagging of either substrate or the target protein itself with 19F-markers, that then report back on turnover and binding, respectively, via an the NMR screen. Taking this one step further, substrate-tagging with fluorine can be done is such a manner as to provide stereochemical information on enzyme mechanism. For example, substitution of one of the terminal hydrogens in phosphoenolpyruvate, provides insight into the, otherwise latent, facial selectivity of C-C bond formation in KDO synthase. Perhaps, most importantly, from the point of view of this discussion, appropriately tailored fluorinated functionality can be used to form to stabilized “transition state analogue” complexes with a target enzymes. Thus, 5-fluorinated pyrimidines, α-fluorinated ketones, and 2-fluoro-2-deoxysugars each lead to covalent adduction of catalytic active site residues in thymidylate synthase, serine protease and glycosidase enzymes, respectively. In all such cases, 19F NMR allows the bioorganic chemist to spectrally follow “transition state analogue” formation. Finally, the use of specific fluorinated functionality to engineer “suicide substrates” is highlighted in a discussion of the development of the α-(2′Z-fluoro)vinyl trigger for amino acid decarboxylase inactivation. Here 19F NMR allows the bioorganic chemist to glean useful partition ratio data directly out of the NMR tube.
1. Introduction
The C-F bond is one of the strongest covalent bonds available, with an average bond energy of approximately 105–116 kcal/mol. This contributes significantly to the relative metabolic inertness of carbon-fluorine bonds, particularly those at unactivated sp2-carbon-centers. Moreover, introduction of a C-F bond imposes only modest steric constraints, as the C-F bond (1.41–1.47 Å) is slightly shorter than a C-OH bond (1.52 Å) [1]. And while fluorine is the most electronegative element in the periodic table, it also has a very a small atomic radius, resulting in an exceptionally low polarizability. DiMagno has pointed out that this feature of organically-bound fluorine means that fluoroalkyl are less able to engage in dispersion-based interactions with aqueous solvent than simple alkyl groups. He has proposed the term, “polar hydrophobicity” [2] to describe this phenomenon, and points out that this may provide unique opportunities for enhancing ligand binding to a protein target [3]. In terms of specific interactions with functionalities in proteins, while C-F bonds appear to have rather limited H-bond acceptor ability[4–6], in optimally aligned cases F--H-N-amide interactions may make contributions to binding [7–9]. Additionally, more recent observations by Diederich and Müller [10–12] suggest that the hard C-F bond is able to engage amide carbonyls in specific attractive interactions reminiscent of the sort of trajectory-dependent n-π* (amine-carbonyl) interactions suggested by Bürgi and Dunitz years before [13,14]. Finally, in the context of ionizable groups, such as fluorinated phosphonates as phosphate surrogates, one can use position and degree of organic fluorination to finely tune the pKa of the surrogate [15]. Thus, the α-monofluorophosphonates are generally “isoacidic” with the phosphate monoesters that they mimic [16,17]. For all of these reasons, incorporation of fluorinated functionality into ligands directed at protein targets is often advantageous, and will likely remain an important stratagem in medicinal chemistry for years to come [9,18–21]. An interesting new development along these lines involves the incorporation of the SF5-group, in place of CF3 groups, for example, as has been put forth by Welch [22].
It is the purpose of this article to focus on the advantage offered by specific fluorinated functional groups, in both inhibitor design, and in mechanistic analysis. In this regard, emphasis will placed on the possibility of observing protein-ligand interactions through the use of 19F NMR, and on the development of organofluorine functional groups to target active sites of interest, based upon an understanding of mechanism. We will begin with examples in which fluoroorganics are strategically introduced to serve as NMR-based reporting element-to provide (i) the medicinal chemist with a rapid screen for enzyme inhibition; (ii) the functional proteomics investigator with an assay for function and (iii) the mechanistic enzymologist with information on the stereochemical course of a biocatalytic reaction. From there, our discussion will move into organofluorine functionalities that have been specifically tailored to produce either transition state analogue inhibition or irreversible, enzyme-activated inhibition (i.e. suicide substrates).
2. Emergence of 19F-Based NMR Screens for Inhibitor Development and Functional Proteomics
The past decade or so has seen the coming of age of NMR spectroscopy as a screening tool to facilitate the drug discovery process. This is particularly due to the influential work of Fesik and coworkers in developing so-called SAR by NMR techniques [23,24]. The last few years have seen the emergence of a number of creative 19F-based NMR techniques, that while philosophically similarly motivated, highlight the utility fluorinated functionality in such systems. Notable advantages of the fluorine nucleus include its virtual “bio-orthogonality” [25], and its responsiveness to environmental factors. This particularly true if one considers fluorination of an enzymatic substrate. The 19F isotropic chemical shift is very sensitive to small structural perturbations, resulting in chemical shift changes with substrate turnover, even in cases where the label is distal to the site of the chemistry. Moreover, if one employs CF3 groups as tags, one increases sensitivity, generating sharp singlets in the 19F spectrum and obviating the need for proton-decoupling, so long as the CF3 groups are not scalar-coupled to 1H nuclei. Thus, trifluoromethylated aromatics are ideal platforms for such applications.
This area has really blossomed in past several years, due in no small part to the work of Dalvit and co-workers [26]. As is shown in Figures 2 and 3, for screens of enzyme activity on peptide substrates this technique is particularly well-suited. If one employs trifluoromethylated aromatic amino acids, a single CF3 group suffices to yield clean assays for both peptide phosphorylation, by AKT kinase in this case, or peptide cleavage, by trypsin here. Note that the fluorinated reporting amino acid does not itself undergo chemical transformation for either reaction being screened. Because CF3 groups are employed for the reasons elaborated above, Dalvit labels this method 3-FABS (3 Fluorine Atoms for Biochemical Screening) [27]. A similar approach has been taken by Giralt and coworkers to screen for HIV protease inhibitors [28]. Quite recently, the Dalvit group has pointed out that the installation of reporting amino acids with two symmetrically disposed CF3 groups (Fig. 2) increases sensitivity [29]. Indeed, his group has shown that sensitivity can be improved still further through the application of cryoprobe technology. These studies have established the ability of such CF3-tagged substrate methods to yield accurate IC50 values. Thus, the 3-FABS approach is expected to see wider application for inhibitor screening, in automated manifolds, particularly in combinatorial chemistry applications.
Fig. 2.

Polyfluorinated amino acids to improve sensitivity 19F-NMR-based screening methods.
Fig. 3.

Use of a 19F stereochemical marker to decipher latent facial selectivity in the mechanism of KDO8P synthase (19NMR spectrum adapted from ref. 30)
In addition to this, one of the most promising observations to arise from this work is the notion that one might be able to generate a library of tagged substrates to probe for protein function. Thus, the tetradecapeptide illustrated in Figure 1 gives unambiguous and distinct signature signals for its specific phosphorylation by AKT kinase, and for its cleavage by trypsin (Note that though up to four potential tryptic cleavage sites are present, one appears to be preferred). When used as a test substrate for the PAK-4 protein, one see the signature of serine-kinase activity. It remains to be seen how many such test peptide/signature reactions can be screened in parallel, perhaps in a single NMR tube, but the possibilities are intriguing, to be sure. One can imagine, for example, given optimal chemical shift dispersion, rapidly getting a fingerprint for the substrate specificities of newly isolated kinases, phosphatases, proteases or perhaps even histone acetyltransferases and/or histone deacetylases, by such methods. A principal advantage of such 19F-based functional proteomics techniques is the near “bio-orthogonality” of carbon-bound fluorine [25].
Fig. 1.

The 3-FABS method used to screen for protease and kinase activity simultaneously – pep = substrate peptide; P-pep = kinase product; C-pep = tryptic digestion product; 19F NMR (564 MHz). (NMR figure adapted from ref 27)
3. Use of Fluorinated Functionality to Reveal Latent Enzyme Stereochemistry
Furdui, Anderson and coworkers have provided an elegant current example of how fluorine can be employed as analytical tool to study enzyme mechanism, even illuminating otherwise latent issues of pi-facial selectivity. Thus, both E- and Z-isomers of 3-fluoro-PEP serve as substrates for the enzyme KDO (3-deoxy-D-manno-2-octulosonate) 8-phosphate synthase [30]. And while the product stereochemistry requires that the key C-C-bond forming step involve attack of the nucleophilic-PEP 3-carbon upon the re-face of D-arabinose 5-phosphate, any facial selectivity with respect to PEP would be invisible here. As can be seen in the 19F NMR spectrum of this enzymatic reaction (Fig. 3), Aquifex pyrophilus KDO8PS takes the E-isomer (−138 ppm) of 3-fluoro-PEP noticeably faster than the Z-isomer (−150 ppm), and, in so doing, catalyzes an aldol condensation at what would correspond to the si-face of the PEP substrate. This can be clearly seen from the NMR spectrum, as the new product carries the spectroscopic signature of authentic (3R)-fluoro-KDO-8P (δ19F @ −206 ppm). Interestingly, this A. pyrophilus enzyme and the KDO8PS from E. coli display the same facial selectivity for PEP and the same preference for the E-fluorinated compound, though they belong to different mechanistic classes, with only the latter being metal-dependent.
4. Use of Fluorinated Functionality for “Transition State Analogue” Inhibition
Particular advantage is offered by carefully crafted fluorinated functional groups that, at once, are able to trap out enzyme bound species resembling intermediates or “transition states.” Though the term “transition state analogue” used here must be understood figuratively, it has become common parlance in the field. Even in the most apropos cases, the term is a bit of an oxymoron, when one thinks about the concavity of a free energy surface. A true transition state mimic would sit at a concave downward point on that surface, whereas for tight-binding inhibition, the E-TS analogue complex must rest in a free energy well, i.e. in a concave upward region. That said, in some cases, one, in fact, succeeds in forming a reversible covalent enzyme-inhibitor complex that resembles a high energy intermediate along the normal enzymatic reaction coordinate, but for which no productive pathway forward (to product) is available. And, by a Hammond postulate argument, mimicking a high energy intermediate perhaps justifies the label “transition state” analogue. This could be said of the α-fluorinated ketone inhibitors for serine proteases discussed below, as well as for the 2-fluoro-sugar glycosidase inhibitors. In all of the cases presented here, fluorination serves the dual purpose of finely tuning the functional group to form a stable complex with the target enzyme, and of providing a unique spectroscopic window through with the bioorganic chemist can follow and characterize for the formation of this complex.
4.1 α-Trifluoromethyl Ketones as Serine Protease Inhibitors
Robert Abeles and coworkers pioneered the use of α-fluorinated ketones as serine protease inhibitors. In this design, the α-CFn substituent destabilizes the C-O π-bond to which it is attached. This renders the carbonyl susceptible to hydration, and to addition of the active site serine, when incorporated into a suitable peptide scaffold, mimicking a typical substrate (Fig. 4). The resultant hemiacetal addition complex represents an analogue of the tetrahedral intermediate formed upon the normal reaction coordinate for enzymatic peptide bond cleavage. A secondary effect of the fluoroalkyl substituent is to lower the hemiacetal pKa for this tetrahedral intermediate (vide infra for 19F NMR titration). This, in turn, insures essentially complete oxyanionic character of the addition complex, promoting favorable interactions with the active site “oxyanion hole.”
Fig. 4.

Carbonyls with a high propensity for hydration serve as transition state analogue inhibitors for chymotrypsin.
An early study by Imperiali and Abeles [31] established that for N-acetylleucine-phenylalanyl-fluoroketone dipeptide mimics, both the percent hydration and the potency of chymotrypsin inhibition correlate nicely with the extent of of α-fluorination. Thus, 19F NMR established that the α-monofluoroketone is approximately 50% hydrated, whereas the di- and trifluoroketones are ~100% hydrated in aqueous solution. Ki decreases from 200 to 25 to 1 micromolar, in this series. Subsequent work showed it possible to monitor the formation of the active site serine adduct by 19F NMR [32]. Comparison of chemical shifts with those of model compounds was consistent with hemiketal formation (Fig. 5).
Fig. 5.

Formation of the tetrahedral adduct with the active site serine of chymotrypsin. Lower inset: 19F spectrum (adapted from ref. 31) showing both free inhibitor (hydrate, @ −82 ppm) and the covalent enzyme adduct (broad peak @ −80 ppm)
Moreover, pH titration of the E-I complex, as monitored by 19F NMR (Fig. 6.), allowed for experimental estimation of the bound, α-fluorinated hemiketal pKa at 4.9 [32,33]. Thus, the α-fluorinated hemiketal pKa is apparently lowered by approximately 4 log units, in the active site, suggestive of favorable interactions in the oxyanion hole. These studies firmly established the utility of such fluorinated functionality, both to trap out a stable mimic of the E-S tetrahedral intermediate, and as an analytical tool providing a 19F beacon, allowing the experimentalist to monitor the formation of this binary complex and its chemical environment. The work built on earlier studies by Shah and Gorenstein, that established 19F NMR as an analytical tool to monitor transition state analogue formation with peptidyl aldehydes in which a 19F tag had been inserted at the p-position of a phenylalaninal ring [34,35].
Fig. 6.
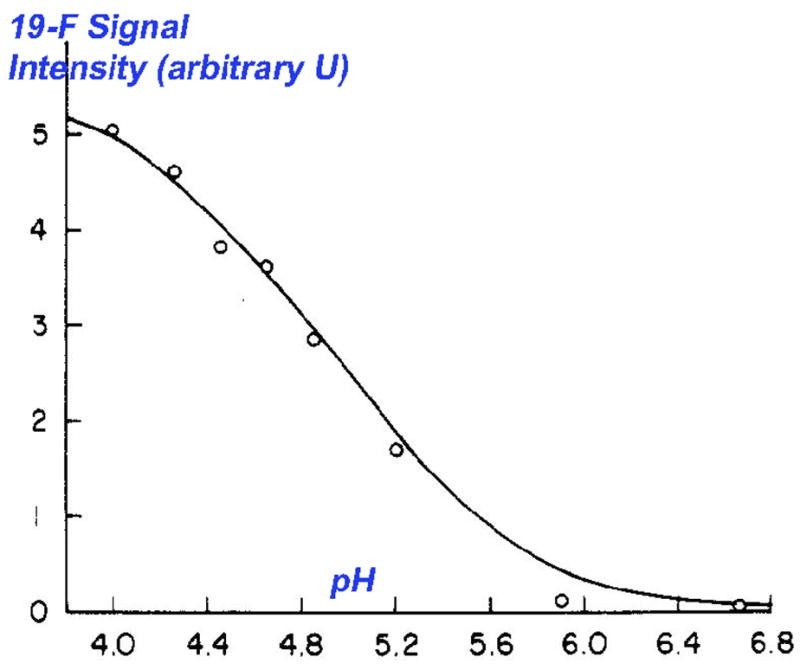
19F NMR titration of the TS-analogue complex shown in Figs. 5 and 7 (adapted from ref. 32)
More recently, the structural details of this protein-bound hemiketal presaged by these nice, NMR-assisted bioorganic studies, have been revealed by X-ray crystallographic studies of the complex [36]. The canonical Ser-195-His-57-Asp-102 catalytic triad is highlighted in Figure 7, with the former residue clearly having added in to the activated carbonyl center. Later, a higher resolution structures of this same complex and the analogous chymotrypsin-N-Ac-phenylalanine-COF3 complex [37] were taken as structural evidence by Frey and coworkers for a “low barrier hydrogen-bond (LBHB)” postualated to form in the chymotrypsion transition state. Specifically, the observed δ-N-His(57)-δ-O-Asp(102) distance of 2.6 Å is less than the sum of the van der Waals radii of N and O (2.7 Å), thereby meeting the definition of a LBHB. More recently, Warshel has called this view of the complex into question, concluding rather that one is seeing tight complexation here as result of dipole preorganization by the protein [38]. All of these studies highlight the value of this fluorinated functionality in mechanistic enzymology, providing insight into factors stabilizing the enzymatic transition state.
Fig. 7.

X-Ray crystallographic view of the same transition state analogue complex shown in Fig. 5 (PDB ID 7GCH).
Trifluoromethyl ketone inhibitors of other serine proteases, such as elastase and α-lytic protease [39] and the notable cardiovascular target chymase [40,41] have been developed. A related a,a-difluoropyruvamide motif is also quite effective in the chymotrypsin active site, as illustrated in Figure 5. The fluorinated ketone transition state analogue approach can be extended to the inhibition of serine esterases, including acetylcholinesterase [42]and phospholipase A2 [43]. 19F NMR can be applied to this class of enzymes as well, to observe E-I complex formation [44]. Interestingly, the zinc-metalloproteases carboxypeptidase A and angiotensin converting enzyme [45] are also susceptible to inhibition based upon this design [46].
4.2 5-Fluorodeoxyuridylate Monophosphate and Thymidylate Synthase
Over the last 25 years, 5-fluorouracil (5-FU) has seen widespread clinical application, in chemotherapy, particularly for colorectal cancer [47]. On the one hand, 5-fluorouracil does get incorporated into both nucleosides and 2′-deoxynucleosides and these nucleosides are fully phosphorylated to the 5′-triphosphates. This leads to the incorporation of 5-FU into both DNA and RNA. However, incorporation into nucleic acid does not generally correlate with effectiveness as a chemotherapeutic agent [48]. On the other other hand, the key 5-FU metabolite, 5-fluoro-dUMP acts, as designed, as an effective inhibitor of the enzyme, thymidylate synthase (TS). This enzyme normally catalyzes the one carbon transfer from N5,N10-methylene-tetrahydrofolate (CH2-THF) to the 5-position of dUMP, producing thymidine-5′-monophosphate and dihydrofolate (DHF). This is an essential step in pyrimidine biosynthesis, and the effectiveness of 5-FU as a chemotherapeutic generally correlates well with TS levels in the target tumor, and with effectiveness in TS activity knockdown upon treatment.
The 5-fluoro-dUMP system is also one of the earliest in which a mechanistically important fluorine atom would report back to the bioorganic chemist on inhibition mechanism. By design, 5-fluorination should promote the conjugate addition of the active site cysteine and allow for the capture of the resultant α-fluoro-enolate with the iminium ion derived from CH2-THF (Fig. 8.). However, the accepted mechanistic third step, α, β-elimination of THF, would necessarily be prevented by α-fluorination. This, by design, should lead to the build-up of a covalent E-I-CH2-THF complex. Indeed, as early as 1976, James, Santi and co-workers described 19F NMR evidence of a successful adduction of the active site nucleophile via conjugate addition [49]. These studies were performed on an peptide fragment of the protein that included the adducted active site cysteine.
Fig. 8.

Formation of the 5F-dUMP-CH2-THF-TS ternary complex: Top - color-coded reaction scheme; Bottom right – 188.8 MHz 19F NMR spectrum, showing progressive formation of the binary, pseudo-ternary and covalent ternary complex (adapted from ref 53); Bottom left – three-dimensional structure of the ternary complex (PDB ID:1TLS).
Several years later, an elegant series of studies by Dunlap and co-workers with the intact thymidylate synthase (TS) protein [50–53] demonstrated that one could follow and differentiate the formation of (i) binary (E-FdUMP); (ii) pseudo-ternary (E-FdUMP + THF) and (iii) covalent ternary (E-FdUMP-CH2-THF) complexes by 19F NMR. The spectroscopic signatures for these complexes are provided in Figure 8. Comparison with the 19F NMR spectrum of the bisulfite adduct of 5-fluoro-dUMP guided the original assignment of the covalent, binary adduct as the 5,6-dihydro derivative of the fluorinated pyrimidine. Independent determinations of the three dimensional structure for the covalent ternary complex with the E. coli enzyme have been provided from the groups of Matthews and Montfort. A rendering of the latter structure with VMD [54] is presented in Fig. 8., demonstrating clearly that the ternary complex has been arrested, following conjugate addition of cysteine-146 and THF-methylenation, as designed.
4.3 Fluoro-sugars as Glycosidase Inhibitors
The Withers group has introduced a strategy for inhibiting glycosidases that exploits an α-fluoro effect not unlike that established by Abeles in the serine protease arena (vide supra). A common feature of “retaining glycosidases” is that they are believed to proceed via the intermediacy of a covalent E-S complex. Moreover, both the formation and hydrolysis of the glycosyl enzyme species proceed via transition states with considerable oxocarbenium ion character. Thus, α-fluorination, just as for a carbonyl center, is expected to destabilize this onium species, slowing both the rates of glycosyl enzyme formation and breakdown. However, the inclusion of a reactive leaving group, such as dinitrophenol or fluoride at the anomeric center of the inhibitor serves to accelerate formation of the glycosyl enzyme intermediate with these inhibitors. Once formed, the adduct is greatly stabilized by the presence of the 2-fluoro-substituent. Such activated 2-deoxy-2-fluorosaccharides have now been shown to trap out active site carboxylate residues in a number of glycosidase active sites. The original work was carried with β-glucosidase [55–57], and both the formation of an active site 2-deoxy-2-fluoroglucoside, and its α-stereochemistry were established by 19F NMR. Figure 9 illustrates a more recent example in which one observes the capture of glutamate-233, with the α-anomeric stereochemistry, in the active site of β-glucanase via 19 F NMR [58]. Subsequent solution of the three dimensional X-ray structure of the E-I complex confirmed these assignments.
Fig. 9.

Use of the 2″,4″-Dinitrophenyl 2-fluoro-2-deoxy-β-cellobioside to trap out the carboxlate residue in the active site of Cellulomonas fimi glucanase. Lower Inset: 19F NMR of complex formation (adapted from ref. 58). The signals at −198.1 and −198.4 ppm are assigned to the unbound and hydrolyzed inhibitor, respectively. The broader signal at −195.4 ppm is due to the covalent intermediate. Note that the chemical shift (−205.5 ppm) of the adduct from the fluoro-sugar possessing the manno-stereochemistry unambiguously establishes the α-stereochemistry of the adduct with Glu-233. Right hand inset: View of the X-ray crystal structure of this complex (PDB ID: 1EXP).
The same principle has now been applied to the inhibition of lysozyme [59], as well as xylanase [60], galactosidase [61], mannosidase [62] enzymes. More recent studies indicate that sialidase enzymes, important bacterial, viral and trypanosomal targets, are also effectively adducted with fluorinated neuraminic acids [63]. Here, X-ray crystallography has indeed, established that the trapped out active site residue is a tyrosine, rather than a glutamate or aspartate residue [64–66]. Even beyond this, success has been achieved in inhibiting 1,3-fucosyltransferase, for example, with guanosine 5′-diphospho-2-deoxy-2-fluoro-beta-L-fucose (GDP-2F-Fuc) [67], suggesting that the Withers approach to the inhibition of glycosyl hydrolase enzymes may be quite broadly extensible to the domain of glycoside synthesis enzymes, as well.
5. Use of Fluorinated Functionality for “Suicide Substrate” Inactivation
Quite a number of tailored fluorinated functional groups serve as the basis for the suicide inactivator development. Among the most notable examples are α-difluoromethylornithine (DFMO), a mechanism-based inactivator of L-ornithine decarboxylase (ODC), the enzyme that controls flux through the polyamine pathway, associated with cell growth and differentiation. Inactivation of this enzyme proceeds via pyridoxal phosphate (PLP)-assisted decarboxylative elimination of fluoride to produce an fluorinated Michael acceptor, that is captured by an active site cysteine residue. This has been established by both mass spectrometric [68] and x-ray crystallographic studies [69] from the groups of Pegg and Phillips, respectively. DFMO inhibition of trypanosomal ODC cures African Sleeping Sickness [70]. The compounds is also under clinical evaluation for the prevention of colon cancer [71,72].
Gemcitabine, 2′-deoxy-2′,2′-difluorocytidine, is widely used in the treatment of advanced pancreatic cancer. The compound is a pro-drug, being 5′-phosphorylated in vivo. The triphoshpate is a DNA polymerase chain terminator, whereas the diphosphate is a suicide substrate for ribonucleotide reductase, an enzyme that operates via a radical mechanism, using an active site tyrosyl radical. Fluoride’s leaving group ability from an electron rich system, once again, is exploited here [73].
Like DFMO, α-monofluorinated amino acids, can be used as suicide substrates for PLP-dependent enzymes, and, once again the leaving group ability of fluoride is important in the inactivation mechanism, in this case. However, the monofluoromethyl trigger generally leads to an enamine-based nucleophilic inhibition mechanism [74,75] first observed by Metzler with serine O-sulfate [76,77]. Another notable PLP enzyme inactivator is α-(1′-fluoro)vinylglycine, a suicide substrate for both bacterial alanine racemase [78,79]and for tryptophan synthase [80]. Once again, the ability of fluoride to leave from an electron rich intermediate is exploited, presumably leading to an allenic imine, following enzymatic α-deprotonation, as originally proposed by Abeles.
Finally, the use of o- or p-(di)fluoromethylphenoxy or thiophenoxy leaving groups, as precursors to (halo)quinone methides has also been exploited in phosphatase, glycosidase and amidase active sites by the groups of Widlanski [81–83], Danzin [84] and Badet [85–87], respectively. Recently, Thompson has shown that arginine deiminase enzymes can be effectively targeted utilizing a fluorinated acetamidine functionality [88,89].
That said, the discussion here will focus on work in these laboratories, specifically on the development of the α-(2′Z-fluoro)vinyl trigger for PLP-dependent amino acid decarboxylase (AADC) inactivation. This approach evolved from initial efforts to stereoselectively install the simple, unsubstituted α-vinyl trigger, itself. α-Vinylglycine is a naturally occurring inactivator of number of PLP-dependent enzymes [90], including transaminases and ACC (1-amino-1-cyclopropanecarboxylate) synthase [91]. The natural occurrence of this Trojan horse functionality presumably inspired its incorporation into γ-vinyl-GABA (Vigabatrin), an effective GABA transaminase inactivator [92,93], and anti-convulsant drug. In both of these cases, x-ray crystallographic studies [92,91] have now confirmed that the active site lysine is captured, presumably via conjugate addition to an α, β-unsaturated iminium ion, following enzyme-mediated azallylic isomerization.
If one wishes to use such a π-trigger to selectively target PLP-dependent AADC’s, as opposed to transaminase, racemase, eliminase or replacement enzymes, then substitution of the α-proton with the vinylic functionality appears to be a good design. This design eliminates the possibility of α-deprotonation [as opposed to situating the π-trigger along the side chain or in place of the α-carboxylate (product analogue)], but still allows for α-decarboxylation, thereby potentially conferring specificity. This design also likely promotes errant protonation, following decarboxylation, another potential advantage discussed below.
With these design advantages, however, comes a synthetic challenge. One must construct quaternary α-vinyl amino acids, ideally with control of stereochemistry at this quaternary center. Because of the accessibility of amino ester-based enolates, two disconnections are most attractive here, either installation of the vinyl group, via formal vinylation of the enolate derived from the cognate amino acid (AA), or installation of the side chain, via formal alkylation of a vinylglycine enolate equivalent. Initially, we chose the former approach, finding that N-benzoyl-protected amino acid esters could be doubly deprotonated and selectively α-alkylated with ethylene oxide, as vinyl cation equivalent [94,95]. Pleasingly, one could carry the protected side chains of even highly functionalized AA’s (His, Lys, Orn, hSer, DOPA) into these dianions, suggesting broader application of such an approach to the synthesis of α-alkylated AA’s, in general. Of course, these quaternary, α-vinyl AA’s, though rapidly assembled, are obtained in racemic form. Coupling this chemistry with an enzymatic resolution via “reverse transesterification” [96] provided a partial, though incomplete, solution to this stereochemical problem.
We next turned to the converse approach, beginning with vinylglycine equivalent and installing the side chain via stereoselective alkylation. On the one hand, we found that N-benzoyl-L-α-vinylglycine methyl ester, available from L-homoserine lactone [97], could be effectively cyclized to the corresponding L-cis and trans-oxazalines, under the aegis of a “PhSe+” equivalent, without loss of absolute stereochemistry (Fig. 10). Separation of these diastereomeric oxazolines give both a D-α-vinyl AA synthon (cis) and an L-α-vinyl AA synthon (trans) [98]. This is because one sees essentially absolute 1,2-stereoinduction in the alkylation event that introduces the AA side chain. Because one destroys the original α-stereocenter in this step, and exploits the newly installed β-stereocenter to direct the α-alkylation, one may regard this as a “self-reproduction of chirality” [99–101] or a “self-regeneration of stereocenters” approach [102], terms introduced by Seebach. One then unmasks the α-vinyl group through a sequence involving elimination (E2 conditions) to the α-(2′E-phenylseleno)vinyl AA, selenium-tin interchange and proto-destannylation. The intermediate conversion of a terminal vinyl selenide to a terminal vinylstannane constituted a fundamentally new method for vinylstannane synthesis. Moreover, the resultant, protected α-(2′E-tributylstannyl)vinyl AA’s could be exploited as building blocks for the construction of α-branch-extended analogues via Pd-mediated cross coupling chemistry [103]. For all of its versatility, this route has the limitation that only particularly reactive SN2 electrophiles can be efficiently captured in the oxazoline enolate alkylation step.
Fig. 10.

Asymmetric synthesis of quaternary, α-vinyl amino acids via a “self-regeneration of stereocenters” approach.
As a solution to this enolate reactivity issue, a complementary approach to the stereocontrolled synthesis of quaternary α-vinyl amino acids was developed. Namely, the AA side chain is introduced via alkylation of a chiral vinylglycine-derived dianionic dienolate, bearing a β-naphthylmenthyl ester auxiliary (Fig. 11) [104]. Enolate geometry is presumably controlled via amidate nitrogen chelation to lithium, and facial selectivity follows an “exo-extended” model for the reactive conformation of the dienolate. High levels of acyclic stereocontrol are achieved, even for unreactive SN2 electrophiles. Moreover, given Comins’ enzyme resolution technology [105], both antipodes of the requisite (desmethyl) chiral auxiliary are available, providing access to either antipode of the target quaternary α-vinyl AA. More recently in the group, good progress has been made toward transition metal-mediated routes into these densely functionalized β, γ-unsaturated AA’s, with the potential for catalytic control of stereochemistry. This includes both allylic amination [106–108] and formal [3,3]-sigmatropic rearrangement approaches [109].
Fig. 11.

Asymmetric synthesis of quaternary, α-vinyl amino acids via alkylation of chiral, vinylglycine-derived dienolates.
It is important to point out at this stage that the vinyl AA’s themselves serve as precursors to other α-branched AA’s with potential as enzyme inactivators, including α-oxiranyl [110], α-chlorovinyl [111] and α-bromovinyl AA’s. But, most importantly here, we were able to demonstrate that protected, enantiomerically enriched, α-vinyl AA’s, available from the chemistry described in Figs. 10 and 11, could be smoothly transformed into the corresponding α-(2′Z-fluoro)vinyl AA’s, with preservation of stereochemical purity [112]. As outlined in Figure 12, this transformation involves an ozonolysis/fluoromethylenation sequence, that avails itself of the McCarthy reagent [113,114].
Fig. 12.

Synthesis of quaternary, α-(2′Z-fluoro)vinyl amino acids from the corresponding α-vinyl amino acids.
In light of earlier success with the simple α-vinyl trigger and Hafnia alvei L-lysine decarboxylase (LDC) [115], we set to examine the α-(2′Z-fluoro)vinyl trigger [116,117] for AADC inactivation in this active site. Figure 13 illustrates the proposed mechanism(s) of AADC inactivation with this new AADC suicide substrate trigger. Transaldimination is expected to give external aldimine I, thereby releasing the enzymatic lysine from the internal aldimine linkage. Decarboxylation, the normal second mechanistic step, then provides the quinonoid intermediate, II. While the previously isolated fluorovinyl group is now conjugated with the extended π-system of the cofactor, it is not very electrophilic, as it possesses, at once, α, β-unsaturated iminium ion and tetra-enamine substructures. In the third step, protonation at any of three available sites would restore pyridinium ion aromaticity. On the one hand, α-protonation would re-isolate the fluorovinyl group, and represents the simple turnover pathway. However, C4′-protonation would have the effect of protonating the tetra-enamine substructure, thereby producing a potent electrophile, the β-fluoro-α, β-unsaturated iminium ion IV. In this species, the β-fluoro substituent serves the dual role of increasing the electrophilicity of the Michael acceptor, and potentially serving as a leaving group, transforming what would be a conjugate addition mechanism with a simple vinyl trigger into an addition-elimination mechanism with the fluorovinyl trigger. Finally, protonation at the γ-position should lead to release of an enamine-type product, VIII, that, most likely would either undergo Mannich condensation with the internal aldimine (Metzler mechanism [76,77]) or expel the γ-fluoride leaving group. This latter pathway would give a second electrophile, X, a potentially diffusible α, β-unsaturated iminium ion.
Fig. 13.

Proposed mechanism of PLP-linked lysine decarboxylase inactivation with the new, quaternary, α-(2′-fluoro)vinyl trigger. Green boxes highlight two projected electrophilic pathways for this functionality.
Note that errant protonation (i.e. C4′- and/or γ-protonation) is thus required, by design, for inactivation with this trigger. Consistent with this requirement, early studies by O’Leary and others on AADC mechanism reveals that α-substitution, specifically, replacement of the α-proton with a methyl substituent, tends to promote C4′ protonation. Thus, this so-called abortive transamination pathway is known to be increased by 1–2 orders of magnitude upon α-methylation [118–121]. As noted, this validates placement of this π-trigger at the α-carbon, so as to create steric hindrance to α-protonation there. Note further that errant protonation is expected to lead to release of fluoride ion, either (i) prior to enzyme alkylation (α, β-unsaturated iminium ion pathway), (ii) concomitant with enzyme alkylation (β-fluoro-α, β unsaturated iminium ion pathway) or (iii) subsequent to enzyme adduction (Metzler enamine pathway). This means that 19F NMR should allow the experimentalist to directly monitor partitioning in the protonation step itself.
An enantioselective synthesis of each antipode of α-(2′Z-fluoro)vinyllysine (FVL) was achieved as outlined in Figures 11 and 12 [122]. 4-Iodo-N-SES-N-PMB-butylamine was used as the SN2 electrophile for introduction of the lysine side chain. Examinaton of the individual FVL-antipodes shows the D-enantiomer to be exclusively a substrate, whereas the L-antipode is a suicide substrate, displaying time dependent inactivation of LDC. A Kitz-Wilson analysis of the data for L-FVL, yields kinact = 0.26 ± 0.07 min−1 and KI = 86 ± 22 mM. This is comparable to the kinact values seen for both antipodes of DFMO and (S)-Vigabatrin. The KI value is significantly higher than that seen for L-DFMO (~1.3 mM) [123], but significantly lower than that seen for (S)-Vigabatrin (~3 mM) [124]. Inactivation was seen to be functionally irreversible (exhaustive dialysis). Curvature in ln(Et/E0) vs. t plots was observed, suggestive of significant partitioning into natural or unnatural turnover pathways. A titration of the enzyme, with varying I:E ratios, provided an estimate of the partition ratio (total turnovers per inactivation event) of ~20 ±3.
Independent synthesis of α-(2′Z-fluoro)vinylcadaverine (FVC) established the viablility of following the enzymatic reaction course by 19F NMR (see Fig. 14 and the FVC- and fluoride-spiking experiments shown). Inactivation of 15 μM LDC produces 168 ± 14 μM turnover product (FVC) and 70 ± 13 μM fluoride. The fluoride value was confirmed with an ion-specific electrode (78 ± 5 μM). Since all errant protonations are projected to release fluoride, we can estimate that 1 in 3.2 decarboxylations leads to errant protonation (29%!), with 1 in 5 errant protonation events leading to LDC inactivation. This gives an overall partition ratio of 16 ± 2. The high errant protonation rate (~8 times the maximum value produced by α-methylation) seen in this model AADC active site is promising, as, by design, altered protonation is required for trigger actuation. Given this and the favorable kinact, it will be of interest to examine this trigger in other AADC active sites. The remarkable enantiodiscrimination observed contrasts with the case of DFMO, in which similar behavior has been reported for both antipodes [123].
Fig. 14.

Observation of turnover and aberrant protonation by 564 MHz 19F NMR (a) 19F NMR spectrum following inactivation of LDC with L-α-(2′Z-fluoro)vinyllysine (L-FVL); (b) Addition of KF to (a); (c) Addition of α-(2′Z-fluoro)vinylcadaverine (FVC) to (b).
In summary, synthetic chemistry has been developed that allows for the enantioselective construction of quaternary amino acids, outfitted with an α-(2′Z-fluoro)vinyl trigger. In the first test of this trigger, in the LDC active site, one sees both efficient enzyme inactivation, and the fringe benefit of the 19F beacon, built into the trigger. Thus, 19F NMR can be exploited to provide information on trigger partitioning, particularly in the important quinonoid protonation step. Thus, whereas the traditional titration experiment provides information on the number of inhibitor molecules “turning over” per inactivation event, it provides no information on how those turnovers take place. The 19F experiment supplies additional fine structure, showing here that approximately 30% of those turnovers do result in the desired errant protonation, with the remaining 70% going on to the usual decarboxylation product (α-protonation). This example, when considered along with the fluoromethyl ketone (serine proteases), 5-fluoropyrimidine (thymidylate synthase) and 2-deoxy-2-fluoro-sugar (retaining glycosidases and glycosyl transferases) cases discussed earlier in this article, highlights the dual utility of appropriately tailored fluorinated functionality, for both enzyme inhibition and the mechanistic study of that inhibition, in situ, with 19F NMR spectroscopy.
Acknowledgments
We thank the NSF and the NIH (CA 62034 and RR016544-01) for support of unnatural amino acid synthesis, fluorinated trigger development and mechanism-based enzyme inhibition efforts in our laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.O’Hagan D. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a. [DOI] [PubMed] [Google Scholar]
- 2.Biffinger JC, Kim HW, DiMagno SG. ChemBioChem. 2004;5:622–627. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 3.Kim HW, Rossi P, Shoemaker RK, DiMagno SG. J Am Chem Soc. 1998;120:9082–9083. [Google Scholar]
- 4.Plenio H, Diodone R. Chem Ber/Recl. 1997;130:633–640. [Google Scholar]
- 5.Dunitz JD, Taylor R. Chem--Eur J. 1997;3:89–98. [Google Scholar]
- 6.Howard JAK, Hoy VJ, O’Hagan D, Smith GT. Tetrahedron. 1996;52:12613–12622. [Google Scholar]
- 7.Hyla-Kryspin I, Haufe G, Grimme S. Chem Eur J. 2004;10:3411–3422. doi: 10.1002/chem.200305584. [DOI] [PubMed] [Google Scholar]
- 8.Chen L, Wu L, Otaka A, Smyth MS, Roller PP, Burke TR, Jr, den Hertog J, Zhang ZY. Biochem Biophys Res Commun. 1995;216:976–984. doi: 10.1006/bbrc.1995.2716. [DOI] [PubMed] [Google Scholar]
- 9.Mueller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 10.Fischer FR, Schweizer WB, Diederich F. Angew Chem, Int Ed. 2007;46:8270–8273. doi: 10.1002/anie.200702497. [DOI] [PubMed] [Google Scholar]
- 11.Schweizer E, Hoffmann-Roder A, Scharer K, Olsen JA, Fah C, Seiler P, Obst-Sander U, Wagner B, Kansy M, Diederich F. ChemMedChem. 2006;1:611–621. doi: 10.1002/cmdc.200600015. [DOI] [PubMed] [Google Scholar]
- 12.Paulini R, Mueller K, Diederich F. Angew Chem, Int Ed. 2005;44:1788–1805. doi: 10.1002/anie.200462213. [DOI] [PubMed] [Google Scholar]
- 13.Burgi HB, Dunitz JD, Shefter E. J Amer Chem Soc. 1973;95:5065–5067. [Google Scholar]
- 14.Buergi HB, Dunitz JD. Acc Chem Res. 1983;16:153–161. [Google Scholar]
- 15.Berkowitz DB, Bose M. J Fluorine Chem. 2001;112:13–33. [Google Scholar]
- 16.Berkowitz DB, Bose M, Pfannenstiel TJ, Doukov T. J Org Chem. 2000;65:4498–4508. doi: 10.1021/jo000220v. [DOI] [PubMed] [Google Scholar]
- 17.Nieschalk J, Batsanov AS, O’Hagan D, Howard JAK. Tetrahedron. 1996;52:165–76. [Google Scholar]
- 18.Kirk KL. Org Process Res Dev. 2008;12:305–321. [Google Scholar]
- 19.Begue JP, Bonnet-Delpon D. Actualite Chimique. 2006;301–302:83–87. [Google Scholar]
- 20.O’Hagan D, Rzepa HS. Chem Commun. 1997:645–652. [Google Scholar]
- 21.Pongdee R, Liu H-w. Bioorg Chem. 2004;32:393–437. doi: 10.1016/j.bioorg.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 22.Welch JT, Lim DS. Bioorg Med Chem. 2007;15:6659–6666. doi: 10.1016/j.bmc.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 23.Fesik SW, Shuker SB, Hajduk PJ, Meadows RP. Protein Eng. 1997;10:73. [Google Scholar]
- 24.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 25.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 26.Dalvit C. Prog Nucl Magn Reson Spectrosc. 2007;51:243–271. doi: 10.1016/j.pnmrs.2023.07.001. [DOI] [PubMed] [Google Scholar]
- 27.Dalvit C, Ardini E, Flocco M, Fogliatto Gian P, Mongelli N, Veronesi M. J Am Chem Soc. 2003;125:14620–14625. doi: 10.1021/ja038128e. [DOI] [PubMed] [Google Scholar]
- 28.Frutos S, Tarrago T, Giralt E. Bioorg Med Chem Lett. 2006;16:2677–2681. doi: 10.1016/j.bmcl.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 29.Papeo G, Giordano P, Brasca MG, Buzzo F, Caronni D, Ciprandi F, Mongelli N, Veronesi M, Vulpetti A, Dalvit C. J Am Chem Soc. 2007;129:5665–5672. doi: 10.1021/ja069128s. [DOI] [PubMed] [Google Scholar]
- 30.Furdui CM, Sau AK, Yaniv O, Belakhov V, Woodard RW, Baasov T, Anderson KS. Biochemistry. 2005;44:7326–7335. doi: 10.1021/bi047282q. [DOI] [PubMed] [Google Scholar]
- 31.Imperiali B, Abeles RH. Biochemistry. 1986;25:3760–3767. doi: 10.1021/bi00361a005. [DOI] [PubMed] [Google Scholar]
- 32.Liang TC, Abeles RH. Biochemistry. 1987;26:7603–7608. doi: 10.1021/bi00398a011. [DOI] [PubMed] [Google Scholar]
- 33.Brady K, Liang TC, Abeles RH. Biochemistry. 1989;28:9066–9070. doi: 10.1021/bi00449a017. [DOI] [PubMed] [Google Scholar]
- 34.Shah DO, Gorenstein DG. Biochemistry. 1983;22:6096–7101. [Google Scholar]
- 35.Gorenstein DG, Shah DO. Biochemistry. 1982;21:4679–4686. doi: 10.1021/bi00262a025. [DOI] [PubMed] [Google Scholar]
- 36.Brady K, Wei A, Ringe D, Abeles RH. Biochemistry. 1990;29:7600–7607. doi: 10.1021/bi00485a009. [DOI] [PubMed] [Google Scholar]
- 37.Neidhart D, Wei Y, Cassidy C, Lin J, Cleland WW, Frey PA. Biochemistry. 2001;40:2439–2447. doi: 10.1021/bi002535a. [DOI] [PubMed] [Google Scholar]
- 38.Schutz CN, Warshel A. Proteins. 2004;55:711–723. doi: 10.1002/prot.20096. [DOI] [PubMed] [Google Scholar]
- 39.Govardhan CP, Abeles RH. Arch Biochem Biophys. 1990;280:137–146. doi: 10.1016/0003-9861(90)90528-7. [DOI] [PubMed] [Google Scholar]
- 40.Deguchi T, Shiratake R, Sato F, Fujitani T, Honda Y, Kiyoshi A, Notake M, Showell GA, Boyle RG, Klair SS. Dainippon Pharmaceutical Co., Ltd; Japan: 2003. p. 44. Application: JP JP. [Google Scholar]
- 41.Deguchi T, Shiratake R, Sato F, Fujitani B, Kiyoshi A, Honda Y, Notake M. Abstracts of Papers, 223rd ACS National Meeting; Orlando, FL, United States. April 7–11, 2002; 2002. MEDI-061. [Google Scholar]
- 42.Allen KN, Abeles RH. Biochemistry. 1989;28:8466–8473. doi: 10.1021/bi00447a029. [DOI] [PubMed] [Google Scholar]
- 43.Street IP, Lin HK, Laliberte F, Ghomashchi F, Wang Z, Perrier H, Tremblay NM, Huang Z, Weech PK, Gelb MH. Biochemistry. 1993;32:5935–40. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 44.Rosell G, Herrero S, Guerrero A. Biochem Biophys Res Commun. 1996;226:287–292. doi: 10.1006/bbrc.1996.1347. [DOI] [PubMed] [Google Scholar]
- 45.Natesh R, Schwager SLU, Sturrock ED, Acharya KR. Nature. 2003;421:551–554. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 46.Gelb MH, Svaren JP, Abeles RH. Biochemistry. 1985;24:1813–1817. doi: 10.1021/bi00329a001. [DOI] [PubMed] [Google Scholar]
- 47.Longley DB, Harkin DP, Johnston PG. Nat Rev Cancer. 2003;3:330–338. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 48.Noordhuis P, Holwerda U, Van der Wilt CL, Van Groeningen CJ, Smid K, Meijer S, Pinedo HM, Peters GJ. Annals of Oncology. 2004;15:1025–1032. doi: 10.1093/annonc/mdh264. [DOI] [PubMed] [Google Scholar]
- 49.James TL, Pogolotti AL, Jr, Ivanetich KM, Wataya Y, Lam SSM, Santi DV. Biochem Biophys Res Commun. 1976;72:404–410. doi: 10.1016/s0006-291x(76)80057-5. [DOI] [PubMed] [Google Scholar]
- 50.Lewis CA, Jr, Ellis PD, Dunlap RB. Biochem Biophys Res Commun. 1978;83:1509–1517. doi: 10.1016/0006-291x(78)91392-x. [DOI] [PubMed] [Google Scholar]
- 51.Lewis CA, Jr, Ellis PD, Dunlap RB. Biochemistry. 1980;19:116–123. doi: 10.1021/bi00542a018. [DOI] [PubMed] [Google Scholar]
- 52.Lewis CA, Jr, Ellis PD, Dunlap RB. Biochemistry. 1981;20:2275–2285. doi: 10.1021/bi00511a032. [DOI] [PubMed] [Google Scholar]
- 53.Zapf JW, Weir MS, Emerick V, Villafranca JE, Dunlap RB. Biochemistry. 1993;32:9274–9281. doi: 10.1021/bi00087a003. [DOI] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. J Molec Graphics. 1996;14.1:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 55.Withers SG, Rupitz K, Street IP. J Biol Chem. 1988;263:7929. [PubMed] [Google Scholar]
- 56.Wicki J, Rose David R, Withers Stephen G. Methods Enzymol. 2002;354:84–105. doi: 10.1016/s0076-6879(02)54007-6. [DOI] [PubMed] [Google Scholar]
- 57.Street IP, Kempton JB, Withers SG. Biochemistry. 1992;31:9970–9978. doi: 10.1021/bi00156a016. [DOI] [PubMed] [Google Scholar]
- 58.White A, Tull D, Johns K, Withers SG, Rose DR. Nat Struct Biol. 1996;3:149–154. doi: 10.1038/nsb0296-149. [DOI] [PubMed] [Google Scholar]
- 59.Vocadlo DJ, Withers SG. Carbohydr Res. 2005;340:379–388. doi: 10.1016/j.carres.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 60.Miao S, Ziser L, Aebersold R, Withers SG. Biochemistry. 1994;33:7027–7032. doi: 10.1021/bi00189a002. [DOI] [PubMed] [Google Scholar]
- 61.Ly HD, Howard S, Shum K, He S, Zhu A, Withers SG. Carbohydr Res. 2000;329:539–547. doi: 10.1016/s0008-6215(00)00214-7. [DOI] [PubMed] [Google Scholar]
- 62.Howard S, He S, Withers SG. J Biol Chem. 1998;273:2067–2072. doi: 10.1074/jbc.273.4.2067. [DOI] [PubMed] [Google Scholar]
- 63.Watts AG, Damager I, Amaya ML, Buschiazzo A, Alzari P, Frasch AC, Withers SG. J Am Chem Soc. 2003;125:7532–7533. doi: 10.1021/ja0344967. [DOI] [PubMed] [Google Scholar]
- 64.Newstead SL, Potter JA, Wilson JC, Xu G, Chien CH, Watts AG, Withers SG, Taylor GL. J Biol Chem. 2008;283:9080–9088. doi: 10.1074/jbc.M710247200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Watts AG, Oppezzo P, Withers SG, Alzari PM, Buschiazzo A. J Biol Chem. 2006;281:4149–4155. doi: 10.1074/jbc.M510677200. [DOI] [PubMed] [Google Scholar]
- 66.Amaya MF, Watts AG, Damager I, Wehenkel A, Nguyen T, Buschiazzo A, Paris G, Frasch AC, Withers SG, Alzari PM. Structure. 2004;12:775–784. doi: 10.1016/j.str.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 67.Murray BW, Wittmann V, Burkart MD, Hung SC, Wong CH. Biochemistry. 1997;36:823–831. doi: 10.1021/bi962284z. [DOI] [PubMed] [Google Scholar]
- 68.Poulin R, Lu L, Ackermann B, Bey P, Pegg AE. J Biol Chem. 1992;267:150–158. [PubMed] [Google Scholar]
- 69.Grishin NV, Osterman AL, Brooks HB, Phillips MA, Goldsmith EJ. Biochemistry. 1999;38:15174–15184. doi: 10.1021/bi9915115. [DOI] [PubMed] [Google Scholar]
- 70.McCann PP, Bacchi CJ, Clarkson AB, Jr, Bey P, Sjoerdsma A, Schecter PJ, Walzer PD, Barlow JL. The American journal of tropical medicine and hygiene. 1986;35:1153–6. doi: 10.4269/ajtmh.1986.35.1153. [DOI] [PubMed] [Google Scholar]
- 71.Wallace HM, Fraser AV. Amino Acids. 2004;26:353–365. doi: 10.1007/s00726-004-0092-6. [DOI] [PubMed] [Google Scholar]
- 72.Meyskens FL, Jr, Gerner EW. Clinical Cancer Research. 1999;5:945–951. [PubMed] [Google Scholar]
- 73.van der Donk WA, Yu G, Perez L, Sanchez RJ, Stubbe J, Samano V, Robins MJ. Biochemistry. 1998;37:6419–6426. doi: 10.1021/bi9729357. [DOI] [PubMed] [Google Scholar]
- 74.Bhattacharjee MK, Snell EE. J Biol Chem. 1990;265:6664–6668. [PubMed] [Google Scholar]
- 75.Hayashi H, Tanase S, Snell EE. Journal of Biological Chemistry. 1986;261:11003–9. [PubMed] [Google Scholar]
- 76.Likos JJ, Ueno H, Feldhaus RW, Metzler DE. Biochemistry. 1982;21:4377–4386. doi: 10.1021/bi00261a029. [DOI] [PubMed] [Google Scholar]
- 77.Ueno H, Likos JJ, Metzler DE. Biochemistry. 1982;21:4387–4393. doi: 10.1021/bi00261a030. [DOI] [PubMed] [Google Scholar]
- 78.Thornberry NA, Bull HG, Taub D, Wilson KE, Gimenez-Gallego G, Rosegay A, Soderman DD, Patchett AA. J Biol Chem. 1991;266:21657–21665. [PubMed] [Google Scholar]
- 79.Thornberry NA, Bull HG, Taub D, Greenlee WJ, Patchett AA, Cordes EH. J Am Chem Soc. 1987;109:7543–7544. [Google Scholar]
- 80.Xu Y, Abeles RH. Biochemistry. 1993;32:806–811. doi: 10.1021/bi00054a010. [DOI] [PubMed] [Google Scholar]
- 81.Born TL, Myers JK, Widlanski TS, Rusnak F. J Biol Chem. 1995;270:25651–25655. doi: 10.1074/jbc.270.43.25651. [DOI] [PubMed] [Google Scholar]
- 82.Myers JK, Cohen JD, Widlanski TS. J Am Chem Soc. 1995;117:11049–11054. [Google Scholar]
- 83.Myers JK, Widlanski TS. Science. 1993;262:1451–1453. doi: 10.1126/science.8248785. [DOI] [PubMed] [Google Scholar]
- 84.Halazy S, Berges V, Ehrhard A, Danzin C. Bioorg Chem. 1990;18:330–344. [Google Scholar]
- 85.Yaouancq L, Anissimova M, Badet-Denisot MA, Badet B. Eur J Org Chem. 2002:3573–3579. [Google Scholar]
- 86.Araoz R, Anhalt E, Rene L, Badet-Denisot MA, Courvalin P, Badet B. Biochemistry. 2000;39:15971–15979. doi: 10.1021/bi001408b. [DOI] [PubMed] [Google Scholar]
- 87.Massiere F, Badet-Denisot MA, Rene L, Badet B. J Am Chem Soc. 1997;119:5748–5749. [Google Scholar]
- 88.Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR. J Am Chem Soc. 2006;128:1092–1093. doi: 10.1021/ja0576233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Sato M, Thompson PR. Biochemistry. 2006;45:11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Berkowitz DB, Charette BD, Karukurichi KR, McFadden JM. Tetrahedron: Asymmetry. 2006;17:869–882. doi: 10.1016/j.tetasy.2006.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Capitani G, Tschopp M, Eliot AC, Kirsch JF, Gruetter MG. FEBS Lett. 2005;579:2458–2462. doi: 10.1016/j.febslet.2005.03.048. [DOI] [PubMed] [Google Scholar]
- 92.Storici P, De Biase D, Bossa F, Bruno S, Mozzarelli A, Peneff C, Silverman RB, Schirmer T. J Biol Chem. 2004;279:363–373. doi: 10.1074/jbc.M305884200. [DOI] [PubMed] [Google Scholar]
- 93.Silverman RB, Bichler KA, Leon AJ. J Am Chem Soc. 1996;118:1241–1252. [Google Scholar]
- 94.Pedersen ML, Berkowitz DB. J Org Chem. 1993;58:6966–6975. [Google Scholar]
- 95.Pedersen ML, Berkowitz DB. Tetrahedron Lett. 1992;33:7315–7318. [Google Scholar]
- 96.Berkowitz DB, Pumphrey JA, Shen Q. Tetrahedron Lett. 1994;35:8743–8746. [Google Scholar]
- 97.Berkowitz DB, Smith MK. Synthesis. 1996:39–41. doi: 10.1055/s-1996-4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berkowitz DB, McFadden JM, Chisowa E, Semerad CL. J Am Chem Soc. 2000;122:11031–11032. doi: 10.1021/ja0055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Seebach D, Fadel A. Helv Chim Acta. 1985;68:1243–1250. [Google Scholar]
- 100.Naef R, Seebach D. Liebigs Ann Chem. 1983:1930–1936. [Google Scholar]
- 101.Seebach D, Boes M, Naef R, Schweizer WB. J Am Chem Soc. 1983;105:5390–5398. [Google Scholar]
- 102.Seebach D, Sting AR, Hoffmann M. Angew Chem, Int Ed. 1997;35:2708–2748. [Google Scholar]
- 103.Berkowitz DB, Chisowa E, McFadden JM. Tetrahedron. 2001;57:6329–6343. doi: 10.1016/S0040-4020(01)00499-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Berkowitz DB, McFadden JM, Sloss MK. J Org Chem. 2000;65:2907–2918. doi: 10.1021/jo9918091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Comins DL, Salvador JM. Journal of Organic Chemistry. 1993;58:4656–4661. [Google Scholar]
- 106.Berkowitz DB, Shen W, Maiti G. Tetrahedron: Asymmetry. 2004;15:2845–2851. doi: 10.1016/j.tetasy.2004.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berkowitz DB, Maiti G. Org Lett. 2004;6:2661–2664. doi: 10.1021/ol049159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Berkowitz DB, Bose M, Choi S. Angew Chem Int Ed. 2002;41:1603–1607. doi: 10.1002/1521-3773(20020503)41:9<1603::aid-anie1603>3.0.co;2-d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Berkowitz DB, Wu B, Li H. Org Lett. 2006;8:971–974. doi: 10.1021/ol060019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Berkowitz DB, Pedersen ML. J Org Chem. 1995;60:5368–9. doi: 10.1021/jo00122a002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Berkowitz DB, Pedersen ML, Jahng WJ. Tetrahedron Lett. 1996;37:4309–4312. doi: 10.1016/0040-4039(96)00832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Berkowitz DB, De la Salud-Bea R, Jahng WJ. Org Lett. 2004;6:1821–1824. doi: 10.1021/ol049422u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McCarthy JR, Matthews DP, Stemerick DM, Huber EW, Bey P, Lippert BJ, Snyder RD, Sunkara PS. J Am Chem Soc. 1991;113:7439–7440. [Google Scholar]
- 114.McCarthy JR, Matthews DP, Edwards ML, Stemerick DM, Jarvi ET. Tetrahedron Lett. 1990;31:5449–5452. [Google Scholar]
- 115.Berkowitz DB, Jahng WJ, Pedersen ML. Bioorg Med Chem Lett. 1996;6:2151–2156. doi: 10.1016/0960-894X(96)00366-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Leon AJ, Silverman RB. Bioorg Med Chem Lett. 1996;6:1319–1322. [Google Scholar]
- 117.Silverman RB, Bichler KA, Leon AJ. J Am Chem Soc. 1996;118:1253–1261. [Google Scholar]
- 118.Sukhareva BS, Dunathan HC, Braunshtein AE. FEBS Letters. 1971;15:241–4. doi: 10.1016/0014-5793(71)80321-6. [DOI] [PubMed] [Google Scholar]
- 119.O’Leary MH, Baughn RL. J Biol Chem. 1977;252:7168–7173. [PubMed] [Google Scholar]
- 120.O’Leary MH, Herreid RM. Biochemistry. 1978;17:1010–1014. doi: 10.1021/bi00599a011. [DOI] [PubMed] [Google Scholar]
- 121.Akhtar M, Stevenson DE, Gani D. Biochemistry. 1990;29:7648–7660. doi: 10.1021/bi00485a014. [DOI] [PubMed] [Google Scholar]
- 122.Karukurichi KR, De la Salud-Bea R, Jahng WJ, Berkowitz DB. J Am Chem Soc. 2007;129:258–259. doi: 10.1021/ja067240k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qu N, Ignatenko NA, Yamauchi P, Stringer DE, Levenson C, Shannon P, Perrin S, Gerner EW. Biochemical Journal. 2003;375:465–470. doi: 10.1042/BJ20030382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pan Y, Qiu J, Silverman RB. J Med Chem. 2003;46:5292–5293. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]