

Abstract
The aim of this study was to investigate the preparation, characterization, and encapsulation/release performance of an electrospun composite nanofiber mat. The hypothesis was that the composite nanofiber mat with nano-scaled drug particles impregnated in biocompatible and biodegradable polymer nanofibers can serve as an innovative type of tissue engineering scaffold with desired and controllable drug encapsulation/release properties. To test the hypothesis, the composite nanofiber mat electrospun from an emulsion consisting of poly (lactic-co-glycolic acid) (PLGA) Rhodamine B (a model compound to simulate drugs), sorbitan monooleate (Span-80, a non-ionic emulsifier/surfactant that is presumably non-toxic/safe for cell-growth), chloroform, DMF, and distilled water was prepared and characterized; and the Rhodamine B encapsulation/release profile in phosphate buffered saline (pH = 7.4) was recorded and analyzed. For comparison purposes, two additional nanofiber mats electrospun from (1) a solution containing PLGA and Rhodamine B, and (2) a solution containing PLGA, Rhodamine B, and Span-80 were also prepared and assessed as the control samples. The results indicated that the composite nanofiber mat electrospun from the emulsion had the most desired and controllable Rhodamine B encapsulation/release profile and the excellent morphological sustainability; thus, it could be utilized as both a drug encapsulation/release vehicle and a tissue engineering scaffold.
Keywords: Drug release, Electrospinning, Nanofiber, Non-woven mats, PLGA
1. Introduction
The encapsulation and controllable release of bioactive agents such as drugs are crucial for maximizing the safety, effectiveness, and reliability of these agents [1]. Among several methods for drug encapsulation, the spray-drying and solvent-evaporation techniques have been well studied due to their simple procedures and the potential for industrial scale-up [2]. The spray-drying technique is a straightforward method. A drug to be encapsulated and an amphipathic carrier are homogenized as a mixture in water (i.e., the slurry). The slurry is then fed into a spray drier heated to a temperature well above the boiling point of water. The spray-drying technique, however, is limited to the drugs that have relatively high molecular weight and are chemically stable at the elevated temperature, since those with small molecular weights tend to boil off in large quantities at the processing temperature. Comparatively, the solvent-evaporation technique is featured by the application of an additional aqueous phase which provides a mild processing condition for encapsulation and the convenience of controlling particle size in a wide range. However, this method makes the leakage of hydrophilic drugs unavoidable and thus becomes an obstacle to achieving the high encapsulation efficiency [3]. No matter which technique is adopted for drug encapsulation, the resultant products are usually in the powder form; and they cannot directly serve as tissue engineering scaffolds unless incorporated into a polymer matrix or sintered together [4,5]. On the other hand, biocompatible and biodegradable polymer fiber mats (particularly electrospun polymer nanofiber mats) are capable of serving as both drug encapsulation vehicles and tissue engineering scaffolds [6].
Electrospinning is a technique that utilizes the electric force to drive the spinning process and to produce polymer fibers [7-9]. Unlike conventional spinning methods that produce fibers with diameters in the micrometer range (5−50 μm), electrospinning is capable of producing fibers with diameters in the nanometer range (10−1000 nm). Unlike nanorods, nanotubes, and nanowires that are produced mostly by bottom-up methods, electrospun nanofibers are produced through a top-down nano-manufacturing process, which results in low-cost and continuous nanofibers that are relatively easy to align, assemble, and process into applications. Electrospun polymer nanofibers possess extraordinary properties including small diameters and the concomitant large specific surface areas, a high degree of structural perfection and the resultant superior mechanical properties [9]. Additionally, the non-woven mats made of electrospun polymer nanofibers offer a unique capability to control the pore sizes among nanofibers.
One promising application of electrospun polymer nanofiber mats is the cell-growth scaffold for regeneration of tissues [10]. This is due to the following four reasons: (1) the architecture of electrospun polymer nanofiber mats generally mimics the Extra Cellular Matrix (EMC) thus affects cell-growth positively; (2) the biocompatibility of fibers improves with decreasing diameters; it is noteworthy that the electrospun polymer nanofibers have diameters one to three orders of magnitude smaller than their conventional counterparts; (3) the porosity of electrospun polymer nanofiber mats (especially after being soaked in cell-growth fluids) also has a favorable influence on cell-growth; and (4) the mechanical properties of the nanofiber mats are sufficient for being cell-growth scaffolds. Many nanofiber mats electrospun from numerous synthetic and/or natural polymers including, but not limited to, poly (lactic acid) (PLA), poly (glycolic acid) (PGA), poly (ε-caprolactone) (PCL), poly (lactic-co-glycolic acid) (PLGA), proteins (e.g., collagen/gelatin), and polysaccharides (e.g., chitosan) have shown favorable results as scaffolds for growing various kinds of cells [9-11].
The conventional spin dopes for electrospinning are solutions in which all components are dissolved; the obtained nanofibers are structurally homogeneous, and they are unlikely to encapsulate bioactive agents as nano-scaled particles. Although the co-axial electrospinning technique can result in the formation of nanofibers having core-sheath structures with sheaths being biocompatible/biodegradable polymers and cores being encapsulated bioactive agents [12,13], the operational conditions have to be judiciously adjusted and it is often difficult to ensure desired results. Recently, electrospinning of emulsions has attracted growing interests [14-16]. The emulsions (particularly water-in-oil type of emulsions) usually contain a continuous phase that is a polymer (e.g., a biocompatible/biodegradable polymer) dissolved in organic solvent(s), and a separated/isolated phase (emulsion particles/droplets with sizes ranging from submicrons to microns) that is a bioactive agent (e.g., a drug) dissolved in water. Electrospinning of emulsions can produce composite nanofibers with nano-scaled drug particles surrounded/coated by emulsifiers/surfactants and impregnated in biocompatible and/or biodegradable polymers. Such type of composite nanofiber mats possesses the combined characteristics of a tissue engineering scaffold and a controllable drug encapsulation/release vehicle. The properties of the composite nanofiber mats (including fiber morphologies and structures, pore sizes among nanofibers, and others) can be tailored by carefully selecting polymers, drugs, solvents, emulsifiers/surfactants, and electrospinning processing conditions.
In this study, a composite nanofiber mat was prepared from electrospinning an emulsion made of PLGA, Rhodamine B, sorbitan monooleate (Span-80), chloroform, N,N-dimethylformamide (DMF), and distilled water. PLGA was selected because (1) it is one of the most well-studied polymeric materials for biomedical applications due to its excellent biocompatibility and biodegradability [17]; (2) the biodegradation time of PLGA can be controlled by adjusting the ratio of lactic versus glycolic acid units in the copolymer [17]; and (3) numerous publications have repeatedly demonstrated that the electrospun PLGA nanofiber mat is an excellent tissue engineering scaffold for growing various kinds of cells [18-22]. Rhodamine B was selected as a model compound to simulate drugs because its amount in solution can be accurately detected/measured by fluorescent spectroscopy; therefore, the encapsulation/release profiles of Rhodamine B from the electrospun nanofiber mats can be readily acquired. Span-80 was selected because it is a non-ionic emulsifier/surfactant widely used in food products and oral pharmaceuticals; therefore, we presume that it would be non-toxic/safe for cell-growth. For comparison purposes, two additional nanofiber mats electrospun from (1) a solution containing PLGA and Rhodamine B, and (2) a solution containing PLGA, Rhodamine B, and Span-80 were also prepared and assessed as the control samples. The morphologies of the nanofiber mats were examined by scanning electron microscopy (SEM); the surface hydrophilicity was measured by the water contact angle method; and the Rhodamine B encapsulation/release profiles from the electrospun nanofiber mats immersed in phosphate buffered saline (pH = 7.4) for various time periods were recorded and analyzed. The results indicated that the composite nanofiber mat electrospun from the emulsion had the most desired and controllable Rhodamine B encapsulation/release profile and the excellent morphological sustainability. Thus, we envision that it could be utilized as both a drug encapsulation/release vehicle and a tissue engineering scaffold.
2. Experimental
2.1. Materials
PLGA (weight-average molecular weight Mw = 50,000−75,000 g/mol) with the mass ratio of lactic versus glycolic acid units being 85/15, Rhodamine B, Span-80, DMF, chloroform, and phosphate buffered saline (PBS, pH = 7.4) were purchased from the Sigma-Aldrich Co. (Milwaukee, WI). The distilled water used in this study was made using a Barnstead™ FI-Streem™ III 4LPH Bi-Distiller purchased from the Thermo Fisher Scientific Inc. (Waltham, MA). The chemicals were used without further purification.
2.2. Preparation of spin dopes
To prepare the emulsion (spin dope “C” in Figure 1), Rhodamine B was first dissolved in distilled water to make a 5 % (mass fraction) solution; Span-80 (25 mg) and PLGA (1.5 g) was then dissolved in a mixture solvent made of chloroform (8.25 g) and DMF (2.75 g). Subsequently, the aqueous solution was added into the organic solution dropwise, while the mixture was mechanically stirred at 200 rpm. The mass fraction of Rhodamine B in the prepared emulsion was 0.1 %, and the emulsion was uniform and stable. The control samples of a solution containing 12 % PLGA and 0.1 % Rhodamine B (spin dope “A” in Figure 1) and a solution containing 12 % PLGA, 0.1 % Rhodamine B, and 0.2 % Span-80 (spin dope “B” in Figure 1) were prepared by simply dissolving the chemicals in the mixture solvent made of chloroform and DMF with the mass ratio being 3/1.
Figure 1.
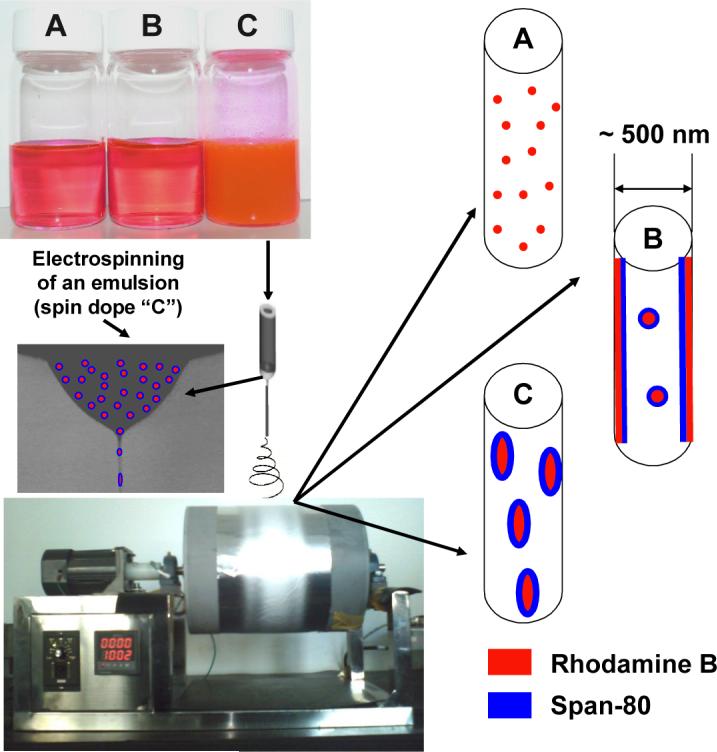
The prepared spin dopes, the adopted electrospinning setup, and the schematic representation of the three types of nanofibers.
2.3. Electrospinning of nanofiber mats
The experimental set-up for conducting electrospinning is schematically shown in Figure 1. A specially designed spinneret was utilized for conducting electrospinning. The spinneret consisted of a high-density polypropylene tube (an inner diameter of 1.0 inch) and a stainless steel hemispherical head which had an orifice of 0.4 mm diameter at the center. The electrospinning setup also consisted of a high voltage power supply purchased from the Gamma High Voltage Research, Inc. (Ormond Beach, FL) and a laboratory-built roller with a diameter of 10 inches. During electrospinning, a positive high voltage of 27 kV was applied through a metal rod to the spin dope held inside the spinneret. Electrospun nanofibers were collected on the roller covered with electrically grounded aluminum foil, which was placed 10 inches below the spinneret. The rotational speed of the roller during electrospinning was set at 100 rpm. In this manner, electrospinning was extremely stable; and the electrospinning jet could run continuously without breaking for several hours. The obtained mat on the aluminum foil was thus hypothetically a single nanofiber loosely aligned along the rolling direction [8,9]. The electrospinning was carried out under the ambient conditions; and the collected nanofiber mats had a thickness around 50 μm and a mass per unit area of approximately 30 g/m2.
2.4. Water contact angle measurements
Water contact angles of electrospun nanofiber mats were measured using a Contact Angle Analyzer made by the Data Physics Corp. (San Jose, CA). During the measurements, samples of the nanofiber mats were first cut into square specimens with the size of 1 cm x 1 cm, followed by placing them on a testing plate. Subsequently, 0.05 ml distilled water was carefully dropped onto the prepared specimens. The contact angles between water droplets and the nanofiber mat specimens were measured using photos taken at various time periods (1, 5, 10, 30, and 60 seconds). Five measurements at different positions were carefully conducted for each specimen; and the reported data were the mean value with the error bar representing one standard deviation.
2.5. Encapsulation/release studies
The encapsulation/release profiles of the composite nanofiber mat electrospun from the emulsion as well as the two control samples were acquired by measuring the fluorescent emissions of Rhodamine B in PBS (pH = 7.4). During the studies, three kinds of electrospun nanofiber mats were first cut into pieces with the same mass of 0.675 g and then immersed into 1 L PBS individually. The specimens for the fluorescent measurements were prepared by collecting 1 ml solutions after various immersion time periods followed by diluting them to 10 ml using PBS. A Perkin-Elmer L-55 fluorometer equipped with a Xe lamp as the light source was employed for the study. The excitation wavelength was set at 510 nm, and the spectra were obtained by scanning the samples from 520 to 800 nm at the speed of 500 nm/min. The band-pass of the monochromator was set at 5 nm, and a 515 nm emission cutoff filter was utilized. The emission areas were calculated by integrating the spectra from 520 to 800 nm. Since the integrated areas were proportional to the concentrations of Rhodamine B in PBS, they were utilized for plotting the encapsulation/release profiles.
2.6. Morphological examinations
A Zeiss Supra 40VP field-emission SEM was employed to examine morphologies of the three kinds of electrospun nanofiber mats before and after immersion in PBS for various time periods (1, 7, and 50 days). During the SEM examinations, the specimens were not sputter-coated with gold/carbon; so that the acceleration voltage was set at a low level of 2.0 kV to minimize charge accumulations. This was due to the reason that the heat generated by the sputter-coating process could vary the nanofiber mat morphology.
3. Results and discussion
3.1. Preparation and morphological examination
The detailed methods and procedures for preparation of the electrospun nanofiber mats have been described in the Experimental section. The spin dopes “A”, “B”, and “C” inside the three bottles in Figure 1 had no distinguishable variations during the experimental time period of several days. This suggested that the prepared emulsion (in bottle “C”) was relatively uniform and stable. Although Rhodamine B is soluble in both water and chloroform/DMF, we believe that the majority was in the aqueous phase because of the procedure for preparing the emulsion. The chloroform/DMF mixture was selected as the solvent for the organic phase because chloroform is an excellent solvent for PLGA while DMF is an excellent solvent for electrospinning probably due to its high dielectric constant [8]. If chloroform alone was used as the solvent, the electrospinning process would be unstable; and the obtained nanofibers would have diameters larger than 1 μm with many beads and/or beaded nanofibers [23]. During this study, several mixtures with different chloroform/DMF mass ratios were explored as the solvents for preparation of the spin dopes; and the mixture solvent with the chloroform/DMF mass ratio of 3/1 was identified as the optimal one based upon the stability of the electrospinning process and the morphology of the nanofibers. The optical microscopy (pictures not shown) indicated that the emulsion particles/droplets in spin dope “C” had diameters ranging from submicrons to microns. Presumably, those spherically shaped emulsion particles/droplets would be significantly elongated and then broken into smaller particles/droplets during the process of electrospinning (particularly during “bending instability” [8]); and would eventually result in the formation of the composite nanofibers as shown schematically in Figure 1. It is noteworthy that the electrospinning jet can be elongated up to 10,000 times in tens of milliseconds during electrospinning [8]. This is accompanied by the rapid jet-solidification, i.e., the evaporation of solvents (such as DMF, chloroform, and water in this study) is extremely fast; over 99 % of the solvent(s) is removed during or shortly after (< 0.1 second) “bending instability” starts [8]. The rapid solvent evaporation during electrospinning occurs under special conditions including (1) the jet has micron- or submicron-scaled diameter, (2) the jet carries excess charges, and (3) the solvents evaporate under the influence of a strong electric field. Nonetheless, the volatilities of solvents/solutes still significantly affect the jet solidification process, and further influence the morphological properties of electrospun nanofibers. Span-80 is a viscous liquid at the room temperature, and it is expected to have relatively high mobility in the electrospinning jet. Since the Span-80 molecule has a highly hydrophilic end that can strongly interact with the ionic molecule of Rhodamine B, we believe that Span-80/Rhodamine B complexes would carry excess charges and move to the outer region of the electrospinning jet due to the charge repulsion. Therefore, unlike the nanofibers electrospun from the solution containing PLGA and Rhodamine B (spin dope “A”) that are structurally homogeneous, the nanofibers electrospun from the solution containing PLGA, Rhodamine B, and Span-80 (spin dope “B”) are expected to have much higher concentration of Span-80/Rhodamine B at the outer region. Such a situation is not applicable to the composite nanofibers electrospun from the emulsion (spin dope “C”) because emulsion particles/droplets (i.e., water droplets containing Rhodamine B and surrounded/stabilized by Span-80) are two to three orders of magnitude larger/heavier than the Span-80/Rhodamine B complexes, and the movement of the emulsion particles/droplets in the electrospinning jet is considerably slower than that of the complexes. As described before, the jet solidifies in tens of milliseconds during electrospinning; thus the dispersion of nano-scaled Rhodamine B particles (surrounded/coated by Span-80 molecules) in the composite nanofibers electrospun from the emulsion is reasonably uniform. The representative morphologies of the nanofiber mats electrospun from the spin dopes “A”, “B”, and “C” are shown in Figure 2. It is evident that the diameters of nanofibers were 500 nm or less, and the nanofibers were smooth (without microscopically identifiable beads and/or beaded-nanofibers [23]) and uniform (with relatively small variations in diameter). It is noted that the electrospun nanofiber mats as shown in Figure 2 were deliberately collected for the SEM examinations; the actual nanofiber mats used for the following studies were much denser.
Figure 2.

SEM images showing representative morphologies of the nanofiber mats electrospun from the spin dopes “A”, “B”, and “C”.
3.2. Water contact angle characterization
The hydrophilicity of electrospun nanofiber mats could play an important role in determination of their overall performances as tissue engineering scaffolds, and was characterized in this study by the water contact angle method. The detailed procedures have been described in the Experimental section. The inset in Figure 3 shows the optical observations of the water contact angles immediately (approximately 1 second) after the water droplets were placed on the nanofiber mats. It was obvious that the nanofiber mat electrospun from the spin dope “A” had the lowest hydrophilicity (i.e., the largest water contact angle), while the one electrospun from the spin dope “B” had the highest hydrophilicity (i.e., the smallest water contact angle). Although the hydrophilicity of the nanofiber mat electrospun from the emulsion (spin dope “C”) was similar to that of the one electrospun from the spin dope “B”, it took a few seconds for the water droplets to reach the equilibrium stage. As shown in Figure 3, approximately 30 seconds after the water droplets were placed on the nanofiber mats, the contact angle of the nanofiber mat “A” was close to 90°, while those of the nanofiber mats “B” and “C” were only a few degrees. These results from the water contact angle characterizations further support our previous speculations about the morphological structures of the three types of electrospun nanofiber mats, since Span-80 molecules and Span-80/Rhodamine B complexes are much more hydrophilic than PLGA. Because scaffolds with higher hydrophilicity are generally more favorable for cell-growth, we believe that the composite nanofiber mat electrospun from the emulsion would outperform the neat PLGA nanofiber mat, assuming Span-80 is not toxic to cells. Additionally, the results also suggested that the electrospinning of emulsions could be utilized as a general approach for tailoring the hydrophilicity of nanofiber mats.
Figure 3.

The water contact angle variations of the nanofiber mats electrospun from the spin dopes “A” (symbol “▲”), “B” (symbol “■”), and “C” (symbol “○”). Each datum is the mean value of five measurements with the error bar representing one standard deviation. The inset shows the optical observations of the water contact angles immediately (approximately 1 second) after the water droplets were placed on the nanofiber mats.
3.3. Encapsulation/release and morphological sustainability studies
An important objective of this study was to investigate whether the composite nanofiber mat electrospun from the emulsion could serve not only as a cell-growth scaffold but also as a drug encapsulation/release vehicle. Figure 4 shows the Rhodamine B encapsulation/release profiles acquired from the three types of electrospun nanofiber mats that were immersed in PBS for various time periods. Because Rhodamine B was uniformly encapsulated as individual and/or aggregated molecules in the electrospun nanofiber mat “A”, it could not be released until PLGA started to degrade. As shown in Figure 4, there was almost no Rhodamine B that could be detected until the mat was immersed in PBS for approximately 200 hours. Such an encapsulation/release profile is certainly not desired, because the first 200 hours could be critical for cell-growth and the lack of bioactive agents would not benefit the regeneration of tissues. The encapsulation/release profile of the electrospun nanofiber mat “B” is not desired either. As shown in Figure 4, the Rhodamine B in the electrospun nanofiber mat “B” was rapidly released (burst) within several hours after the mat was immersed in PBS; whereas after that, almost no more Rhodamine B could be further released as evidenced by the amount/concentration of Rhodamine B stayed almost constantly for a month period. This can be explained as follows: (1) the electrospun nanofiber “B” has an outer region that contains a high concentration of Span-80 and Rhodamine B, therefore Rhodamine B could be quickly released from the surface layer of the nanofiber shortly after the mat was immersed in PBS; and (2) after the initial burst, the Span-80 on the surface could prevent the penetration/diffusion of water molecules into the nanofiber and further prevent the degradation of PLGA, therefore hinder the release of the Rhodamine B impregnated inside the nanofiber. Contrastively, the encapsulation/release profile of the electrospun nanofiber mat “C” is much more desired. As shown in Figure 4, a controllable amount of Rhodamine B in the electrospun nanofiber mat “C” was quickly released within a few hours after the mat was immersed in PBS; after that, more Rhodamine B could be further controllably released as evidenced by the amount/concentration of Rhodamine B kept increasing for at least a month. The initial releasing amount as well as the following releasing rate could be judiciously controlled through adjusting the amount of aqueous phase in the emulsion and/or through adjusting the Rhodamine B concentration in the aqueous phase (results not shown).
Figure 4.
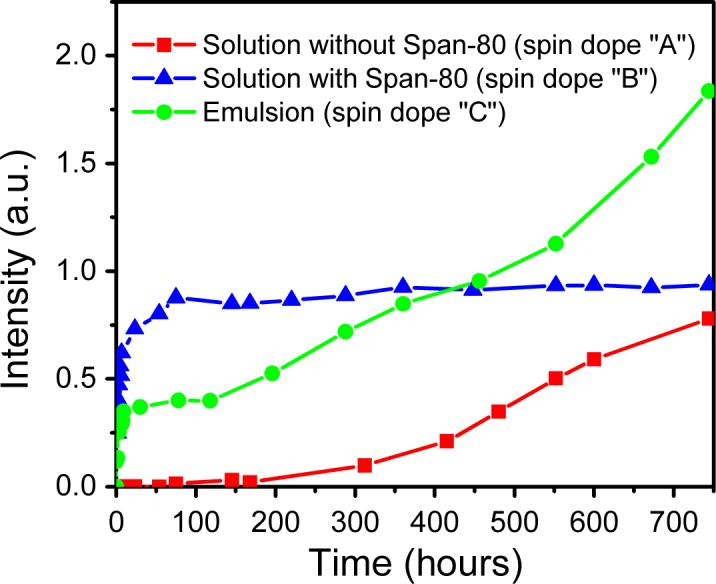
The Rhodamine B encapsulation/release profiles of the nanofiber mats electrospun from the spin dopes “A” (symbol “■”), “B” (symbol “▲”), and “C” (symbol “•”) after various hours’ immersion in PBS (pH = 7.4).
Additionally, we also investigated the morphological sustainability of the electrospun nanofiber mats after immersion in PBS for various time periods. As shown in Figure 5, unlike the electrospun nanofiber mat “A” that noticeably degraded after 50 days’ immersion in PBS (image “A”), the electrospun nanofiber mats “B” and “C” retained the nanofiber mat morphology (images “B” and “C”). The degradation of the mat “A” could be easily understood since 50 days’ immersion in PBS would allow PLGA to partially degrade. For the mat “B”, as described above, Span-80 molecules covered on the nanofiber surface could prevent the penetration/diffusion of water molecules, thus could prevent the degradation of PLGA. For the mat “C”, Span-80 acted as the stabilizer for the impregnated Rhodamine B nanoparticles in the composite nanofibers. After immersion in PBS, the Span-80 molecules would be released together with the Rhodamine B molecules for the nanoparticles that were close to the surface of the nanofibers. Since Span-80 molecules have both hydrophilic and hydrophobic ends, they would attach on the surface of the nanofibers and delay the degradation of PLGA. As described in the Introduction section, numerous publications have repeatedly demonstrated that the electrospun PLGA nanofiber mat is an excellent tissue engineering scaffold for growing various kinds of cells [18-22]; we therefore believe that the composite nanofiber mat electrospun from the emulsion can serve as an innovative type of tissue engineering scaffold with desired and controllable drug encapsulation/release properties. It is noteworthy that several parallel experiments were carried out during this study and the three encapsulation/release profiles as shown in Figure 4 are representative. The preparation and assessment of the three types of electrospun nanofiber mats in each of the parallel experiments were carried out under exactly the same condition. The reason that there is no standard deviation included in Figure 4 is because the actual time spots (after the nanofiber mats being immersed in PBS) to collect the samples for the fluorescent analyses were not exactly the same in each of the parallel experiments.
Figure 5.

SEM images showing representative morphologies of the nanofiber mats electrospun from the spin dopes “A”, “B”, and “C” after immersion in PBS (pH = 7.4) for 50 days.
4. Conclusions
In this study, a composite nanofiber mat was prepared by electrospinning an emulsion made of PLGA, Rhodamine B, Span-80, chloroform, DMF, and distilled water. Its morphology was examined by SEM, its surface hydrophilicity was measured by the water contact angle method, and its Rhodamine B encapsulation/release profile in PBS (pH = 7.4) was recorded and analyzed. For comparison purposes, two additional nanofiber mats electrospun from (1) a solution containing PLGA and Rhodamine B, and (2) a solution containing PLGA, Rhodamine B, and Span-80 were also prepared and assessed as the control samples. The results indicated that the composite nanofibers electrospun from the emulsion had diameters of 500 nm or less, and they were smooth (without microscopically identifiable beads and/or beaded-nanofibers) and uniform (with relatively small variations in diameter). The encapsulation/release studies of the three types of electrospun nanofiber mats revealed that the composite nanofiber mat electrospun from the emulsion exhibited the most desired and controllable release performance as well as the excellent morphological sustainability. This is because the composite nanofibers possess the unique morphological structure of nano-scaled Rhodamine B particles surrounded/coated with Span-80 and impregnated in PLGA matrix. Compared with the control samples of the PLGA nanofiber mats, which have been repeatedly demonstrated by numerous publications to be excellent tissue engineering scaffolds for growing various kinds of cells, the prepared PLGA-containing composite nanofiber mat can not only serve as an innovative type of tissue engineering scaffold, but also possess the desired and controllable drug encapsulation/release properties. We believe that the electrospinning of emulsions (particularly water-in-oil type of emulsions) containing bioactive agents and biocompatible/biodegradable polymers could be a general approach for preparation of innovative tissue engineering scaffolds with desired and controllable drug encapsulation/release properties.
Acknowledgements
This research was supported by the National Institute of Dental and Craniofacial Research (R03 DE16042), and by the “BioMedical Engineering (BME) Program” at the South Dakota School of Mines and Technology (SDSM&T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langer R. Biomaterials in drug delivery and tissue engineering: one laboratory's experience. Acc Chem Res. 2003;3:94–101. doi: 10.1021/ar9800993. [DOI] [PubMed] [Google Scholar]
- 2.Park TG, Alonso MJ, Langer R. Controlled-release of proteins from poly (L-lactic acid) coated polyisobutylcyanoacrylate microcapsules. J Appl Polym Sci. 1994;52:1797–1807. [Google Scholar]
- 3.Freitas S, Merkle HP, Gander B. Microencapsulation by solvent extraction/evaporation: reviewing the state of the art of microsphere preparation process technology. J Controlled Release. 2005;102:313–332. doi: 10.1016/j.jconrel.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 4.Hu Y, Hollinger JO, Marra KG. Incorporation of polymer microspheres within fibrin scaffolds. J Drug Targeting. 2001;9:431–438. doi: 10.3109/10611860108998777. [DOI] [PubMed] [Google Scholar]
- 5.Borden M, Attawia M, Laurencin CT. The sintered microsphere matrix for bone tissue engineering: in vitro osteoconductivity studies. J Biomed Mater Res. 2002;61:421–429. doi: 10.1002/jbm.10201. Part A. [DOI] [PubMed] [Google Scholar]
- 6.Venugopal J, Ramakrishna S. Applications of polymer nanofibers in biomedicine and biotechnology. Appl Biochem Biotechnol. 2005;125:147–157. doi: 10.1385/abab:125:3:147. [DOI] [PubMed] [Google Scholar]
- 7.Ramakrishna S, Fujihara K, Teo W, Lim T, Ma Z. An introduction to electrospinning and nanofibers. World Scientific Publishing Co. Pte. Ltd.; Singapore: 2005. [Google Scholar]
- 8.Fong H. Electrospun polymer, ceramic, carbon/graphite nanofibers and their applications. In: Nalwa HS, editor. Polymeric nanostructures and their applications. American Scientific Publishers; Stevenson Ranch, California: 2007. pp. 451–474. [Google Scholar]
- 9.Greiner A, Wendorff JH. Electrospinning: a fascinating method for the preparation of ultrathin fibers. Angew Chem Int Ed. 2007;46:5670–5703. doi: 10.1002/anie.200604646. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Laurencin CT, Caterson EJ, Tuan RS, Ko FK. Electrospun nanofibrous structure: a novel scaffold for tissue engineering. J Biomed Mater Res. 2002;60:613–621. doi: 10.1002/jbm.10167. Part A. [DOI] [PubMed] [Google Scholar]
- 11.Barnes CP, Sell SA, Boland ED, Simpson DG, Bowlin GL. Nanofiber technology: designing the next generation of tissue engineering scaffolds. Adv Drug Deliv Rev. 2007;59:1413–1433. doi: 10.1016/j.addr.2007.04.022. [DOI] [PubMed] [Google Scholar]
- 12.Sun ZC, Zussman E, Yarin AL, Wendorff JH, Greiner A. Compound Core-Shell Polymer Nanofibers by Co-Electrospinning. Adv Mater. 2003;15:1929–1932. [Google Scholar]
- 13.Zhang YZ, Wang X, Feng Y, Li J, Lim CT, Ramakrishna S. Coaxial electrospinning of fluorescein isothiocyanate-conjugated bovine serum albumin-encapsulated poly(ε-caprolactone) nanofibers for sustained release. Biomacromolecules. 2006;7:1049–1057. doi: 10.1021/bm050743i. [DOI] [PubMed] [Google Scholar]
- 14.Sanders EH, Kloefkorn R, Bowlin GL, Simpson DG, Wnek GE. Two-phase electrospinning from a single electrified jet: microencapsulation of aqueous reservoirs in poly(ethylene-co-vinyl acetate) fibers. Macromolecules. 2003;36:3803–3805. [Google Scholar]
- 15.Xu X, Yang L, Xu X, Wang X, Chen X, Liang Q, Zeng J, Jing X. Ultrafine medicated fibers electrospun from W/O emulsions. J Controlled Release. 2005;108:33–42. doi: 10.1016/j.jconrel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 16.Qi H, Hu P, Xu J, Wang A. Encapsulation of drug reservoirs in fibers by emulsion electrospinning: morphology characterization and preliminary release assessment. Biomacromolecules. 2006;7:2327–2330. doi: 10.1021/bm060264z. [DOI] [PubMed] [Google Scholar]
- 17.Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 18.Zong X, Ran S, Kim KS, Fang D, Hsiao BS, Benjamin Chu. Structure and morphology changes during in vitro degradation of electrospun poly (glycolide-co-lactide) nanofiber membrane. Biomacromolecules. 2003;4:416–423. doi: 10.1021/bm025717o. [DOI] [PubMed] [Google Scholar]
- 19.Zong X, Bien H, Chung CY, Yin L, Fang D, Hsiao BS, Chu B, Entcheva E. Electrospun fine-textured scaffolds for heart tissue constructs. Biomaterials. 2005;26:5330–5338. doi: 10.1016/j.biomaterials.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 20.You Y, Min BM, Lee SJ, Lee TS, Park WH. In vitro degradation behavior of electrospun polyglycolide, polylactide, and poly (lactide-co-glycolide) J Appl Polym Sci. 2005;95:193–200. [Google Scholar]
- 21.Sahoo S, Ouyang H, Goh JCH, Tay TE, Toh SL. Characterization of a novel polymeric scaffold for potential application in tendon/ligament tissue engineering. Tissue Engineering. 2006;12:91–99. doi: 10.1089/ten.2006.12.91. [DOI] [PubMed] [Google Scholar]
- 22.Yoshimoto H, Shin YM, Terai H, Vacanti JP. A biodegradable nanofiber scaffold by electrospinning and its potential for bone tissue engineering. Biomaterials. 2003;24:2077–2082. doi: 10.1016/s0142-9612(02)00635-x. [DOI] [PubMed] [Google Scholar]
- 23.Fong H, Chun I, Reneker DH. Beaded nanofibers formed during electrospinning. Polymer. 1999;40:4585–4592. [Google Scholar]