

Abstract
Cleft lip with or without cleft palate is the most common facial birth defect and it is caused by a complex interaction between genetic and environmental factors. The purpose of this review is to provide an overview of the spectrum of the genetic causes for cleft lip and cleft palate using both syndromic and nonsyndromic forms of clefting as examples. Although the gene identification process for orofacial clefting in humans is in the early stages, the pace is rapidly accelerating. Recently, several genes have been identified that have a combined role in up to 20% of all clefts. While this is a significant step forward, it is apparent that additional cleft causing genes have yet to be identified. Ongoing human genome-wide linkage studies have identified regions in the genome that likely contain genes that when mutated cause orofacial clefting, including a major gene on chromosome 9 that is positive in multiple racial groups. Currently, efforts are focused to identify which genes are mutated in these regions. In addition, parallel studies are also evaluating genes involved in environmental pathways. Furthermore, statistical geneticists are developing new methods to characterize both gene-gene and gene-environment interactions to build better models for pathogenesis of this common birth defect. The ultimate goal of these studies is to provide knowledge for more accurate risk counseling and the development of preventive therapies.
Introduction
In humans, orofacial clefts are common congenital anomalies with a prevalence of 1–2/1000 live births. They can be separated into two different phenotypes: (1) cleft lip with or without cleft palate (CL/P); and (2) cleft palate only (CPO). Orofacial clefts can be further classified as nonsyndromic (isolated) or syndromic based upon the presence of other anomalies. Approximately 30% of CL/P and 50% of CPO patients have one of over 400 described syndromes.1–4 The focus of this review is primarily nonsyndromic cleft lip (CL/P) since this trait has been studied the most in humans, while the etiology of CPO has been studied more in animal models.
It is generally accepted that CL/P and CPO are genetically distinct phenotypes in terms of their inheritance patterns. CPO is less common, with a prevalence of approximately 1/1500–2000 births in Caucasians, while CL/P is more common, 1–2/1000 births. The prevalence of CPO does not vary in different racial backgrounds, while the prevalence of CL/P varies considerably, with Asian and American Indians having the highest rate and Africans the lowest.5,6 There are also gender ratio differences with more males having CL/P and more females having CPO. Finally, families with one type of clefting segregating in the family do not have the other cleft type occur at a rate higher than the population prevalence.7 It will be interesting from a genetic perspective to determine the basis for the different inheritance patterns in CL/P and CPO. For instance, it will be important to determine whether the difference is due to locus or allelic heterogeneity, meaning that the differences are due to different genes (loci) or different mutations (alleles) within the same gene. Given that both primary and secondary palatogenesis involve fusion between facial processes, it is expected that some genes may be involved in both disorders.
Simple versus Complex Genetic Traits
It is important to recognize that human traits (some of which are diseases or developmental anomalies) can be caused by a variety of genetic mechanisms and phenomena, which can be reflected by the inheritance patterns within families. These mechanisms include whether the disorder is inherited in a Mendelian pattern such as autosomal dominant, autosomal recessive, or X-linked (Figure 1–Figure 3).
Figure 1.
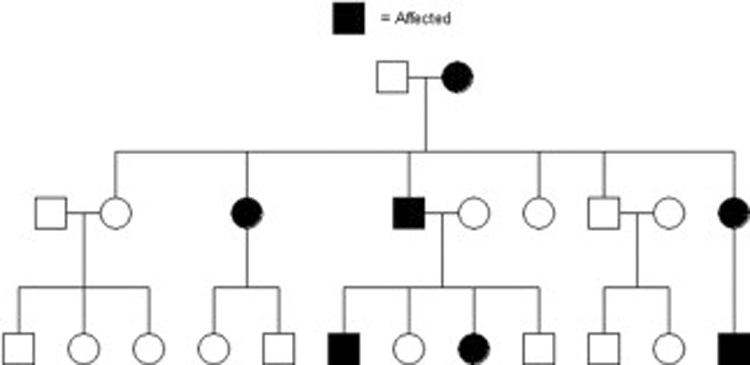
Pedigree showing a dominant pattern of inheritance. Dominance means only 1 of the 2 copies of a gene needs to be mutated to cause the disease or trait. Hence at least 1 parent is affected and 50% of descendants from affected parents are also affected. For mapping purposes, in a trait (disease) with high penetrance, every normal person can be assumed to not carry the trait (disease) gene. Thus this pedigree is much more powerful for mapping than a disease with incomplete penetrance as shown in figure 5. Shaded individuals are affected with the trait (disease) of interest.
Figure 3.
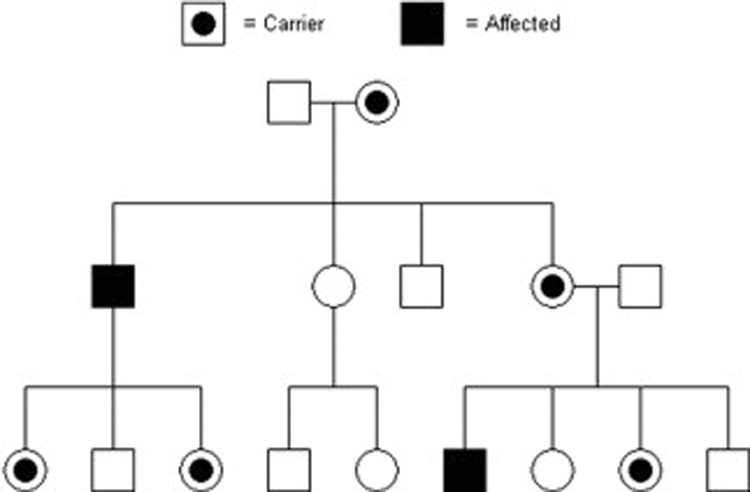
Pedigree showing an X-Linked pattern of inheritance. Male descendants from carrier females are usually affected since they only have one X chromosome. Females may be variably affected depending upon the X-inactivation pattern. Shaded individuals are affected and individuals with dots are carriers of the specific trait (disease).
The penetrance of a mutation, defined as the frequency of the disease trait in individuals carrying the disease mutation, will also affect the inheritance pattern. Expressivity, which describes the severity or variation of the trait, can also vary considerably among affected individuals. Expressivity can affect the perceived inheritance pattern if the severity is so mild or below a certain stipulated threshold such that the person is considered normal when in fact they carry the disease mutation, and could be identified as being affected with careful examination or highly sensitive diagnostic techniques.
Finally, the number of genes underlying a specific trait (disease) can also vary. For example, some traits (diseases) are all caused by the same gene, meaning everyone with the trait (disease) has a mutation in the same gene. This situation is called genetic homogeneity. Interestingly, there are examples in which mutations in any one of several genes will result in the same trait (disease). This is termed locus heterogeneity. Finally, some traits (diseases) only become apparent when multiple genes are mutated in the same individual. Thus traits (diseases) can be caused by simple to complex mechanisms (Figure 4).8
Figure 4.
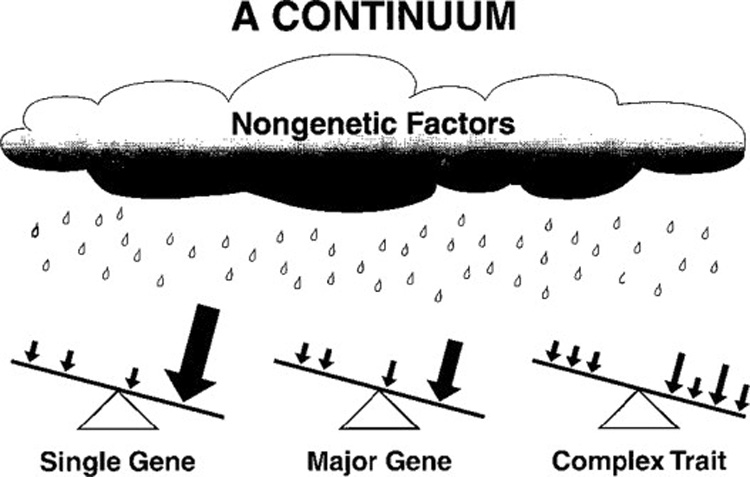
The spectrum of underlying genetic causes for diseases, ranging from single gene to multiple genes. All diseases and traits are also under influence of modifier genes (small arrows) and nongenetic or environmental factors. These influences can be protective or negative and ultimately the balance of all factors determines the outcome. Printed with permission from.8
A simple genetic trait implies that it is inherited in a Mendelian pattern, and that all individuals with the trait have a mutation in the same gene, meaning a major gene for the trait exists. A complex genetic trait is one that does not conform to simple Mendelian inheritance patterns. It is presumably caused by the combination of multiple mutations in different genes interacting with environmental factors.
Inheritance patterns, penetrance, expressivity and genetic homogeneity can significantly impact the ability to identify causative genes. The simplest scenario is when all affected individuals have mutations in the same gene, the penetrance is high and the expressivity is consistent such that one can assume that all affected individuals within a family share the same DNA mutation and DNA markers near the mutation.
The approach in this situation involves scanning of the human genome to look for the shared region between affected members within a family. When this is found, it is said that linkage exists for a specific marker or region with the disease. This can be readily accomplished by using approximately 400 DNA markers. The sharing is statistically evaluated under the assumption that each child has a 50:50 chance of inheriting a specific chromosome carrying the mutated or normal copy of the gene since the genome consists of pairs of chromosomes, and only one is transferred from a given parent to an offspring.
The statistical output of these genetic tests is usually the LOD score, which is the log10 of the odds that a trait (disease) and DNA marker are linked versus the odds that they are not linked, assuming a 50:50 chance for each individual. LOD scores can be positive or negative, meaning there is evidence for or against linkage. LOD scores greater than 3.0 are considered significant evidence for linkage, and less than −2.0 excludes linkage (for more on linkage analysis see “Investigation of Genetic Factors Affecting Complex Traits Using External Apical Root Resorption as a Model” by Abass and Hartsfield in this volume).
Assuming every affected family has mutations in the same trait (disease) gene, the LOD scores can be summed across all families. This will both increase the power to detect linkage and narrow the region of linkage. The ability to sum LOD scores also aids studies that involve families too small to independently yield significant results. Thus it is relatively easy to identify the disease gene location region for simple traits. The next step is to identify what genes map (are located) within the linked region. This is easily accomplished by utilizing a variety of genetic databases and maps that are available from the successful sequencing of the human genome. The genes can then be sequenced to identify possible causal mutations, with success being obtained upon finding mutations only in affected individuals.
Complex traits are much more difficult to map since not everyone has contributory mutations in the same gene. Thus families may map to different trait (disease) genes, such that combining the LOD scores will result in negative results for one family canceling out the positive results for another family. Furthermore, the decreased penetrance commonly observed in complex traits means some people without the trait (disease) carry mutations, thus breaking the linkage between a given gene or marker with the disease trait. In the early stages of gene mapping, it was commonly believed that a researcher must know the inheritance pattern, and also have some justification to assume trait (disease) homogeneity; otherwise the study was doomed to fail.
These beliefs, limited technology and statistical methods, and the relative ease for mapping simple genetic traits (diseases) resulted in early success for simple traits and a delayed start for complex traits. However with the development of high throughput technology and improved statistical methods that take into account both genetic heterogeneity and decreased penetrance, successful mapping of complex traits occurred in the mid-1990’s for type 1 diabetes9,10 and multiple sclerosis.11–14
With this as background, we will present examples of CL/P diseases that cover the spectrum of simple to complex genetic traits. Specifically, we will review the data indicating nonsyndromic CL/P is a genetically complex trait involving genetic heterogeneity, low penetrance and the influence of various environmental factors.
Syndromic forms of CL/P
Syndromic forms of CL/P often have simple Mendelian inheritance patterns and are thus more suitable for conventional genetic mapping strategies.15
Van der Woude Syndrome
Van der Woude syndrome (VWS) is an autosomal dominant (Figure 1) form of orofacial clefting with an estimated prevalence of 1/34,000 live births.16 Autosomal indicates that the observed inheritance pattern excludes the sex chromosomes. VWS has a variety of features that distinguish it from nonsyndromic CL/P, including the presence of lower lip pits (Figure 5), hypodontia, and either CL/P or CPO. Furthermore, the penetrance is very high, approximately 97%. The disease gene was localized by linkage mapping to a large region on the long arm of chromosome 1, 1q32–q41.17 Subsequent genetic studies identified several patients with deletions in the area that greatly narrowed the critical region to 350 kilo-bases (Kb = 1 thousand base pairs), containing over 20 known genes.18 An elegant strategy was implemented in which monozygotic twins, one with VWS and the other normal, were sequenced and a mutation discovered in the interferon regulatory factor 6 (IRF6) gene.19
Figure 5.

Patient with Van der Woude syndrome with a repaired right cleft lip and two abnormal mounds on the vermilion of the lower lip that are indicative of lower lip pits.
IRF6 is a transcription factor that contains DNA binding and protein interaction domains. IRF6 is expressed in the medial edge epithelia of the secondary palatal shelves. Thus it appears to regulate the expression of other genes during palatogenesis. Interestingly, VWS is an example of an orofacial syndrome in which CL/P and CPO can occur in the same family, suggesting that it is likely involved in the fusion process that occurs in both primary and secondary palatogenesis.
CL/P-Ectodermal Dysplasia Syndrome
CL/P ectodermal dysplasia (CLPED1) syndrome is characterized by cleft lip, cleft palate, partial syndactyly of the fingers and toes, dental anomalies and sparse hair.20 It is a rare autosomal recessive trait (Figure 2). However there is a very high prevalence on Margarita Island, suggesting a founder effect in the small and relatively isolated population. The disease gene was mapped to a 1–2 mega-base (Mb = 1 million base pairs) region on chromosome 11,21 and subsequently mutations were identified in the Poliovirus Receptor-Like 1 (PVRL1) gene.22 Again, like IRF6, the PVRL1 gene name is misleading, suggesting an infectious function rather than an important facial developmental gene.
Figure 2.

Pedigree showing a recessive pattern of inheritance. Recessive means that both copies of a gene need to be mutated to cause the disease or trait. In this situation both parents are not affected, but descendants of parents who are both carriers will be affected, unaffected (but trait or disease gene carriers like the parents), or unaffected with normal genes in proportions of 25%:50%:25% on average. These are also the respective likelihoods of a child being one of the three outcomes at conception. Once it is known that a child of carrier parents is not affected, then the chance of the child being a carrier is 2 out of 3. Shaded individuals are affected and individuals with dots are carriers of the specific trait (disease).
PVRL1 encodes a cell-cell adhesion molecule that is expressed in the epithelia of the palatal shelves, nose and skin, as well as the dental ectoderm. Thus it appears PVRL1 is a molecule important for cell-cell contact in these tissues. Interestingly, PVRL1 is used by various viruses, including the herpes simplex viruses, as a method to gain entry into cells.23
X-Linked Cleft Palate and Ankyloglossia
Cleft palate occurring with ankyloglossia (CPX) has been reported segregating in an X-linked recessive pattern (Figure 5) in large families in Iceland and British Columbia, Canada. CPX was the first orofacial cleft syndrome mapped, with linkage being identified to a large region on the long arm of chromosome X.24 Recently, the causal gene was identified as TBX22,25 which is expressed in the palatal shelves and tongue during development.26.27 TBX22 functions by binding to specific DNA elements to regulate the expression of target genes. X-linked diseases are interesting since males have one X chromosome and females two X chromosomes. If a male inherits a mutated TBX22 it is highly likely that he will have the disease since this is the only copy of the TBX22 gene.
In females, it is important to compensate the dose effect of having two X chromosomes. This is accomplished by X-inactivation (Lyonization) in which one X chromosome is inactivated such that most genes are expressed only from the active X chromosome. This is normally a random 50:50 process occurring early in development in each cell, with subsequent daughter cells having the same X inactivated. There is some normal range of X-inactivation if the total number of cells are not split 50:50 in regard to which X is inactivated in each cell. Thus it is possible for a female to inherit a mutated X-linked disease gene and either not have the disease or have a milder form of the disease depending upon the ratio and tissue distribution of X-inactivation. For example, if the X chromosome containing the disease gene is more often inactivated or inactivated in the affected tissues, the female will likely not have the disease, although each son of hers has a 50:50 chance of being affected. For CPX it is hypothesized that X-inactivation would explain the reduced penetrance or milder phenotype in females,28 though this has not been formally tested.
In summary, there has been significant success in identifying etiologic genes for syndromic forms of orofacial clefting, with approximately 15–20% cloned to date.29 Identifying syndromic disease genes will provide insight about the molecular processes involved in facial development and other affected tissues. Furthermore, these genes can be analyzed to determine if different mutations are associated with the more common nonsyndromic form of CL/P.
Genetics of Nonsyndromic CL/P
Nonsyndromic CL/P is an example of a genetically complex trait.30 The majority of affected patients have no positive family history and the evaluation of inheritance patterns in the familial cases has not revealed a simple Mendelian mode of inheritance (Figure 6). It is also clear that there is reduced penetrance. However, there is solid evidence that CL/P is a genetic trait since there is a 40 fold risk for CL/P amongst first degree relatives of an affected individual and there is greater concordance in identical (monozygotic) compared to fraternal (dizygotic) twins. However, the concordance rate in monozygotic twins is only 40–60%, suggesting the influence of environmental factors is also important.
Figure 6.
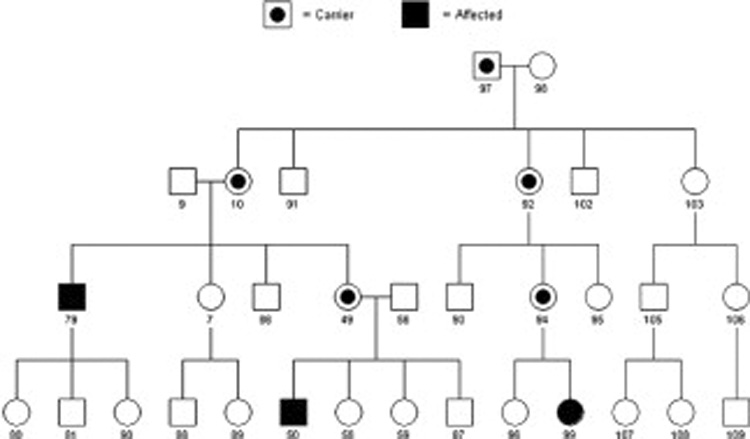
Pedigree showing a complex inheritance pattern. Note that the number of affected descendants in the pedigree does not match expected proportions from any possible Mendelian pattern. One can assume that the linking relatives between affected individuals are disease gene carriers and at least one of the original parents is also a carrier. Because of the reduced penetrance, it is not possible to know for certain that an unaffected person is not a disease gene carrier and hence unaffected people do not add to the mapping power of the pedigree. Therefore this pedigree has significantly reduced power compared to those in figure 1 and figure 2 even though it contains more people. Shaded individuals are affected and individuals with dots are carriers of the specific trait.
Studies have estimated that 3–14 genes interacting multiplicatively may be involved, indicating that CL/P is a heterogeneous disorder,31 making it more difficult to map these genes since only a portion of affected individuals will have a mutation in the same gene and currently there is not any method to identify different genetic subsets a priori. However the impact for a given CL/P gene is estimated to be sufficiently large enough to be mapped using a variety of strategies.32,33 Another limitation is that very large families with CL/P are rare, thus it is necessary to combine the LOD scores across families when using a linkage approach, reducing power and increasing the likelihood for missing a gene.
Human studies have used both association and linkage analyses to evaluate the role of candidate genes in the etiology of CL/P. Candidate genes have been chosen based on expression patterns during facial development, cleft phenotype in transgenic or knockout mouse models, association with syndromic forms of clefting, previous positive findings in humans, role in nutritional or xenobiotic pathways, and cytogenetic location adjacent to chromosomal anomalies associated with orofacial clreft phenotypes. Association mapping is the identification of nonrandom correlations (associations) between alleles at two loci in a population. A simple way to envision this is the higher occurrence of a given allele in cases as compared to controls. Association approaches have more study power especially in the presence of genetic heterogeneity. Using a combination of both linkage and association can also be successful for complex traits.34
Candidate Genes
Initial efforts to identify genes for nonsyndromic CL/P relied on candidate gene approaches.5,35 Genes at 1q32 (IRF6), 2p13 (TGFA), 4p16 (MSX1), 6p23–25, 14q24 (TGFB3), 17q21 (RARA) and 19q13 (BCL3, TGFB1) have the most supporting data (Table 1). Below we will highlight several of these genes.
Table 1.
Selected candidate genes with positive evidence for a role in nonsyndromic cleft lip or palate
| Candidate Region | Candidate Gene | Linkage | Association | Animal Model | Chromosome Anomaly | Cleft Syndrome |
|---|---|---|---|---|---|---|
| 1p36 | MTHFR, SKI, PAX7 | X | X | X | ||
| 1q32 | IRF6 | X | X | X | X | X |
| 2p13 | TGFA | X | X | |||
| 4p16 | MSX1 | X | X | X | X | |
| 6p23–25 | TFAP2A, OFCC1 | X | X | X | X | |
| 14q24 | TGFB3, BMP4, PAX9 | X | X | X | ||
| 17q12–21 | RARA, Clf1 | X | X | X | ||
| 19q13 | BCL3, CLPTM1, PVRL2, TGFB1 | X | X | X |
The VWS Gene, IRF6 is associated with Nonsyndromic CL/P
As mentioned previously, mutations in IRF6 cause VWS. Since VWS has a very similar presentation to isolated CL/P, IRF6 was evaluated as a candidate gene, and a highly significant association between IRF6 variants and CL/P was identified.36 Estimates suggest that genetic variation in IRF6 contributes to 12% of CL/P and triples the recurrence risk in some families. These results have been replicated in additional populations,37–39 although the specific mutations have not yet been identified. This discovery constitutes one of the most exciting discoveries so far in the field of isolated CL/P.
MSX1
MSX1 is a DNA binding transcription factor that when inactivated in mice results in cleft palate and tooth agenesis.40 This finding greatly aided the identification of a MSX1 mutation in a family with hereditary tooth agenesis that was recruited through an orthodontic clinic.41 Simultaneously, DNA variations in MSX1 were shown to be associated with CL/P.5,42–46 Knowing these findings, a MSX1 mutation was found in a family with tooth agenesis, CL/P and/or CPO.47 Recently, mutations in MSX1 have been identified in 2% of patients with nonsyndromic orofacial clefting.48,49 These findings indicate that MSX1 is involved in both primary and secondary palatogenesis, even though the mouse phenotype only involved the latter. This is supported by the strong mesenchymal expression in the nasal and maxillary processes during primary palatogenesis (Figure 7).
Figure 7.

Expression of the MSX1 messenger RNA in the medial nasal (MNP), lateral nasal (LNP), and maxillary (MxP) processes at the time of primary palate fusion on gestational day 11.5 in a mouse embryo.
Transforming Growth Factor Beta 3 (TGFB3)
Studies of TGFB3 further underscore the importance of animal studies as the observation of cleft palate in mice missing TGFB350,51 led to the discovery of associations with CL/P in humans.5 Furthermore, linkage studies have showed positive results for the region containing TGFB3.52 Nevertheless, only 15% of the families were linked to this region, which may in part explain the observed inconsistencies in some previous studies. It is plausible that the nearby BMP4 and PAX9 genes, both associated with orofacial clefting when inactivated in mice53,54 may be the cause of the positive findings, or are also involved.
19q13.1 (BCL3, CLPTM1, PVRL2, TGFB1)
Several studies have found linkage or association with candidate genes on the long arm of chromosome 19.5,55,56 Furthermore, a chromosomal anomaly involving this region was found in a family with CL/P.57 Candidate genes in this area include BCL3, PVRL2, CLPTM1 and TGFB1. Of these, PVRL2 is of interest since it is similar to PVRL1 that causes CLPED1, and carriers for a PVRL1 mutation are thought to have increased risk for nonsyndromic CL/P.58 Hence it is possible that PVRL2 has an analogous role.
Syndromic Orofacial Clefts Provide Important Clues
In addition to the IRF6 and PVRL1 examples of syndromic genes playing a role in nonsyndromic clefting, efforts are underway to determine whether variants in other cleft syndrome genes have similar roles. Mutations and deletions in the FGFR1 gene account for 10% of Kallman syndrome patients, which have orofacial clefting, dental anomalies, hypogonadism and anosmia59 Suggestive association and linkage to CL/P has been found for markers within the FGFR1 gene60 Also, mutations in the CPX gene, TBX22, have been identified in 4% of patients with CPO or ankyloglossia.61 These findings highlight the importance of studying rare forms of diseases since in addition to identifying genes and pathways involved in a disease process, variants in the same gene may be associated with more common forms of the disease.62
Scanning the Genome for Additional CL/P Genes
As mentioned earlier, the application of linkage to complex traits is complicated by genetic heterogeneity and low penetrance. One approach around this is to collect families with multiple affected individuals to look for sharing of genetic markers between only these related affected people. This circumvents the low penetrance issue of not knowing if an unaffected person is a disease gene carrier or not. Yet at the same time this approach has limited power, necessitating the study of hundreds of families. Previously, technology limited this approach to the evaluation of candidate genes. However, with the emergence of high-throughput genotyping technologies and powerful statistical approaches, this approach has been expanded to scan the entire genome to identify additional disease genes.
The first CL/P scan was published in 200063 and subsequently 5 additional scans of varying size have been published. In general the results have been modest with the exception of a LOD score of 3.0 at 17p13.1 in a scan of two large Syrian families64 The most consistent loci are 2p13 (TGFA), 2q35–q37, 3p21–p24, 4q32–q33, 6p23–p25, 9q22–q33, 14q12–q31 and 18q11–q12. These results reflect genetic heterogeneity both within and between populations, limited study power and a likely high false positive rate for loci with low levels of significance.
A meta-analysis of these 6 published and 7 ongoing genome scans revealed significant results for 11 regions at 1q32, 2q32–q35, 3p25, 6q23–q25, 8p21, 8q23, 12p11, 14q21–q24, 17q21, 18q21, and 20q13.52 Also, linkage analysis was performed allowing for genetic heterogeneity, and the summed results from 7 populations revealed significant heterogeneity LOD scores for chromosomal regions 1p12–p13, 6p23, 6q23–q25, 9q22–q33, 14q21–q24 and 15q15. Of these, the 9q22–q33 region was the most striking with a heterogeneity LOD score equal to 6.6, which is the most significant result ever reported for CL/P. This is a new discovery and the region likely contains a major gene for CL/P.
Gene-Environment Interactions
Epidemiologic studies have revealed an increased risk for CL/P with alcohol65 and smoking66 exposure during pregnancy. Furthermore, some studies suggest periconceptional folate or multivitamin supplementation has a protective effect against CL/P.67 However, not all mothers who drink or smoke have children with CL/P, nor do all mothers taking multivitamins have normal children. Thus it is likely that certain genes that interact with these environmental factors and that genetic variation within these genes affect the risk for CL/P. Researchers have tested this hypothesis by looking at both candidate genes for CL/P and genes involved in the environmental pathways.68
Given that neural tube defects can be prevented by folate supplementation and similar evidence exists for CL/P, some investigators have evaluated genes in the folate pathway with some success. However, it is not clear whether one should look at the DNA of the children, mothers or both. Clearly, environmental agents interact with maternal gene products, but it is not always clear if the same is true for fetal gene products, although it is likely in some situations. It is plausible that a fetus may have a low risk for CL/P due to its genes, but that this risk increases due to maternal environmental exposures and her genetic susceptibility to these exposures.
This uncertainty has impacted the results for folate pathway genes.67 For example functional variations in the methylenetetrahydrofolate reductase (MTHFR) and reduced folate carrier (RFC1) genes revealed no association with CL/P in a South American population.69 However, a method looking at the infants genotype and maternal environmental exposures revealed significant gene-environment interactions between CP infants with certain variations in MTHFR and maternal folic acid consumption70 and these results were also found to be true for CL/P.71 Alternatively, an increased risk has been observed for maternal MTHFR variants.72,73
Genetic variation has been identified in a variety of genes involved in the biotransformation of agents found in tobacco smoke including common deletions of the Glutathione S-transferase theta 1-1 (GSTT1) and Glutathione S-transferase Mu 1 (GSTM1) genes. Mothers carrying the GSTT1-null allele and who smoked had a higher although not significant risk of having a child with CL/P.74 A 6 fold risk increase was found for fetuses lacking both GSTT1 and GSTM1 and whose mothers smoked.75 Furthermore, variations in NAT1, another gene involved in detoxification of cigarette smoke, resulted in a two or four fold increased risk for CL/P among infants homozygous for this polymorphism if their mothers did not use multivitamins or smoked respectively.76,77 These studies highlight the importance of identifying such interactions so that individuals with disease susceptibility alleles can modify their risk by ceasing detrimental exposures and improving their nutritional status.
Summary
In general, the gene identification process for CL/P is still in the early stages, especially compared to other common diseases. However, the candidate gene approaches have identified variations that are associated with up to 20% of patients with CL/P. Furthermore, the genome wide linkage scans have identified the location of several genes, including the previously unknown locus on chromosome 9. Current efforts are ongoing to narrow these regions and identify disease causing mutations. Overall, the published findings support the hypothesis that multiple genes are involved in the etiology of CL/P. Future studies will determine how these genes interact with each other and the environment to develop models for improved genetic counseling and public health policies.
Acknowledgements
Our work has been greatly aided by discussions with Mauricio Arcos-Burgos, Sandy Daack-Hirsch, Mike Dixon, David FitzPatrick, Brion Maher, Mary Marazita, Brian Schutte, Alex Vieira and George Wehby. Also we would like to thank families that over the years have participated in our research studies.
Dr. Moreno is supported by a Fogarty International Maternal and Child Health Research and Training Fellowship (1D43 TW-05503) and Dr. Lidral is supported by NIH Grants RO1DE14677, KO2DE015291 and P50DE016215 with additional funding from both the University of Iowa Craniofacial Anomalies Research Center, brilliantly directed by Jeff Murray, and the College of Dentistry. While this work was not directly supported by the American Association of Orthodontists Foundation, Dr. Lidral’s career has been greatly aided by three AAOF Faculty Development Awards.
Glossary
- Allele
A genetic variation for a given marker or locus
- cM
Centimorgan. 1cM is equal to 1% recombination and approximately 1 million base pairs.
- Expressivity
The severity of a disease in individuals carrying the disease mutation. For diseases that involve multiple features, the expressivity can describe both the number of affected features as well as the severity for each feature.
- Gene
Typically, this describes the DNA sequence encoding a protein. But this is also used to describe the DNA sequence that is used to make the message RNA (mRNA). The term may also be expanded to describe the entire gene, meaning the coding sequence, the mRNA sequence and all the regulatory DNA elements that control the expression of the mRNA and protein.
- Genome
The entire genetic material for a given organism. For humans this consists of 22 pairs of chromosomes and two sex chromosomes either XX or XY.
- Genotyping
A laboratory technique used to determine which alleles exist for a marker in a person.
- Haplotype
A combination of alleles for markers as they occur on a chromosome
- Heterozygosity
The presence of two different alleles for a given marker
- Homozygosity
The presence of the same alleles for a given marker
- Locus
A given region of the human genome,;specifically, the location of a gene or particular DNA sequence on a chromosome (plural loci)
- LOD Score
The statistic used to measure genetic linkage
- Marker
A region of the human genome that contains genetic variation within it. The variation is used as a marker to follow inheritance of the genetic material within the locus.
- Penetrance
The frequency that the disease phenotype is expressed when the disease mutation is present.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Statements and opinions expressed in the articles and communications herein are those of the authors and not necessarily those of the Editor or Publisher, and the Editor and Publisher disclaim any responsibility or liability for such material. Neither the Editor(s) nor the Publisher guarantee, warrant, or endorse any product or service advertised in this publication; neither do they guarantee any claim made by the manufacturer of such product or service. Each reader must determine whether to act on the information contained in the publication, and neither the journal nor the Editor(s) shall be liable for any injury due to the publication of erroneous information.
References
- 1.Cohen MM. Syndromes with cleft lip and cleft palate. Cleft Palate J. 1978;15:306–328. [PubMed] [Google Scholar]
- 2.Shprintzen RJ, Siegel VL, Amato J, et al. Anomalies associated with cleft lip, cleft palate, or both. American Journal of Medical Genetics. 1985;20:585–595. doi: 10.1002/ajmg.1320200404. [DOI] [PubMed] [Google Scholar]
- 3.Gorlin RJ. Syndromes of the head and neck. New York: Oxford University Press; 1990. [Google Scholar]
- 4.Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 5.Marazita ML, Mooney MP. Current concepts in the embryology and genetics of cleft lip and cleft palate. Clin Plast Surg. 2004;31:125–140. doi: 10.1016/S0094-1298(03)00138-X. [DOI] [PubMed] [Google Scholar]
- 6.Vanderas AP. Incidence of cleft lip, cleft palate, and cleft lip and palate among races: a review. Cleft Palate J. 1987;24:216–225. [PubMed] [Google Scholar]
- 7.Fraser FC. The genetics of cleft lip and palate: yet another look. In: Pratt Christiansen., editor. Current trends in prenatal craniofacial development. 1980. pp. 357–366. [Google Scholar]
- 8.Beaudet AL. 1998 ASHG presidential address. Making genomic medicine a reality. Am J Hum Genet. 1999;64:1–13. doi: 10.1086/302217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies JL, Kawaguchi Y, Bennett ST, et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature. 1994;371:130–136. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto L, Habita C, Beressi JP, et al. Genetic mapping of a susceptibility locus for insulin-dependent diabetes mellitus on chromosome 11q. Nature. 1994;371:161–164. doi: 10.1038/371161a0. [DOI] [PubMed] [Google Scholar]
- 11.Sawcer S, Jones HB, Feakes R, et al. A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat Genet. 1996;13:464–468. doi: 10.1038/ng0896-464. [DOI] [PubMed] [Google Scholar]
- 12.Haines JL, Ter-Minassian M, Bazyk A, et al. A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. Nat Genet. 1996;13:469–471. doi: 10.1038/ng0896-469. [DOI] [PubMed] [Google Scholar]
- 13.Ebers GC, Kukay K, Bulman DE, et al. A full genome search in multiple sclerosis. Nat Genet. 1996;13:472–476. doi: 10.1038/ng0896-472. [DOI] [PubMed] [Google Scholar]
- 14.Kuokkanen S, Gschwend M, Rioux JD, et al. Genomewide scan of multiple sclerosis in Finnish multiplex families. Am J Hum Genet. 1997;61:1379–1387. doi: 10.1086/301637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray JC. Face facts: genes, environment, and clefts. Am J Hum Genet. 1995;57:227–232. [PMC free article] [PubMed] [Google Scholar]
- 16.Rintala AE, Ranta R. Lower lip sinuses: I. Epidemiology, microforms and transverse sulci. Br J Plast Surg. 1981;34:26–30. doi: 10.1016/0007-1226(81)90090-4. [DOI] [PubMed] [Google Scholar]
- 17.Murray JC, Nishimura DY, Buetow KH, et al. Linkage of an autosomal dominant clefting syndrome (Van der Woude) to loci on chromosome 1q. Am J Hum Genet. 1990;46:486–491. [PMC free article] [PubMed] [Google Scholar]
- 18.Schutte BC, Bjork BC, Coppage KB, et al. A preliminary gene map for the Van der Woude syndrome critical region derived from 900 kb of genomic sequence at 1q32–q41. Genome Res. 2000;10:81–94. [PMC free article] [PubMed] [Google Scholar]
- 19.Kondo S, Schutte BC, Richardson RJ, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zlotogora J. Syndactyly, ectodermal dysplasia, and cleft lip/palate. J Med Genet. 1994;31:957–959. doi: 10.1136/jmg.31.12.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki K, Bustos T, Spritz RA. Linkage disequilibrium mapping of the gene for Margarita Island ectodermal dysplasia (ED4) to 11q23. Am J Hum Genet. 1998;63:1102–1107. doi: 10.1086/302072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suzuki K, Hu D, Bustos T, et al. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate-ectodermal dysplasia. Nat Genet. 2000;25:427–430. doi: 10.1038/78119. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian RP, Dunn JE, Geraghty RJ. The nectin-1[alpha] transmembrane domain, but not the cytoplasmic tail, influences cell fusion induced by HSV-1 glycoproteins. Virology. 2005;339:176–191. doi: 10.1016/j.virol.2005.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moore GE, Ivens A, Chambers J, et al. Linkage of an X-chromosome cleft palate gene. Nature. 1987;326:91–92. doi: 10.1038/326091a0. [DOI] [PubMed] [Google Scholar]
- 25.Braybrook C, Doudney K, Marcano AC, et al. The T-box transcription factor gene TBX22 is mutated in X-linked cleft palate and ankyloglossia. Nat Genet. 2001;29:179–183. doi: 10.1038/ng730. [DOI] [PubMed] [Google Scholar]
- 26.Braybrook C, Lisgo S, Doudney K, et al. Craniofacial expression of human and murine TBX22 correlates with the cleft palate and ankyloglossia phenotype observed in CPX patients. Hum Mol Genet. 2002;11:2793–2804. doi: 10.1093/hmg/11.22.2793. [DOI] [PubMed] [Google Scholar]
- 27.Bush JO, Lan Y, Maltby KM, et al. Isolation and developmental expression analysis of Tbx22, the mouse homolog of the human X-linked cleft palate gene. Dev Dyn. 2002;225:322–326. doi: 10.1002/dvdy.10154. [DOI] [PubMed] [Google Scholar]
- 28.Stanier P, Forbes SA, Arnason A, et al. The localization of a gene causing X-linked cleft palate and ankyloglossia (CPX) in an Icelandic kindred is between DXS326 and DXYS1X. Genomics. 1993;17:549–555. doi: 10.1006/geno.1993.1370. [DOI] [PubMed] [Google Scholar]
- 29.Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2:458–468. doi: 10.1038/35076601. [DOI] [PubMed] [Google Scholar]
- 30.Wyszynski DF. Cleft Lip and Palate: From Origin to Treatment. Oxford: Oxford University Press; 2002. [Google Scholar]
- 31.Schliekelman P, Slatkin M. Multiplex relative risk and estimation of the number of loci underlying an inherited disease. Am J Hum Genet. 2002;71:1369–1385. doi: 10.1086/344779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell LE, Christensen K. Analysis of the recurrence patterns for nonsyndromic cleft lip with or without cleft palate in the families of 3,073 Danish probands. Am J Med Genet. 1996;61:371–376. doi: 10.1002/(SICI)1096-8628(19960202)61:4<371::AID-AJMG12>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 33.Farrall M, Buetow KH, Murray JC. Resolving an apparent paradox concerning the role of TGFA in CL/P. Am J Hum Genet. 1993;52:434–436. [PMC free article] [PubMed] [Google Scholar]
- 34.Horikawa Y, Oda N, Cox NJ, et al. Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet. 2000;26:163–175. doi: 10.1038/79876. [DOI] [PubMed] [Google Scholar]
- 35.Carinci F, Pezzetti F, Scapoli L, et al. Recent developments in orofacial cleft genetics. J Craniofac Surg. 2003;14:130–143. doi: 10.1097/00001665-200303000-00002. [DOI] [PubMed] [Google Scholar]
- 36.Zucchero TM, Cooper ME, Maher BS, et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]
- 37.Houdayer C, Bonaiti-Pellie C, Erguy C, et al. Possible relationship between the van der Woude syndrome (VWS) locus and nonsyndromic cleft lip with or without cleft palate (NSCL/P) Am J Med Genet. 2001;104:86–92. doi: 10.1002/1096-8628(20011115)104:1<86::aid-ajmg10053>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 38.Scapoli L, Palmieri A, Martinelli M, et al. Strong Evidence of Linkage Disequilibrium between Polymorphisms at the IRF6 Locus and Nonsyndromic Cleft Lip With or Without Cleft Palate, in an Italian Population. Am J Hum Genet. 2005;76:180–183. doi: 10.1086/427344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Srichomthong C, Siriwan P, Shotelersuk V. Significant association between IRF6 820G->A and non-syndromic cleft lip with or without cleft palate in the Thai population. J Med Genet. 2005;42:e46. doi: 10.1136/jmg.2005.032235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- 41.Vastardis H, Karimbux N, Guthua SW, et al. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet. 1996;13:417–421. doi: 10.1038/ng0896-417. [DOI] [PubMed] [Google Scholar]
- 42.Marazita ML, Field LL, Tuncbilek G, et al. Genome-scan for loci involved in cleft lip with or without cleft palate in consanguineous families from Turkey. Am J Med Genet. 2004;126A:111–122. doi: 10.1002/ajmg.a.20564. [DOI] [PubMed] [Google Scholar]
- 43.Moreno LM, Arcos-Burgos M, Marazita ML, et al. Genetic analysis of candidate loci in non-syndromic cleft lip families from Antioquia-Colombia and Ohio. Am J Med Genet. 2004;125A:135–144. doi: 10.1002/ajmg.a.20425. [DOI] [PubMed] [Google Scholar]
- 44.Schultz RE, Cooper ME, Daack-Hirsch S, et al. Targeted scan of fifteen regions for nonsyndromic cleft lip and palate in Filipino families. Am J Med Genet. 2004;125A:17–22. doi: 10.1002/ajmg.a.20424. [DOI] [PubMed] [Google Scholar]
- 45.Vieira AR, Orioli IM, Castilla EE, et al. MSX1 and TGFB3 Contribute to Clefting in South America. J Dent Res. 2003;82:289–292. doi: 10.1177/154405910308200409. [DOI] [PubMed] [Google Scholar]
- 46.Suazo J, Santos JL, Carreno H, et al. Linkage Disequilibrium between MSX1 and Non-syndromic Cleft Lip/Palate in the Chilean Population. J Dent Res. 2004;83:782–785. doi: 10.1177/154405910408301009. [DOI] [PubMed] [Google Scholar]
- 47.van den Boogaard MJ, Dorland M, Beemer FA, et al. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans [published erratum appears in Nat Genet 2000 May;25:125] Nat Genet. 2000;24:342–343. doi: 10.1038/74155. [DOI] [PubMed] [Google Scholar]
- 48.Jezewski PA, Vieira AR, Nishimura C, et al. Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet. 2003;40:399–407. doi: 10.1136/jmg.40.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suzuki Y, Jezewski PA, Machida J, et al. In a Vietnamese population, MSX1 variants contribute to cleft lip and palate. Genet Med. 2004;6:117–125. doi: 10.1097/01.gim.0000127275.52925.05. [DOI] [PubMed] [Google Scholar]
- 50.Proetzel G, Pawlowski SA, Wiles MV, et al. Transforming growth factor-B3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaartinen V, Voncken JW, Shuler C, et al. Abnormal lung development and cleft palate in mice lacking TGF-B3 indicates defects of epithelial-mesenchymal interaction. Nature Genetics. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 52.Marazita ML, Murray JC, Lidral AC, et al. Meta-Analysis of 13 Genome Scans Reveals Multiple Cleft Lip/Palate Genes with Novel Loci on 9q21 and 2q32–35. Am J Hum Genet. 2004;75:161–173. doi: 10.1086/422475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu W, Sun X, Braut A, et al. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005:dev.01676. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- 54.Peters H, Neubuser A, Kratochwil K, et al. Pax9-deficient mice lack pharyngeal pouch derivatives and teeth and exhibit craniofacial and limb abnormalities. Genes Dev. 1998;12:2735–2747. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blanco R, Suazo J, Santos JL, et al. Association between 10 microsatellite markers and nonsyndromic cleft lip palate in the Chilean population. Cleft Palate Craniofac J. 2004;41:163–167. doi: 10.1597/02-147. [DOI] [PubMed] [Google Scholar]
- 56.Fujita H, Nagata M, Ono K, et al. Linkage analysis between BCL3 and nearby genes on 19q13.2 and non-syndromic cleft lip with or without cleft palate in multigenerational Japanese families. Oral Diseases. 2004;10:353–359. doi: 10.1111/j.1601-0825.2004.00995.x. [DOI] [PubMed] [Google Scholar]
- 57.Yoshiura K-i, Machida J, Daack-Hirsch S, et al. Characterization of a Novel Gene Disrupted by a Balanced Chromosomal Translocation t(2;19)(q11.2;q13.3) in a Family with Cleft Lip and Palate*1. Genomics. 1998;54:231–240. doi: 10.1006/geno.1998.5577. [DOI] [PubMed] [Google Scholar]
- 58.Sozen MA, Suzuki K, Tolarova MM, et al. Mutation of PVRL1 is associated with sporadic, non-syndromic cleft lip/palate in northern Venezuela. Nat Genet. 2001;29:141–142. doi: 10.1038/ng740. [DOI] [PubMed] [Google Scholar]
- 59.Sato N, Katsumata N, Kagami M, et al. Clinical Assessment and Mutation Analysis of Kallmann Syndrome 1 (KAL1) and Fibroblast Growth Factor Receptor 1 (FGFR1, or KAL2) in Five Families and 18 Sporadic Patients. J Clin Endocrinol Metab. 2004;89:1079–1088. doi: 10.1210/jc.2003-030476. [DOI] [PubMed] [Google Scholar]
- 60.Schultz RE. Genetics Program. Iowa City: University of Iowa; 2004. Identification of genetic loci involved in nonsyndromic cleft lip with or without cleft palate. [Google Scholar]
- 61.Marcano ACB, Doudney K, Braybrook C, et al. TBX22 mutations are a common cause of cleft palate. J Med Genet. 2004;000:1–8. doi: 10.1136/jmg.2003.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of nonsyndromic clefts. Hum. Mol. Genet. 2004:ddh052. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- 63.Prescott NJ, Lees MM, Winter RM, et al. Identification of susceptibility loci for nonsyndromic cleft lip with or without cleft palate in a two stage genome scan of affected sib-pairs. Hum Genet. 2000;106:345–350. doi: 10.1007/s004390051048. [DOI] [PubMed] [Google Scholar]
- 64.Wyszynski DF, Albacha-Hejazi H, Aldirani M, et al. A genome-wide scan for loci predisposing to non-syndromic cleft lip with or without cleft palate in two large Syrian families. Am J Med Genet. 2003;123A:140–147. doi: 10.1002/ajmg.a.20283. [DOI] [PubMed] [Google Scholar]
- 65.Jones KL, Smith DW, Ullelaand CN, et al. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet. 1973;9:1267–1271. doi: 10.1016/s0140-6736(73)91291-9. [DOI] [PubMed] [Google Scholar]
- 66.Wyszynski DF, Duffy DL, Beaty TH. Maternal cigarette smoking and oral clefts: a meta-analysis. Cleft Palate Craniofac J. 1997;34:206–210. doi: 10.1597/1545-1569_1997_034_0206_mcsaoc_2.3.co_2. [DOI] [PubMed] [Google Scholar]
- 67.Prescott NJ, Malcolm S. Folate and the face: evaluating the evidence for the influence of folate genes on craniofacial development. Cleft Palate Craniofac J. 2002;39:327–331. doi: 10.1597/1545-1569_2002_039_0327_fatfet_2.0.co_2. [DOI] [PubMed] [Google Scholar]
- 68.Murray J. Gene/environment causes of cleft lip and/or palate. Clin Genet. 2002;61:248–256. doi: 10.1034/j.1399-0004.2002.610402.x. [DOI] [PubMed] [Google Scholar]
- 69.Vieira AR, Murray JC, Trembath D, et al. Studies of reduced folate carrier 1 (RFC1) A80G and 5,10-methylenetetrahydrofolate reductase (MTHFR) C677T polymorphisms with neural tube and orofacial cleft defects. Am J Med Genet A. 2005;135:220–223. doi: 10.1002/ajmg.a.30705. [DOI] [PubMed] [Google Scholar]
- 70.Jugessur A, Wilcox AJ, Lie RT, et al. Exploring the Effects of Methylenetetrahydrofolate Reductase Gene Variants C677T and A1298C on the Risk of Orofacial Clefts in 261 Norwegian Case-Parent Triads. Am J Epidemiol. 2003;157:1083–1091. doi: 10.1093/aje/kwg097. [DOI] [PubMed] [Google Scholar]
- 71.van Rooij IALM, Vermeij-Keers C, Kluijtmans LAJ, et al. Does the Interaction between Maternal Folate Intake and the Methylenetetrahydrofolate Reductase Polymorphisms Affect the Risk of Cleft Lip with or without Cleft Palate? Am J Epidemiol. 2003;157:583–591. doi: 10.1093/aje/kwg005. [DOI] [PubMed] [Google Scholar]
- 72.Shotelersuk V, Ittiwut C, Siriwan P, et al. Maternal 677CT/1298AC genotype of the MTHFR gene as a risk factor for cleft lip. J Med Genet. 2003;40:e64. doi: 10.1136/jmg.40.5.e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pezzetti F, Martinelli M, Scapoli L, et al. Maternal MTHFR variant forms increase the risk in offspring of isolated nonsyndromic cleft lip with or without cleft palate. Hum Mutat. 2004;24:104–105. doi: 10.1002/humu.9257. [DOI] [PubMed] [Google Scholar]
- 74.van Rooij IALM, Ocke MC, Straatman H, et al. Periconceptional folate intake by supplement and food reduces the risk of nonsyndromic cleft lip with or without cleft palate. Preventive Medicine. 2004;39:689–694. doi: 10.1016/j.ypmed.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 75.Lammer EJ, Shaw GM, Iovannisci DM, et al. Maternal smoking, genetic variation of glutathione s-transferases, and risk for orofacial clefts. Epidemiology. 2005;16:698–701. doi: 10.1097/01.ede.0000172136.26733.4b. [DOI] [PubMed] [Google Scholar]
- 76.Lammer EJ, Shaw GM, Iovannisci DM, et al. Periconceptional multivitamin intake during early pregnancy, genetic variation of acetyl-N-transferase 1 (NAT1), and risk for orofacial clefts. Birth Defects Res A Clin Mol Teratol. 2004;70:846–852. doi: 10.1002/bdra.20081. [DOI] [PubMed] [Google Scholar]
- 77.Lammer EJ, Shaw GM, Iovannisci DM, et al. Maternal smoking and the risk of orofacial clefts: Susceptibility with NAT1 and NAT2 polymorphisms. Epidemiology. 2004;15:150–156. doi: 10.1097/01.ede.0000112214.33432.cc. [DOI] [PubMed] [Google Scholar]