

Abstract
We seek to determine: 1) whether a long-term high salt diet induces hypertension and renal injury in Sprague-Dawley (SD) rats and 2) whether the high salt diet-induced hypertension and renal injury are associated with decreased renal VEGF expression. Twelve 10-wk-old male SD rats received a high salt diet (HS, 8%) and twelve SD rats received a normal salt diet (NS, 0.5 %) for 8 weeks. Using a tail cuff, weekly monitoring showed that blood pressure increased significantly after 6, 7, & 8 wks in HS group, compared to NS group (P<0.01). At 4 wks and 8 wks of diet, mean arterial pressure (MAP) was determined in conscious rats by continuous monitoring through a catheter placed in the carotid artery. MAP was not significantly different between HS and NS group in 4 wks, but was significantly higher in HS than NS group (140±5.3 vs.112±2.2 mmHg; P<0.01) in 8 wks. Increased proteinuria and albuminuria were associated with marked renal histological abnormalities in HS group, compared to those in NS group. Northern blot and ELISA demonstrated that 8 wks of HS diet significantly decreased renal expression of VEGF mRNA and protein, compared to NS group (P<0.01). In 8 wks, total urinary excretion of sFlt-1 was significantly higher in HS than NS group (9.28±1.05 vs. 2.05±0.55 ng/day; P<0.01) whereas the plasma levels of sFlt-1 remained stable. These results suggest that a long-term HS diet induces renal injury and hypertension, which are associated with decreased renal VEGF expression in normotensive rodent animals.
Keywords: long-term high salt diet, Sprague-Dawley rat, hypertension, renal VEGF
Introduction
Hypertension has been recognized as a multifactorial trait resulting from the effects of a combination of both environmental and genetic factors. An excess of dietary salt is the most common environmental factor that contributes to the pathogenesis of hypertension (1–5). The kidneys play a central role in regulating blood pressure, which has been well characterized by Guyton and colleagues on the pressure natriuresis and diuresis relationship (6–7). An increase in blood pressure causes the kidneys to excrete more salt and water, thereby decreasing the extracellular and plasma volume. Therefore, over a long period, blood pressure is controlled primarily by salt and water balance because of the infinite gain property of the kidneys to rapidly eliminate excess fluid and salt. Neither sinoaortic denervation (8) nor renal nerve denervation (9) prevents the generation of salt dependent forms of hypertension. Much evidence from epidemiological, migration, intervention, and genetic studies suggest that a high salt intake is almost uniformly associated with an increase in blood pressure in normotensive and hypertensive humans (10–14). A long-term high salt intake also increased blood pressure in normotensive pigs (15), baboons (16), African green monkeys (17), and chimpanzees (18). Although many studies indicated that a short-term high salt diet caused hypertension and renal injury in salt-sensitive rodent animals such as Dahl salt-sensitive rats (19–20) and DOCA-salt sensitive rats(21), it is not clear whether a long-term high salt diet can cause hypertension and renal injury in normotensive rodent animals such as Sprague-Dawley rats.
The mechanisms of dietary salt-induced hypertension and renal injury are still not clear, though accumulating evidence has shown how a variety of genetic mutations and polymorphisms of sodium channels and related proteins in the kidney result in the dysregulation of sodium metabolism and/or salt-sensitive hypertension. These include mutations affecting synthesis and circulating levels of mineralocorticoids (22), the role of renal inflammation, oxidative stress, and intra-renal angiotension activity (20, 23–24). Recent clinical evidence has indicated that the inhibition of vascular endothelial growth factor (VEGF) is linked to hypertension. In clinical trials with cancer patients, more than 20% of patients developed hypertension and proteinuria under treatment with anti-VEGF antibody, or VEGF receptor inhibitors such as ZD6474 and SU5416 (25–27). Significantly increased levels of circulating sFlt-1, an endogenous VEGF inhibitor, contribute to hypertension in patients with preeclampsia (28). Many animal studies also suggested that VEGF plays a very important role in maintaining kidney function and structure (29–32). We previously reported that low dietary salt intake stimulated the renal VEGF mRNA in WKY rats, compared to the normal salt group (33). However, there is no study available for examining whether high salt diet-induced renal injury and hypertension are associated with the down-regulation of the renal VEGF expression in an animal model; and for testing whether a long-term high salt diet can cause renal injury and hypertension in normotensive rodent animals such as Sprague-Dawley rats.
Based on all findings mentioned above, we hypothesize that a long-term high salt diet can cause renal injury and hypertension, which are associated with decreased renal VEGF expression in normotensive rodent animals such as Sprague-Dawley rats. In the present study, we seek to determine: 1) whether a long-term high salt diet (8 weeks) induces hypertension and renal injury in Sprague-Dawley (SD) rats and 2) whether the high salt diet-induced hypertension and renal injury are associated with decreased renal VEGF expression.
Methods
Animal Protocol
The protocols were carried out according to the guidelines for the care and use of laboratory animals implemented by the National Institutes of Health and the Guidelines of the Animal Welfare Act, and were also approved by the University of Mississippi Medical Center's Institutional Animal Care and Use Committee. Male Sprague-Dawley (SD) rats were received at 9 weeks of age and were allowed to acclimate for 1 week with standard rat diet (Teklad, Harlan Sprague Dawley; Indianapolis, IN) and tap water before beginning the experiment. Rats were placed in a temperature-controlled room with a 12:12 light-dark cycle. During the 8-week experimental period, the NS group (n = 12) received a normal salt (0.5%) diet (Teklad, Harlan Sprague Dawley; Indianapolis, IN) and the HS group (n = 12) received a high-salt (8%) diet (Teklad, Harlan Sprague Dawley; Indianapolis, IN). Systolic blood pressure was measured weekly by a tail cuff method. During the last week of the experimental period, the rats were placed in metabolic cages, where water intake and urinary volume output were measured daily. Twenty-four-hour urine samples were collected daily, and urine sodium concentration was determined by flame photometry. The urine protein concentration was determined using a Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Inc., Hercules, CA). Urine albumin concentration was measured using a Nephrat ELISA Kit (Exocell Inc., Philadelphia, PA). Mean arterial pressure (MAP) was determined in the conscious rats 4 hours/d for 2 days at 4 weeks or 8 weeks of the experiment. At the end of experiment, the kidneys were removed for various analyses.
Measurements of Blood Pressure
During the 8-week experimental period, the systolic blood pressure was measured weekly in the NS and HS SD rats (n = 12 each group) by tail plethysmography (IITV, Inc). After 4 weeks or 8 weeks of the experiment, both groups of SD rats (n = 6 each group) were anesthetized by isoflurane inhalation using a gas vaporizer (Harvard Apparatus, Holliston, MA). With the use of aseptic techniques, a micro-catheter was placed into the carotid artery and routed under the skin to the back of the neck and secured. Two days after the rats recovered from the surgery, the catheter was connected to a computerized monitoring system, called PowerLab (AD Instruments, Castle Hill, Australia) to continuously record MAP and heart rate (HR) in the conscious SD rats 4 hours/d for 2 days. The measurements of MAP and HR in each rat were made simultaneously, and all of the values for each rat represented the average of the 4 hours/d for 2 days measurements.
Northern Blot Analysis
Total RNA was prepared from the kidneys of SD rats (n = 6 each group) after 8 weeks of normal salt or high salt diet using a modified guanidinium-based method (Total RNA isolation kit, Ambion, Austin, TX), as previously described (34). The genomic DNA was removed by treatment with RNase-free DNase I followed by phenol /chloroform extraction and ethanol precipitation. RNA integrity was verified on a MOPS-formaldehyde agarose gel prior to cDNA synthesis. Expression of VEGF mRNA in the kidneys was examined by Northern blotting, as previously described (34). The VEGF cDNA probe was a 580-bp EcoR I-BamH I fragment of the murine VEGF cDNA that was cloned into pBluescrip plasmid. The cDNA probe was labeled with α-32P-dCTP (New England Nuclear, Boston, MA) using a Multiprime random-primed DNA labeling kit (Amersham, UK). Quantization of VEGF mRNA expression was performed on phosphor images of blots that were collected using a Phosphor-Imager (Molecular Dynamics; software: ImageQuant 3.3, Molecular Dynamics). To verify the relative amounts of total RNA, filters were hybridized with a 32P-labeled 28S rRNA antisense oligonucleotide probe (Ambion). The VEGF mRNA expression was normalized against 28S rRNA in each sample.
Measurements of Protein Levels of VEGF and sFlt-1 by ELISA
Protein levels of VEGF and sFlt-1 in the plasma, urine, and whole kidney tissue of SD rats were determined using mouse VEGF and mouse sFlt-1 ELISA kits (R&D Systems, Minneapolis, MN), according to the manufacturer’s instructions. Tissue samples of whole kidneys were homogenized in ice-cold PBS buffer containing protease inhibitor cocktail and the total proteins were extracted using NE-PER Cytoplasmic Extraction Reagents (Pierce, Rockford, IL), according to the manufacturer’s protocol. These extraction reagents did not affect the measurements of tissue proteins of those cytokines compared with other regular protein extraction methods. The total protein concentration of tissue supernatant was determined using a Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). The protein levels of VEGF in the whole kidney tissues were normalized and expressed as picograms per milligram total tissue protein.
Histological Analysis by Light Microscopy
Mid-coronal sections of the left kidneys collected from both normal salt-fed and high salt-fed groups were fixed in buffered formalin and embedded in paraffin for histological studies as previously described (20, 35). Samples were cut into 3-µm sections and stained with periodic acid-Schiff reagent (PAS) followed by hematoxylin counterstaining. All sections were analyzed by semiquantitative histological grading (0 = absent, 1+ = mild, 2+ = moderate, and 3+ = severe) for the severity of tubulointerstitial infiltration with inflammatory cells, focal segmental glomerulosclerosis, tubulointerstitial injury, and extracellular matrix expansion. The average glomerular area in the PAS-stained sections was determined from 30 to 40 glomeruli in each kidney on the computer screen using Image-Pro Plus Software (Media Cybernetics, Inc., Silver Spring, MD).
Statistical Analyses
All determinations were performed in duplicated sets. Where indicated, data is presented as mean ± SE. Statistically significant differences in mean values between two groups were tested by an unpaired Student’s t-test. ANOVA was used to analyze the differences between two groups with multiple comparisons. A value of P < 0.05 was considered statistically significant. All statistical calculations were performed using SPSS software (SPSS Inc., Chicago, IL).
Results
Increased systolic blood pressure and MAP due to a long-term HS diet
Figure 1 A. demonstrates that by weekly monitoring blood pressure using a tail cuff, there is no significant difference in systolic blood pressure during 1 to 5 weeks between NS and HS groups of SD rats, but systolic blood pressure significantly increases after 6, 7, and 8 weeks in HS group, compared to NS group (P < 0.01, respectively). Figure 1B indicates that 4 weeks of a high salt diet (8%) does not significantly change MAP in SD rats in comparison with SD rats having a normal salt diet (123±6.1 vs. 118±2.7 mmHg; n = 6; P >0.05); however, after 8 weeks of the dietary program, MAP is significantly higher in HS than NS group (140±5.3 vs. 112±2.2 mmHg; n = 6; P < 0.01). In addition, there were no significant changes in heart rate between HS and NS groups after 8 weeks of dietary program in SD rats (443±39 vs. 415±53 beats/min; n = 6; P > 0.05). There was no significant difference in the body weight between HS and HS groups (370.0±8.9 vs. 359.3±8.0 g; n = 6; P = 0.41).
Figure 1. A long-term HS diet significantly increases systolic blood pressure (A) and MAP (B) in SD rats.

Using a tail cuff, weekly monitoring blood pressure showed that systolic blood pressure increased significantly after 6, 7, and 8 weeks in HS group, compared to NS group of SD rats, respectively (n = 6; P < 0.01). PowerLab measurement indicated that 4 weeks of a high salt diet (8%) did not significantly change MAP in SD rats in comparison with SD rats having a normal salt diet (n = 6; P >0.05); however, after 8 weeks of the dietary program, MAP was significantly higher in HS than NS group of SD rats (n = 6; P < 0.01).
Increased proteinuria and albuminuria in response to a long-term HS diet
The renal damage was monitored by measuring the urinary protein and albumin secretion in SD rats with HS and NS diet. Figure 2A shows that the urinary protein secretion does not significantly change between HS and NS groups in SD rats after 4 weeks of dietary program (27.3±3.9 vs. 25.2±6.9 mg/day; n = 6; P>0.05). However, the urinary protein secretion was significantly higher in HS than NS group in SD rats after 8 weeks of dietary program (55.6±4.8 vs. 33.1±4.7 mg/day; n = 6; P < 0.01). Figure 2B demonstrates that there is a significant increase in albuminuria in HS group, compared with NS group in both 4 weeks (24.1±2.2 vs. 17.3±2.6 mg/day; n = 6; P < 0.05) and 8 weeks (41.4±3.2 vs. 20.1±4.5 mg/day; n = 6; P < 0.01) of the dietary program, respectively.
Figure 2. Increased proteinuria and albuminuria in response to a long-term HS diet.

Figure 2A shows that the urinary protein secretion does not significantly change between HS and NS groups in SD rats after 4 weeks of dietary program (n = 6; P>0.05), but it is significantly higher in HS than NS group of SD rats after 8 weeks of dietary program (n = 6; P < 0.01). Figure 2B demonstrates that there is a significant increase in albuminuria in HS group, compared with NS group in both 4 weeks (n = 6; P < 0.05) and 8 weeks (n = 6; P < 0.01) of dietary program, respectively.
Decreased renal expression of VEGF mRNA and protein due to a long-term HS diet
Northern blot (Figure 3A) demonstrated that 8 weeks of a high salt diet significantly reduced renal expression of VEGF mRNA by 45%, compared to NS group (0.55±0.12 vs. 1.0±0.16; n = 6; P < 0.01). Consistent with the reduction of renal VEGF mRNA expression, ELISA (Figure 3B) indicated that 8 weeks of a high salt diet significantly decreased renal VEGF protein levels, compared to NS group in SD rats (68.1±3.4 vs. 84.3±4.2 pg/mg total protein; n = 6; P < 0.01).
Figure 3. Decreased renal expression of VEGF mRNA and protein in HS group, compared to NS group of SD rats.
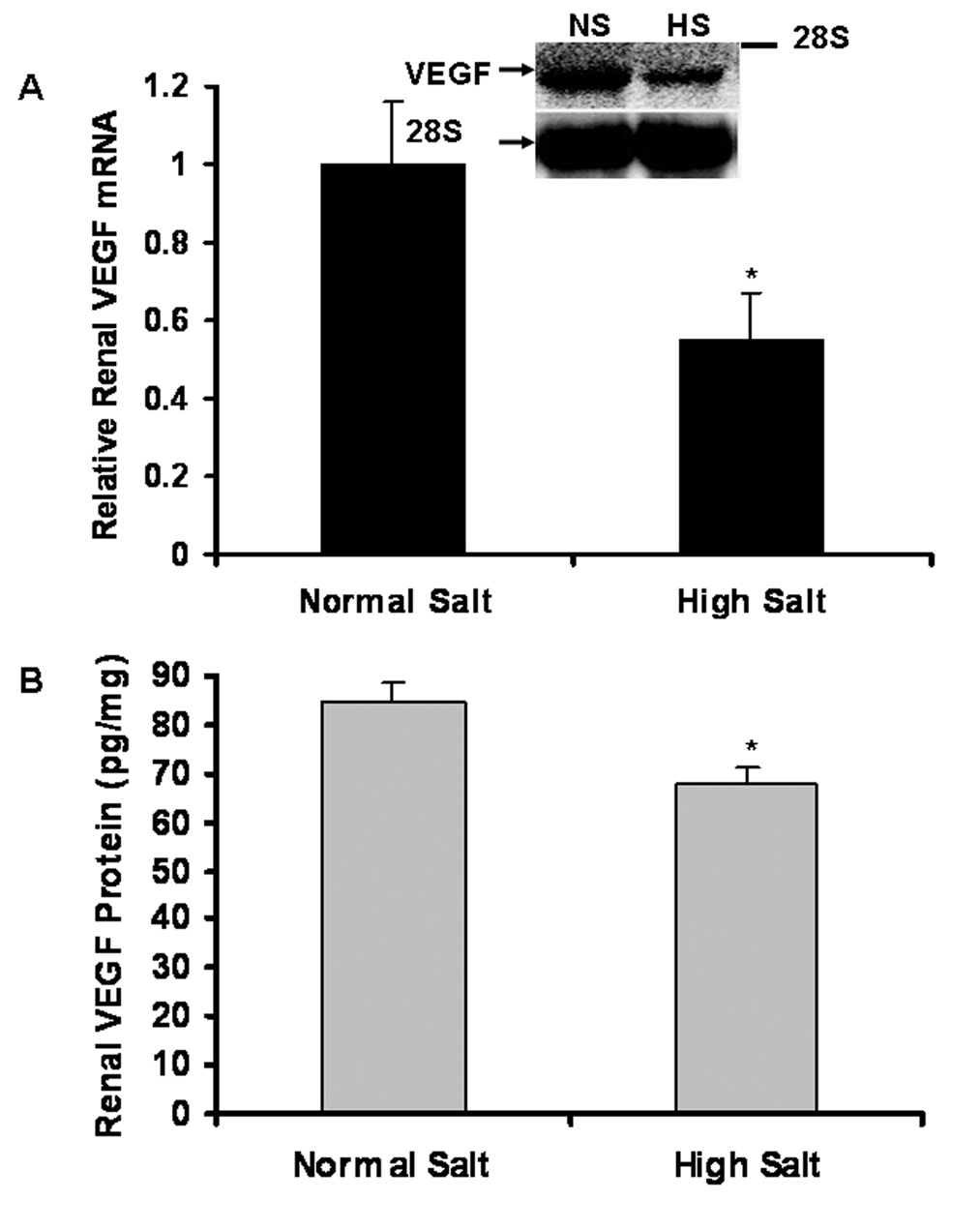
Northern blot (Figure 3A) demonstrated that 8 weeks of a high salt diet significantly reduced renal expression of VEGF mRNA by 45%, compared to NS group (n = 6; P < 0.01). ELISA (Figure 3B) indicated that renal VEGF protein levels significantly decreased in HS group, compared to NS group (n = 6; P<0.01).
Effects of a HS diet on plasma levels and total urinary excretion of VEGF and sFlt-1
ELISA demonstrated that there were no significant changes in plasma levels of VEGF and sFlt-1 (an endogenous VEGF inhibitor) between HS and NS groups. Figure 4A demonstrates that plasma levels of VEGF in NS and HS groups of SD rats are 40.8±8.3 and 45.9±8.3 pg/ml, respectively (n = 6; P >0.05), and that plasma levels of sFlt-1 in NS and HS groups of SD rats are 701±47 and 738±29 pg/ml, respectively (P > 0.05), after 8 weeks of dietary program. There was no significant difference in total urinary excretion of VEGF in NS and HS groups of SD rats after 8 weeks of dietary program (1.45±0.28 vs. 1.42±0.27 ng/day; P > 0.05). Interestingly, as indicated in Figure 4B, total urinary excretion of sFlt-1 was significantly higher in HS than NS group of SD rats after 8 weeks of dietary program (9.28±1.05 vs. 2.05±0.55 ng/day; n = 6; P<0.01).
Figure 4. Effects of a HS diet on plasma levels and total urinary excretion of VEGF and sFlt-1 in SD rats.

ELISA demonstrated that there were no significant changes in plasma levels of VEGF and sFlt-1 and total urinary excretion of VEGF between HS and NS groups in SD rats after 8 weeks of dietary program, respectively (n = 6; P<0.01). However, total urinary excretion of sFlt-1 was significantly higher in HS than NS group of SD rats after 8 weeks of dietary program (n = 6; P<0.01).
Renal Histological Changes due to a High Salt Diet
Histological analysis by light microscopy of PAS-stained sections of the kidneys showed that there were no marked renal abnormalities in SD rats with a normal salt diet (Figures 5A). In contrast (5B), SD rats with a long-term high salt diet developed moderate glomerulosclerosis, extracellular matrix expansion, tubulointerstitial injury, and tubulointerstitial infiltration with inflammatory cells. The average glomerular area in the PAS-stained sections was increased by 34% in HS group, compared to NS group (22,085±4222 vs. 16,432±3689 µm2; n = 6; P < 0.01). The increased glomerular area in HS group indicated that glomerular hypertrophy and hyperfiltration. These renal pathological changes may explain what the mechanisms contribute to proteinuria or albuminuria after a long-term sodium load in SD rats.
Figure 5.

Representative pathological changes in the kidneys of SD rats with a long-term (8 weeks) high-salt diet and normal-salt diet. Kidneys were removed after 8 weeks of the dietary program. PAS stain was used. A: normal salt diet. Normal glomeruli, arterioles, and tubules. B: high salt diet. Glomerulosclerosis, tubulointerstitial injury, inflammatory cells infiltration, extracellular matrix expansion, and increased glomerular area. Magnification × 100.
Discussion
The major new findings in this study include: 1) a long-term high salt intake induces hypertension and renal damage such as albuminuria and histological renal injury in normotensive rodent animals such as Sprague-Dawley rats, and 2) high salt intake decreases renal VEGF expression; and increases renal sFlt-1 secretion suggested by increased urinary excretion of sFlt-1 whereas plasma sFlt-1 remained stable. These findings first suggest that a long-term high salt diet can induce hypertension and renal injury in normotensive rodent animals, in which dietary salt-induced hypertension and renal injury are associated with the down-regulation of the renal VEGF expression.
Many clinical studies suggest that a long-term high salt intake can increase blood pressure in both normotensive and hypertensive humans (10–14). However, it is not clear whether a long-term high salt diet can cause hypertension and renal injury in normotensive rodent animals such as Sprague-Dawley rats. The previous reports had been focused on the short-term effect of a high salt diet on blood pressure in Sprague-Dawley rats. These studies indicated that there were no significant changes in blood pressure in male Sprague-Dawley rats with a high salt diet (4% or 8% sodium) for 10 days (36), 3 weeks (37), and 5 weeks (38), compared to low or normal salt group, respectively. In the present study, male Sprague-Dawley rats received a high salt (8%) or a normal salt (0.5%) diet for 8 weeks. The systolic blood pressure was monitored weekly using a tail cuff method, and MAP was determined in conscious rats using the PowerLab system after 4 weeks and 8 weeks of the dietary program. Systolic blood pressure was not significantly changed in SD rats with a high salt diet, compared to normal salt group, after 1 to 5 weeks of the dietary program. Also, a high salt diet did not significantly increase MAP in SD rats in 4 weeks of the dietary program, compared to normal salt SD rats. These results are consistent with the previous findings that a short term high salt intake did not cause hypertension in SD rats. However, we have demonstrated that systolic blood pressure in SD rats after 6, 7, and 8 weeks of a high salt diet, as well as MAP in SD rats after 8 weeks of high salt diet, are both significantly increased, compared to the normal salt group. These findings suggest that a long-term high salt diet can induce hypertension in normotensive rodent animals. SD rats are a good rodent animal model for studying the effect of long-term high salt intake on hypertension because this model mimics the conditions of high salt-induced hypertension in normotensive humans.
Previous studies (39–40) demonstrated that excessive salt intake for longer periods caused renal injury and abnormal kidney function in both doca-salt hypertensive rats and spontaneous hypertensive rats. In the present study, we found that a long-term high salt diet induced renal injury manifested by albuminuria and renal histological abnormalities in normotensive SD rats. The renal histological abnormalities included increased glomerular area, moderate glomerulosclerosis, extracellular matrix expansion, tubulointerstitial injury, and tubulointerstitial infiltration with inflammatory cells. These pathological changes may explain why a long-term high salt diet causes albuminuria or proteinuria in SD rats. We also found that albuminuria significantly increased in HS group, compared with NS group, but no significant changes in blood pressure at 4 weeks. The blood pressure significantly increased after 6 weeks of high salt diet. It seems that the renal effects may occur before hypertension. It is also conceivable that proteinuria further contributes to renal damage and inflammatory response due to the induction of proinflammatory substances by filtered proteins (41). We believe that a long-term high salt diet-induced renal damage contributes to hypertension that can further enhance renal injury. Further studies are needed to understand the mechanisms of salt sensitive hypertension and the relationship between high salt intake, hypertension, renal injury, and kidney function.
A question that arises from the current study is what causes the attenuation of renal VEGF expression in relation to a high salt diet? Previous studies have shown the major role of hypoxia inducible factor-1α (HIF-1α) in regulating VEGF expression in most tissues (42). In the kidney however, recent in vitro (43) and in vivo (44–45) studies demonstrated that angiotensin II increased renal VEGF expression. Angiotensin II is a powerful sodium-retaining hormone and vasoconstrictor that is increased by low dietary sodium intake and decreased by high dietary sodium intake. We previously reported that low dietary salt intake enhanced renal VEGF mRNA expression in WKY rats, compared to renal VEGF mRNA in rats on the normal diet (33). In the present study, we have demonstrated that a long-term high salt diet decreases renal expression of VEGF mRNA and protein in Sprague-Dawley rats. When these findings are considered together, they suggest that low angiotensin II levels may contribute to the down-regulation of renal VEGF expression in Sprague-Dawley rats with high salt intake. It will be interesting to determine whether an angiogension II infusion could prevent the down-regulation of renal VEGF expression in this model of salt-sensitive hypertension.
What role does VEGF play in maintaining renal function and structure as well as blood pressure? Clinical evidence links the inhibition of vascular endothelial growth factor (VEGF) to hypertension (25–28). Cancer patients treated with anti-VEGF therapy develop hypertension and renal damage such as proteinuria, which dissipates after the treatment is stopped. In addition, there is mounting evidence suggesting that VEGF exerts a very important and unique role in maintaining renal structure and functions (29–32). For example, the two receptors to which VEGF binds, Flt-1 and KDR/Flk-1 (46–47), are expressed in glomerular and peritubular endothelial cells (47). VEGF is critical for maintaining the functions and structure of endothelial cells, but it also exerts autocrine and paracrine actions which regulate the functions and structure of renal cells such as podocytes, mesangial cells, and renal tubular epithelial cells (48). While these findings suggest that VEGF may play a very important role in regulating kidney function and structure and blood pressure, its role in the renal mechanisms related to hypertension is unclear. To our knowledge, this study is the first to demonstrate that a long-term high salt diet decreases renal expression of VEGF, which is associated with the development of hypertension and renal damage in SD rats. We will use this model to further test the hypothesis that attenuated renal VEGF expression contributes to renal damage, which contributes to the development of hypertension. In the future studies, we will use this animal model to test: 1) whether the administration of VEGF receptor inhibitor causes hypertension and renal injury; and 2) whether the administration of VEGF reduces blood pressure and renal damage in salt sensitive hypertension.
The soluble VEGF receptor-1 (sFlt-1) is considered an endogenous VEGF inhibitor because secreted sFlt-1 can directly inhibit VEGF activity by binding to VEGF. In a different setting, pre-eclampsia is associated with elevated sFlt-1, endothelial cell dysfunction and proteinuria. More recently, some studies demonstrated that the administration of sFlt1 induced proteinuria in rats (49–50). In this study, we have demonstrated that a long-term high salt diet increases renal secretion of sFlt-1 related to renal damage in SD rats. We believe that increased renal secretion of sFlt-1 will further suppress renal VEGF activity. Hornig, et al showed that sFlt-1 was up-regulated by hypoxia in cultured human placental villous cells (51). Thus, it is likely that the increased renal secretion of sFlt-1 in SD rats on HS diet could be due to the renal injury as evident in the histological analysis.
In conclusion, our results indicate that a long-term high salt diet causes hypertension and renal damage in normotensive rodent animals such as SD rats. During the 8 weeks of the high salt dietary program, systolic blood pressure, MAP, proteinuria or albuminuria, and renal histological changes were monitored. By weekly monitoring blood pressure using a tail cuff, there was no significant difference in systolic blood pressure during 1 to 5 weeks between NS and HS groups of SD rats, but systolic blood pressure significantly increased after 6, 7, and 8 weeks in HS group, compared to NS group. Albuminuria significantly increased in HS group, compared with NS group at 4 weeks, but no significant changes in blood pressure. However, after 8 weeks of the dietary program, MAP was significantly higher in HS than NS group. The renal PAS-stained sections showed increased glomerular area, moderate glomerulosclerosis, extracellular matrix expansion, tubulointerstitial injury, and tubulointerstitial infiltration with inflammatory cells. In addition, we found that a long-term high salt intake attenuated renal VEGF expression and increased renal secretion of sFlt-1. These findings support the hypothesis that a long-term high salt diet can induce hypertension and renal damage in rodent animals, and that high salt-induced hypertension and renal injury are associated with an impaired renal VEGF signaling system. These findings can lead to new insights into the mechanisms of dietary salt-induced hypertension and renal injury.
Acknowledgments
This work was supported by National Heart, Lung, and Blood Institute Grant HL-51971, and National Institute on Alcohol Abuse and Alcoholism Grant AA-013821-01A2. We greatly appreciate Dr. Elizabeth Brandon’s help in editing this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aviv A. Salt and hypertension: the debate that begs the bigger question. Arch Intern Med. 2001;161:507–510. doi: 10.1001/archinte.161.4.507. [DOI] [PubMed] [Google Scholar]
- 2.Frolich ED, Varagic J. Sodium directly impairs target organ function in hypertension. Curr Opin Cardiol. 2005;20:424–429. doi: 10.1097/01.hco.0000175519.34933.a5. [DOI] [PubMed] [Google Scholar]
- 3.Gu JW, Anand V, Shek EW, Moore MC, Brady AL, Kelly WC, Adair TH. Sodium induces hypertrophy of cultured myocardial myoblasts and vascular smooth muscle cells. Hypertension. 1998;31:1083–1087. doi: 10.1161/01.hyp.31.5.1083. [DOI] [PubMed] [Google Scholar]
- 4.Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER, 3rd, Simons-Morton DG, Karanja N, Lin PH SASH-Sodium Collaborative Research Group. Effects on blood pressure of reduced dietary sodium and the dietary approaches to stop hypertension (DASH) diet. N Engl J Med. 2001;344:3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- 5.Meneton P, Jeunemaitre X, de Wardener HE, Macgregor GA. Links between dietary salt intake, renal salt handling, blood pressure, and cardiovascular diseases. Physiol Rev. 2005;85:679–715. doi: 10.1152/physrev.00056.2003. [DOI] [PubMed] [Google Scholar]
- 6.Guyton AC. Blood pressure control-special role of the kidneys and body fluids. Science. 1991;252:1813–1816. doi: 10.1126/science.2063193. [DOI] [PubMed] [Google Scholar]
- 7.Hall JE, Guyton AC, Smith MJ, Jr, Coleman TG. Blood pressure and renal function during chronic changes in sodium intake: role of angiotension. Am J Physiol Renal Fluid Electrolyte Physiol. 1980;239:F271–F280. doi: 10.1152/ajprenal.1980.239.3.F271. [DOI] [PubMed] [Google Scholar]
- 8.Masson GMC, Aoki K, Page IH. Effects of sinoaortic denervation on renal and adrenal hypertension. Am J Physiol. 1966;211:94–104. doi: 10.1152/ajplegacy.1966.211.1.99. [DOI] [PubMed] [Google Scholar]
- 9.Dzielak DJ, Norman RA., Jr Renal nerves are not necessary for onset or maintenance of DOC-salt hypertension in rats. Am J Physiol Heart Circ Physiol. 1985;249:H945–H949. doi: 10.1152/ajpheart.1985.249.5.H945. [DOI] [PubMed] [Google Scholar]
- 10.Weinberger MH. Salt sensitivity of blood pressure in humans. Hypertension. 1996;27:481–490. doi: 10.1161/01.hyp.27.3.481. [DOI] [PubMed] [Google Scholar]
- 11.Intersalt Cooperative Research Group. Intersalt: an international study of electrolyte excretion and blood pressure. Results for 24 hour urinary sodium and potassium excretion. BMJ. 1988;297:319–328. doi: 10.1136/bmj.297.6644.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poulter N, Khaw Kt, Hopwood BEC, Mugambi M, Peart WS, Rose G, Sever PS. The Kenyan Luo migration study: observations on the initiation of a rise in blood pressure. BMJ. 1990;300:967–972. doi: 10.1136/bmj.300.6730.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Forte JG, Miguel JM, Miguel MJ, de Padua F, Rose G. Salt and blood pressure: a community trial. J Hum Hypertens. 1989;3:179–184. [PubMed] [Google Scholar]
- 14.Lifton RP. Molecular genetics of human blood pressure variations. Science. 1996;272:676–680. doi: 10.1126/science.272.5262.676. [DOI] [PubMed] [Google Scholar]
- 15.Corbett WT, Kuller LH, Blaine EH, Damico FJ. Utilization of swine to study the risk factor of an elevated salt diet on blood pressure. Am J Cllin Nutr. 1979;32:2068–2075. doi: 10.1093/ajcn/32.10.2068. [DOI] [PubMed] [Google Scholar]
- 16.Cherchovich GM, Capek K, Jefremova Z, Pohlova I, Jelinek J. High salt intake and blood pressure in lower primates (Papio hamadryas) J Appl Physiol. 1976;40:601–604. doi: 10.1152/jappl.1976.40.4.601. [DOI] [PubMed] [Google Scholar]
- 17.Srinivasan SR, Dalferes ER, Wolf RH, Radhakrishnamurthy B, Foster TA, Berenson GS. Variability in blood pressure response to dietary sodium intake among African green monkeys (Cercopithecus aethiops) Am J Clin Nutr. 1984;39:792–796. doi: 10.1093/ajcn/39.5.792. [DOI] [PubMed] [Google Scholar]
- 18.Denton D, Weisinger R, Mundy NI, Wickings EJ, Dixson A, Moisson P, Pingard AM, Shade R, Carey D, Ardaillou R, Paillard F, Chapman J, Thillet J, Michel JB. The effect of increased salt intake on blood pressure of chimpanzees. Nat Med. 1995;1:1009–1016. doi: 10.1038/nm1095-1009. [DOI] [PubMed] [Google Scholar]
- 19.Manning RD, Jr, Tian N, Meng S. Oxidative stress and antioxidant treatment in hypertension and the associated renal damage. Am J Nephrol. 2005;25:311–317. doi: 10.1159/000086411. [DOI] [PubMed] [Google Scholar]
- 20.Gu JW, Tian N, Shparago M, Tan W, Bailey AP, Manning RD., Jr Renal NF-κB activation and TNF-α upregulation correlate with salt-sensitive hypertension in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1817–R1824. doi: 10.1152/ajpregu.00153.2006. [DOI] [PubMed] [Google Scholar]
- 21.Brooks VL, Freeman KL, Qi Y. Time course of synergistic interaction between DOCA and salt on blood pressure: roles of vasopressin and hepatic osmoreceptors. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1825–R1834. doi: 10.1152/ajpregu.00068.2006. [DOI] [PubMed] [Google Scholar]
- 22.Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556. doi: 10.1016/s0092-8674(01)00241-0. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Iturbe B, Vaziri ND, Herrera-Acosta J, Johnson RJ. Oxidative stress, renal infiltration of immune cells and salt-sensitive hypertension: all for one and one for all. Am J Physiol Renal Physiol. 2004;286:F606–F616. doi: 10.1152/ajprenal.00269.2003. [DOI] [PubMed] [Google Scholar]
- 24.Vaziri ND, Rodriguez-Iturbe B. Mechanisms of disease: oxidative stress and inflammation in the pathogenesis of hypertension. Nature Clin Prac Nephrol. 2006;2:582–593. doi: 10.1038/ncpneph0283. [DOI] [PubMed] [Google Scholar]
- 25.Sandler AB, Johnson DH, Hertst RS. Anti-vascular endothelial growth factor monoclonals in non-small cell lung cancer. Clin Cancer Res. 2004;10:4258s–4262s. doi: 10.1158/1078-0432.CCR-040023. [DOI] [PubMed] [Google Scholar]
- 26.Holden SN, Eckhardt SG, Basser R, de Boer R, Rischin D, Green M, Rosenthal MA, Wheeler C, Barge A, Hurwitz HI. Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and VEGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol. 2005;16:1391–1397. doi: 10.1093/annonc/mdi247. [DOI] [PubMed] [Google Scholar]
- 27.Zangari M, Anaissie E, Stopeck A, Morimoto A, Tan N, Lancet J, Cooper M, Hannah A, Garcia-Manero G, Faderl S, Kantarjian H, Cherrington J, Albitar M, Giles FJ. Phase II study of SU5416, a small molecular vascular endothelial growth factor tyrosine kinase receptor inhibitor, in patients with refractory multiple myeloma. Clin Cancer Res. 2004;10:88–95. doi: 10.1158/1078-0432.ccr-0221-3. [DOI] [PubMed] [Google Scholar]
- 28.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA. Circulating angiogenic factors and the risk of pre-eclampsia. N Engl J Med. 2004;350:672–683. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 29.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal disease. J Clin Invest. 2003;111:707–716. doi: 10.1172/JCI17423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang DH, Kim YG, Andoh TF, Gordon KL, Suga SI, Mazzali M, Jefferson JA, Hughes J, Bennett W, Schreiner GF, Johnson R. Post-cyclosporin-mediated hypertension and nephropathy: amelioration by vascular endothelial growth factor. Am J Physiol Renal Physiol. 2001;280:F727–F736. doi: 10.1152/ajprenal.2001.280.4.F727. [DOI] [PubMed] [Google Scholar]
- 31.Yau HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia inducible factor-1α. Am J Pathol. 2003;163:2289–2301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pillebout E, Burtin M, Yuan HT, Briand P, Woolf AS, Friedlander G, Terzi F. Proliferation remodeling of the peritubular microcirculation after nephron reduction: association with the progression of renal lesion. Am J Pathol. 2001;159:547–560. doi: 10.1016/S0002-9440(10)61726-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu JW, Llnas MT, Wang J, Stockton A, Adair TH. Low dietary salt intake induces mRNA expression of vascular endothelial growth factor in kidneys of WKY rats. Am J Hypertension. 2002;15(4):128A–129A. P271. [Google Scholar]
- 34.Gu JW, Elam J, Sartin A, Li W, Roach R, Adair TH. Moderate levels of ethanol induce expression of vascular endothelial growth factor and stimulation angiogenesis. Am J Physiol Regulatory Integrative Comp Physiol. 2001;281:R368–R372. doi: 10.1152/ajpregu.2001.281.1.R365. [DOI] [PubMed] [Google Scholar]
- 35.Siegel AK, Kossmehl P, Planer M, Schulz A, Wehland M, Stoll M, Bruijin JA, de Heer E, Kreutz R. Genetic linkage of albuminuria and renal injury in Dahl salt-sensitive rats on a high-salt diet: comparison with spontaneously hypertensive rats. Physiol Genomics. 2004;18:218–225. doi: 10.1152/physiolgenomics.00068.2004. [DOI] [PubMed] [Google Scholar]
- 36.Farjah M, Washington TL, Roxas BP, Geenen DL, Danziger RS. Dietary NaCl regulates renal aminopeptidase N: relevance to hypertension in the Dahl rat. Hypertension. 2004;43:282–285. doi: 10.1161/01.HYP.0000111584.15095.8a. [DOI] [PubMed] [Google Scholar]
- 37.Mattson DL, Higgins DJ. Influence of dietary sodium intake on renal medullary nitric oxide synthase. Hypertension. 1996;27:688–692. doi: 10.1161/01.hyp.27.3.688. [DOI] [PubMed] [Google Scholar]
- 38.Titze J, Luft FC, Bauer K, Dietsch P, Lang R, Veelken R, Wagner H, Eckardt KU, Hilgers KF. Extrarenal Na+ balance, volume, and blood pressure homeostasis in intact and ovariectomized deoxycorticosterone-acetate salt rats. Hypertension. 2006;47:1101–1107. doi: 10.1161/01.HYP.0000221039.17735.1a. [DOI] [PubMed] [Google Scholar]
- 39.Dahl LK, Heine M, Tassinari L. Effects of chronic excess salt ingestion. Role of genetic factors in both doca-salt and renal hypertension. J Exp Med. 1963;118:605–617. doi: 10.1084/jem.118.4.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiBona GF, Sawin LL. Exaggerated natriuresis in experimental hypertension. Proc Soc Exp Biol Med. 1986;182:43–51. doi: 10.3181/00379727-182-42306. [DOI] [PubMed] [Google Scholar]
- 41.Praga M, Morales E. Renal damage associated with proteinuria. Kidney Int. 2002;62:S42–S46. doi: 10.1046/j.1523-1755.62.s82.9.x. [DOI] [PubMed] [Google Scholar]
- 42.Roy H, Bhardwaj S, Yla-Herttuala S. Biology of vascular endothelial growth factors. FEBS Letters. 2006;580:2879–2887. doi: 10.1016/j.febslet.2006.03.087. [DOI] [PubMed] [Google Scholar]
- 43.Pupilli C, Lasagni L, Romagnani P, Bellini F, Mannelli M, Misciglia N, Mavilia C, Vellei U, Serio M. Angiotensin II stimulates the synthesis and secretion of vascular permeability factor/vascular endothelial growth factor in human mesangial cells. Am J Soc Nephrol. 1999;10:245–255. doi: 10.1681/ASN.V102245. [DOI] [PubMed] [Google Scholar]
- 44.Shihab FS, Bennett WM, Isaac J, Yi H, Andoh TF. Angiotensin II regulation of vascular endothelial growth factor and receptors Flt-1 and KDR/Flk-1 in cyclosporine nephrotoxicity. Kidney Int. 2002;62:422–433. doi: 10.1046/j.1523-1755.2002.00452.x. [DOI] [PubMed] [Google Scholar]
- 45.Rizkalla B, Forbes JM, Cooper ME, Cao Z. Increased renal vascular endothelial growth factor and angiopoietins by angiotensin II infusion is mediated by both AT1 and AT2 receptors. Am J Soc Nephrol. 2003;14:3061–3071. doi: 10.1097/01.asn.0000099374.58607.c9. [DOI] [PubMed] [Google Scholar]
- 46.Ferrara N. Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int. 1999;56:794–814. doi: 10.1046/j.1523-1755.1999.00610.x. [DOI] [PubMed] [Google Scholar]
- 47.Schrijvers BF, Flyvbjerg A, DeVriese AS. The role of vascular endothelial growth factor (VEGF) in renal pathophysiology. Kidney Int. 2004;65:2003–2017. doi: 10.1111/j.1523-1755.2004.00621.x. [DOI] [PubMed] [Google Scholar]
- 48.Villegas G, Lange-Sperandio B, Tufro A. Autocrine and paracrine functions of vascular endothelial growth factor (VEGF) in renal tubular epithelial cells. Kidney Int. 2005;67:449–457. doi: 10.1111/j.1523-1755.2005.67101.x. [DOI] [PubMed] [Google Scholar]
- 49.Hara A, Wada T, Furuichi K, Sakai N, Kawachi H, Shimizu F, Shibuya M, Matsushima K, Yokoyama H, Egashira K, Kaneko S. Blockade of VEGF accelerates proteinuria, via decrease in nephrin expression in rat crescentic glomerulonephritis. Kidney Int. 2006;69:1986–1995. doi: 10.1038/sj.ki.5000439. [DOI] [PubMed] [Google Scholar]
- 50.Sugimoto H, Hamano Y, Charyta D, Cosgrove D, Kieran M, Sudhakar A, Kalluri R. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem. 2003;278:12605–12608. doi: 10.1074/jbc.C300012200. [DOI] [PubMed] [Google Scholar]
- 51.Hornig C, Barleon B, Ahmad S, et al. Release and complex formation of soluble VEGFR-1 from endothelial cells and biological fluids. Lab Invest. 2000;80:443–454. doi: 10.1038/labinvest.3780050. [DOI] [PubMed] [Google Scholar]