

Abstract
Reported herein is a new approach to prepare biaryl derivatives via a tandem Pd catalyzed boron-Heck and Suzuki reaction. This one-pot tandem process avoided purification or addition of extra catalyst between steps. The resulting biaryl compounds can be prepared with substrate variability in good to moderate yields.
The biaryl moiety has been prevalent as a key structure of the functional molecules in raw materials for LCD (liquid crystal display) and OLED (organic light emitting diode) scaffolds, as well as biologically active compounds.1 Recently, the palladium catalyzed reactions known as Heck-Mizoroki,2 Suzuki-Miyaura,3 Negishi,4 and Stille5 reactions have been employed as powerful and versatile methods for biaryl synthesis. These reactions have offered increased utility because of their mild conditions compared to traditional biaryl synthetic methods such as the Scholl,6 Gomberg-Bachmann,7 or Ullmann-type reactions.8
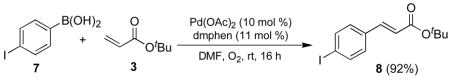
Recently, our group reported general methods for the intermolecular oxidative palladium catalyzed boron-Heck cross coupling between aryl- or alkenylboronic acids and olefins under very mild conditions.9 In particular, we examined several examples of competitive one-pot intermolecular Suzuki, Heck, and our boron-Heck cross coupling reactions. As shown in Scheme 1, we found that under oxidative conditions, the boron-Heck compound (5) was formed exclusively. These results implied that under these coupling conditions, Heck and Suzuki coupling reactions were suppressed due to the absence of base, relatively low temperature, and presence of oxygen atmosphere. For the same competition reaction under nitrogen with sodium carbonate at 100 °C, there was almost completely selective formation of the Heck product (4), and no trace of the boron-Heck coupling product. This suggested that we could simply use different atmospheres to sequentially employ different palladium catalyzed cross-coupling reactions in a three component coupling tandem reaction. Therefore, we examined the oxidative palladium catalyzed cross-coupling reactions of 4-iodophenylboronic acid with olefins such as tert-butyl acrylate (Eq. 1) and 2-cyclohexen-1-one (Eq. 2) because the resulting products would become suitable substrates for ensuing Suzuki couplings.
Scheme 1.

 |
(1) |
 |
(2) |
As expected, boron Heck-type products were exclusively generated, and no other coupling products were obtained. On the basis of these results, herein we report the development of a one-pot chemo- and regioselective method for the preparation of biaryl compounds via a tandem oxidative palladium catalyzed boron-Heck and Suzuki coupling approach.
Initial studies focused on optimization of the boron-Heck type coupling of 4-iodophenylboronic acid (7) and olefins such as tert-butyl acrylate (acyclic example) and 2-cyclohexen-1-one (cyclic example). Table 1 shows the results from the screening of various temperatures, mole ratios, and catalyst/ligand loading amounts for this boron Heck–type coupling using tert-butyl acrylate. The desired product (8) was formed exclusively in a high yield at room temperature using 10 mol % of Pd(II) catalyst and two equivalents of tert-butyl acrylate (entry 3). At 50 °C, the yield of product 8 was diminished due to generation of 4-iodophenol as a side product (entry 4), despite acceleration of the reaction rate.10
Table 1.
Conditions for the selective oxidative boron-Heck reaction with acyclic alkene, tert-butyl acrylate 3a
 | ||||||
|---|---|---|---|---|---|---|
| entry | mole ratio
|
Pd(OAc)2 | T (°C) | time (h) | yield (%)d | |
| 7 | 3 | |||||
| 1b | 1 | 1.1 | 5 mol % | 23 | 16 | 81 |
| 2c | 1 | 1.1 | 10 mol % | 23 | 16 | 85 |
| 3c | 1 | 2 | 10 mol % | 23 | 16 | 92 |
| 4c | 1 | 2 | 10 mol % | 50 | 6 | 87 |
All reactions were carried out with 7 (1.0 mmol) and various mole ratio of tert-butyl acrylate in DMF (2.5 mL).
dmphen [2,9-dimethyl phenanthroline](5.5 mol %).
dmphen (11.0 mol %).
Isolated yields.
Next, experiments directed toward the development of a tandem oxidative boron-Heck/Suzuki reaction were performed using 4-iodophenylboronic acid, tert-butyl acrylate, and phenylboronic acid as coupling partners. As shown in Table 2, the oxidative boron-Heck reaction intermediate product 8 was prepared at room temperature, and then further reacted with phenylboronic acid in a Suzuki coupling in the presence of NaOH under a N2 atmosphere at 50 °C. A tandem oxidative boron-Heck/Suzuki reaction using two equivalents of tert-butyl acrylate as an alkene formed the biaryl product (11) and a disubstituted acryl product (12) in a 2:1 ratio (entry 1), because the Heck reaction took place in the second-step reaction. Therefore, we changed the ratio of tert-butyl acrylate to a molar equivalent, and obtained more of the oxidative boron-Heck/Suzuki reaction product (entry 2). However, in the coupling reaction of disubstituted olefins such as ethyl crotonate and β-methylstyrene (entries 3 and 4), only tandem oxidative boron-Heck/Suzuki reaction products (13 and 14) were obtained in good yields, with excellent (E)-stereoselectivity.
Table 2.
One-pot tandem reactions; oxidative cross-coupling of 7 with various olefinsa and Suzuki reaction with phenylboronic acid b
 | |||
|---|---|---|---|
| entry | olefin | mole equiv. | productc |
| 1 |

|
2 |
|
| 2 |

|
1 |
|
| 3 |

|
2 |

|
| 4 |

|
2 |

|
All reactions were carried out with 7 (1.0 mmol), olefins (2.0 mmol), Pd(OAc)2 (10 mol %), and dmphen (11.0 mol %) in DMF (2.5 mL).
Phenylboronic acid (1.2 mmol) and NaOH (2.0 mmol).
Isolated yields.
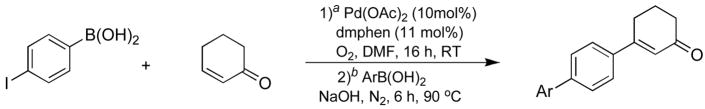
Employing 2-cyclohexen-1-one 9 as an olefin, a similar screening process for the boron-Heck reaction was carried out with 4-iodophenylboronic acid, the results of which are summarized in Table 3. At room temperature, an increase in the amount of Pd(II) complex provided a rise in yield (entries 1 and 2). Increasing the molar ratio of 2-cyclohexen-1-one to 4-iodophenylboronic acid up to 2:1 increased the yield slightly (entry 3). Elevation of the temperature to 50 °C further increased the yield, while also accelerating the reaction rate (entry 4). However, lowering the ratio of 2-cyclohexen-1-one to 4-iodophenylboronic acid at this higher temperature resulted in significant decrease in yield (entry 5). Subsequently, we determined that optimal conditions for the cross-coupling reaction were to use two equivalents of 2-cylcohexen-1-one at 50 °C in the presence of 10 mol % of the Pd(II) catalyst.
Table 3.
Conditions for the selective oxidative boron-Heck reaction with 2-cyclohexen-1-one 9 a
 | ||||||
|---|---|---|---|---|---|---|
| entry | mole ratio
|
Pd/ligand | T (°C) | time (h) | yield (%)d | |
| 7 | 9 | |||||
| 1b | 1 | 1.2 | 5 mol % | 23 | 16 | 61 |
| 2c | 1 | 1.2 | 10 mol % | 23 | 16 | 78 |
| 3c | 1 | 2 | 10 mol % | 23 | 16 | 80 |
| 4c | 1 | 2 | 10 mol % | 50 | 6 | 85 |
| 5c | 1 | 1.2 | 10 mol % | 50 | 6 | 65 |
All reactions were carried out with 7 (1.0 mmol) and various mole ratio of 2-cyclohexen-1-one in DMF (2.5 mL).
dmphen [2,9-dimethyl phenanthroline] (5.5 mol %).
dmphen [2,9-dimethyl phenanthroline] (11.0 mol %).
NMR yields.
Next, we examined the Suzuki reaction of intermediate compound (10) with phenylboronic acid. Thus, intermediate compound 10 was generated from 7, and to the crude reaction mixture containing 10, phenylboronic acid was added and the reaction was stirred at 50 °C under N2 in the presence of NaOH. After six hours, the cross-coupling reaction proceeded to give a 45% yield of the desired biaryl product (15). Although this yield was in fact the overall yield for the two step one-pot tandem reaction, this conversion was not satisfactory compared to the Suzuki reaction of intermediate 8 under similar conditions. Subsequently, we sought optimal conditions by screening various bases and temperatures to increase the Suzuki, and consequently, the overall reaction conversion. In this reaction, the use of carbonate and phosphate bases such as Na2CO3, K2CO3 and K3PO4 at 50 °C provided the biaryl coupling product 15 in low yields. Also, the use of an organic base such as Et3N reduced the yield.11 However, at the elevated temperature of 90 °C in the presence of NaOH, the reaction proceeded well to afford the coupling compound in an increased yield of 67% for the two step tandem reaction (Scheme 2).
Scheme 2.

By utilizing these optimized Suzuki reaction conditions, we examined a tandem palladium catalyzed oxidative boron-Heck and Suzuki reaction with 4-iodophenylboronic acid, 2-cyclohexen-1-one and various arylboronic acids as summarized in Table 4. Reactions with electron donating substituted arylboronic acids such as 4-methoxyphenyl boronic acid, 3,4-dimethoxyphenylboronic acid, and 4-N,N-dimethylaminophenylboronic acid took place smoothly to provide the desired compounds 16, 17 and 18 in 60%, 51%, and 59% yields, respectively (entries 2–4). Also, the reactions with 4-nitrophenylboronic acid, 4-acetylphenylboronic acid, and 4-cyanophenylboronic acid possessing highly electron withdrawing groups afforded the biaryl products 19, 20, and 21 in 77%, 78%, and 63% yields, respectively (entries 5–7). The coupling reaction using 2,6-dimethyl phenylboronic acid gave a 30% yield of the desired product 22 due to steric hindrance (entry 8). In addition, we examined the Suzuki coupling reaction of the alkenylboronic acid, trans-styrenyl boronic acid, which proceeded to afford the corresponding cross-coupling compound 23 in 48% yield. Overall, these tandem reactions offered an efficient method for the preparation of biaryl compounds in a one-pot process.
Table 4.
One-pot tandem reactions; oxidative cross-coupling of 7 with 2-cyclohexen-1-one and Suzuki reaction with various aryl boronic acids.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | arylboronic acid | product | yield (%)c | entry | arylboronic acid | product | yield (%)c |
| 1 |

|

|
67 | 6 |

|

|
78 |
| 2 |

|

|
60 | 7 |

|

|
63 |
| 3 |

|

|
51 | 8 |

|

|
30 |
| 4 |

|

|
59 | 9 |

|

|
48 |
| 5 |

|

|
77 | ||||
All reactions were carried out with 4-Iodophenylboronic acid (1.0 mmol), 2-cyclohexen-1-one (2.0 mmol), Pd(OAc)2 (10 mol%), and dmphen (11 mol%)
in DMF (2.5 mL).
Arylboronic acid (1.2 mmol) and NaOH (2.0 mmol).
Isolated yields.
To investigate the scope and limitation of the final coupling partner, we sought to use different halo-arylboronic acids as the centerpiece component for the tandem reaction. Several different halo-arylboronic acids were used with 2-cyclohexen-1-one and phenylboronic acid as coupling partners, using the established optimal reaction conditions. The results of these reactions are summarized in Table 5. A tandem reaction of 4-iodophenylboronic acid, 2-cyclohexen-1-one, and phenylboronic acid was converted efficiently to biaryl product 15 in 67% yield (Table 4, entry 1) while chloro- and bromo-phenylboronic acid, afforded the desired product 15 % and 45 % yields, respectively (entries 1 and 2). This is because aryl bromide and iodide easily react with phenylboronic acid but chloride does not participate in coupling readily. Furthermore, 3-iodophenylboronic acid reacted with 2-cyclohexen-1-one, and then phenylboronic acid to give a 52% yield of 24 (entry 3). In the case of di-substituted arylboronic acids including 3-chloro-4-methoxyphenyl boronic acid and 5-chloro-2-methoxyphenyl-boronic acid, tandem reactions with 2-cyclohexen-1-one and phenylboronic acid afforded regioselective desired products 25 and 26 in 32% and 36% yields, respectively (entries 4 and 5).
Table 5.
The effect of various haloaryl boronic acids with 2-cyclohexen-1-one 9
 | |||
|---|---|---|---|
| entry | arylboronic acid | product | yield (%)c |
| 1 |

|

|
<5% |
| 2 |

|

|
45 |
| 3 |

|

|
52 |
| 4 |

|

|
32 |
| 5 |

|

|
36 |
All reactions were carried out with Arylboronic acid (1.0 mmol), 2-cyclohexen-1-one (2.0 mmol), and Pd(OAc)2 (10 mol %), and dmphen (11 mol %) in DMF (2.5 mL).
Phenylboronic acid (1.2 mmol) and NaOH (2.0 mmol).
Isolated yields.
In conclusion, a tandem oxidative boron-Heck/Suzuki coupling reaction was developed for the preparation of biaryls in good to moderate yields. The reaction can be performed with variation at all three coupling partners. In addition, the biaryls formed in this study were obtained without the use of long laborious purification or the further addition of more palladium catalyst between coupling reactions. Furthermore, the use of different atmospheric conditions, and consequently, different mechanisms, allows for the longevity of palladium catalysts and opens up possibilities for other tandem oxidative palladium reactions.
Typical Procedure for the tandem reaction: To an oven dried 10 mL round bottom flask equipped with a stir bar was charged 0.10 mmol Pd(OAc)2, and 0.11 mmol 2,9-dimethyl phenanthroline in 2.5 mL DMF. The reaction was stirred at room temperature for 30 minutes, at which time 2.0 mmol alkene was added, followed by 1.0 mmol halo aryl boronic acid. The reaction flask was then fitted with an oxygen balloon and stirred at room temperature for 16 hours, or at 50 °C for 6 hours. Following confirmation of consumption of starting material by TLC, the oxygen balloon was removed and N2 was bubbled through the solution for at least 1 minute. 1.2 mmol aryl boronic acid and 2.0 mmol NaOH were then added to the solution, and the solution was then stirred for up to 6 hours under N2 at 90 °C. The reaction mixture was then dissolved in 50 mL ethyl acetate and washed twice with 50 mL water, once with 50 mL brine, and dried over anhydrous sodium sulfate and concentrated in vacuo. The crude reaction mixture was then subjected to column chromatography using a 10:1 hexane/ethyl acetate eluent system on silica gel (230–400 mesh).
Supplementary Material
Acknowledgments
We acknowledge generous financial support from the National Institutes of General Medical Sciences of the National Institutes of Health (RO1 GM 71495).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Poetsch E. Kontakte. 1988;2:15. [Google Scholar]; (b) Bemis GW, Murcko MA. J Med Chem. 1996;39:2887. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]; (c) Pu L. Chem Rev. 1998;98:2405. doi: 10.1021/cr970463w. [DOI] [PubMed] [Google Scholar]; (d) Hajduk PJ, Bures M, Praestgaard J, Fesik SW. J Med Chem. 2000;43:3443. doi: 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]; (e) Horton DA, Bourne GT, Smythe ML. Chem Rev. 2003;103:893. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; (f) Croom KF, Keating GM. Am J Cardiovasc Drugs. 2004;4:395. doi: 10.2165/00129784-200404060-00008. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hernandez S, SanMartin R, Tellitu I, Dominguez E. Org Lett. 2003;5:1095. doi: 10.1021/ol034148+. [DOI] [PubMed] [Google Scholar]; (b) Alberico D, Scott M, Lautens M. Chem Rev. 2007;107:174. doi: 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]
- 3.(a) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]; (b) Suzuki A. J Organomet Chem. 1999;576:147. [Google Scholar]; (c) Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633. [Google Scholar]; (d) Persichini PJ. Curr Org Chem. 2003;7:1725. [Google Scholar]; (e) Bellina F, Carpita A, Rossi R. Synthesis. 2004:2419. [Google Scholar]
- 4.(a) Negishi E, King AO, Okukado N. J Org Chem. 1977;42:1821. [Google Scholar]; (b) Rottaelander M, Palmer N, Knochel P. Synlett. 1996:573. [Google Scholar]; (c) Tilley JW, Clader JW, Zawoiski S, Wirkus M, LeMahieu RA, O’Donnell M, Crowley H, Welton AF. J Med Chem. 1989;32:1814. doi: 10.1021/jm00128a025. [DOI] [PubMed] [Google Scholar]; (d) Hoye TR, Chen M. J Org Chem. 1996;61:7940. doi: 10.1021/jo960882d. [DOI] [PubMed] [Google Scholar]
- 5.(a) Kosugi M, Hagiwara I, Migita T. Chem Lett. 1983:839. [Google Scholar]; (b) Stille JK. Angew Chem Int Ed Engl. 1986;98:504. [Google Scholar]; (c) Stanforth SP. Tetrahedron. 1998;54:263. [Google Scholar]
- 6.(a) Kovacic P, Jones MB. Chem Rev. 1987;87:357. [Google Scholar]; (b) March J. Advanced Organic Chemistry. 4. Wiley; New York: 1992. p. 539. [Google Scholar]
- 7.(a) Gomberg M, Bachmann WE. J Am Chem Soc. 1924;46:2339. [Google Scholar]; (b) March J. Advanced Organic Chemistry. 4. Wiley; New York: 1992. pp. 715–171. [Google Scholar]
- 8.(a) Ullman F, Bielecki J. Chem Ber. 1901;34:2174. [Google Scholar]; (b) Hassan J, Sevignon M, Gozzi C, Shulz E, Lemaire M. Chem Rev. 2002;102:1359. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- 9.(a) Jung YC, Mishra RK, Yoon CH, Jung KW. Org Lett. 2003;5:2231. doi: 10.1021/ol034458s. [DOI] [PubMed] [Google Scholar]; (b) Yoo KS, Yoon CH, Mishra RK, Jung YC, Yi SW, Jung KW. J Am Chem Soc. 2006;128:16384. doi: 10.1021/ja063710z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreno-Manas M, Perez M, Pleixats R. J Org Chem. 1996;61:2346. doi: 10.1021/jo9514331. [DOI] [PubMed] [Google Scholar]
- 11.Conversion yields of the tandem reaction using various bases at 50 °C for 6 hours under N2 for the Suzuki coupling step; Na2CO3 (29%), K2CO3 (24%), K3PO4 (37%), and triethylamine (25%).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.