

Abstract
Synthesis of a fluorescently labelled (dansylated) linear α(1→6)-linked octamannan, using glycosyl fluoride donors and thioglycosyl acceptors is described. A selective and convergent two-stage activation progression was executed to construct di-, tetra and octa-mannosyl thioglycosides in three glycosylation steps with excellent yield. Further a 5-N,N-Dimethylaminonaphthalene-1-sulfonamidoethyl (dansyl) group was coupled to 1-azidoethyl octamannosyl thioglycoside. Global deprotection of the coupled product afforded the desired dansylated homo-linear α(1→6)-linked octamannan.
Mannans are constituents of many bacterial and fungal cell walls.1 They are functional components of proteins and play important roles in defining the structures of proteins, their stabilization under physiological conditions, tuning of enzymatic activities, cell–cell recognition and in the adhesion of the microorganism to host cells.2 The α(1→6)-linked oligomannans are found in the cell wall polysaccharides of yeast.3 Polysaccharides, especially oligomannan, can be used to deliver drugs, genes and antigens through polysaccharide receptors present in cells and macrophages.4 Macrophages are known to express high levels of specific polysaccharide receptors, e.g. mannan, glucan, and galactin receptors, on their membranes that generally bind neutral or charged polysaccharides and internalize these ligands.5 Receptor mediated delivery of drug–polysaccharide conjugates is an approach that can deliver small and effective amounts of drugs specifically to target organisms, minimizing patient exposure and potential toxic side effects. We have recently demonstrated the selective, receptor–mediated delivery of an antibacterial drug to macrophages infected with Mycobacterium tuberculosis via conjugation of moxifloxacin with 1,3-β-glucan.6
Drug loading on commercially available high molecular weight polysaccharides is very low and it is attributed to their poor solubility in solvents during the drug conjugation reactions. Therefore, we describe here a simple and efficient method for the synthesis of a low molecular weight homo-linear α(1→6)-linked octamannan fluorescent probe 1 (figure 1) that may be utilized to study uptake by macrophages through the mannan receptor present on the plasma membrane. Further, in the future, such conjugation can be exploited for selective receptor–mediated delivery of drugs. We have previously synthesized and reported dansylated disaccharide fluorescent probes to study glycosyltransferases in the biosynthesis of M. tuberculosis cell wall polysaccharides.7 Herein, the assembly of dansylated oligomannan fluorescent probe 1 was approached from synthetic homo-linear α(1→6) thiooctamanosyl glycoside 2, 1-azidoethanol 3 and commercially available dansyl chloride 4 fragments as represented in Figure 1.
Figure 1.
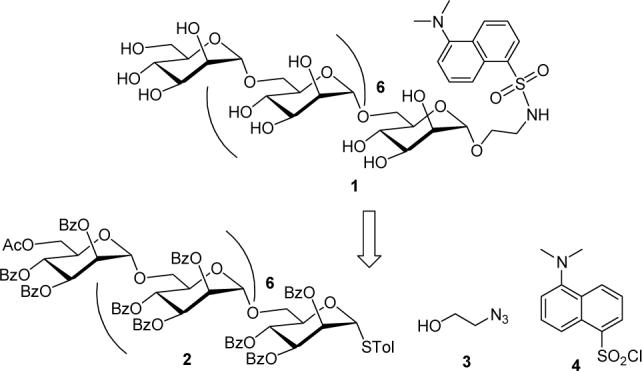
Target α(1→6)-linked octamannan fluorescent probe 1 and its precursors.
Branched oligomannan syntheses are reported in the literature using several different methodologies.8 Specifically, the homo-linear α(1→6) oligomannans were synthesized by glycosylation using a trichloroacetimidate donor glycoside.9 The strategy of combining the chemistry of thioglycosides with that of glycosyl fluorides10 for the synthesis of oligosaccharides known as the two-stage activation procedure (Figure 2) was developed by Nicolaou et. al.11 Briefly, in the activation stage one, the stable thioglycoside is converted to the more reactive glycosyl fluoride donor by treatment with N-bromosuccinimide (NBS) and diethylaminosulfur trifluoride (DAST). In stage two, the glycosyl fluoride is then reacted with a thioglycoside acceptor to produce a thiodisaccahride for further promulgation of the oligosaccharide chain. Reiteration of this process of converting thiooligosaccharide to a glycosyl fluoride and their coupling with acceptor thioglycosides straightforwardly produces chain elongation (figure 2).
Figure 2.
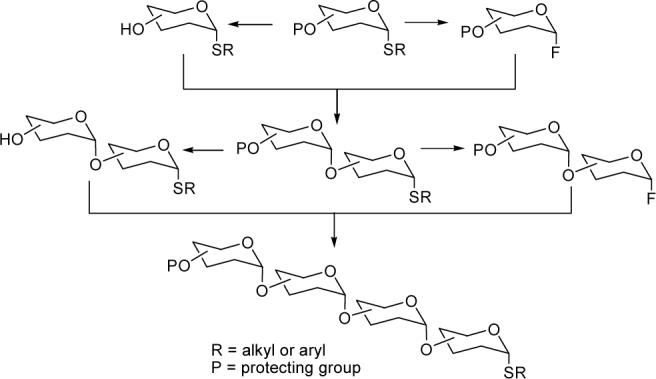
Two stage activation of thioglycoside for glycoside bond formation. Other protected OH's on sugars are omitted for simplification.
We executed the synthesis of the desired p-thiotolyl α(1→6) octamannoside 2 using a convergent two-stage activation and iterative orthogonal glycosylation technique starting from the coupling of thioglycoside acceptor 5 and glycosyl fluoride donor 6 (Figure 3). The synthesis of mannose building blocks 5 and 6 was achieved from thioglycoside 7 as reported earlier by us.12 However, the synthesis of thioglycoside 7 was accomplished from the known synthon 1,6-di-O-acetyl-2,3,4-tri-O-benzoyl-α-D-mannose (8) starting from commercially available D-mannose in an 88% overall yield.13 Selective de-acetylation of 7 to the acceptor glycoside 5 was achieved by the overnight reaction with an AcCl/MeOH/CH2Cl2 (0.1:20:20 v/v, 12.0 mL/mmol) mixture.12
Figure 3.
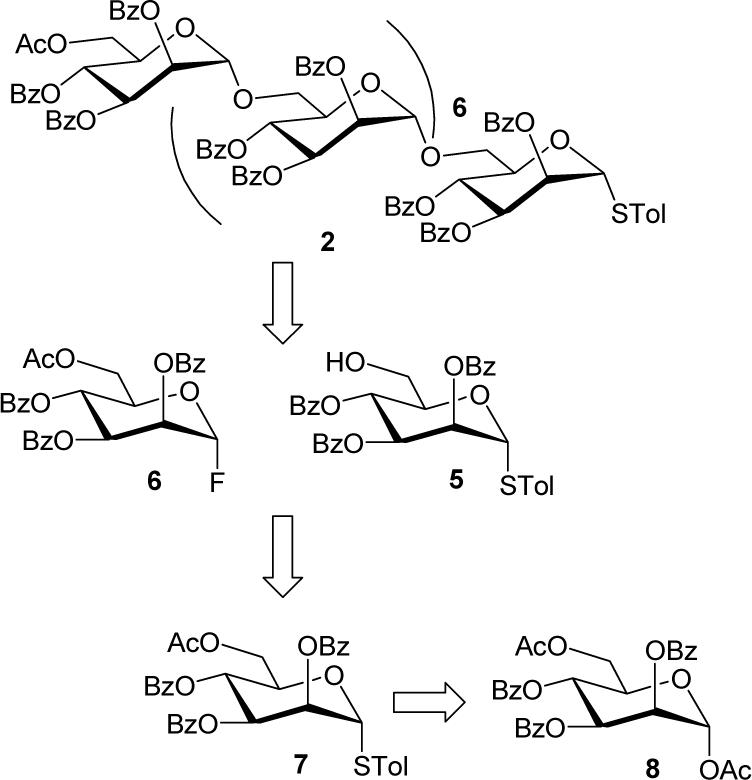
Synthetic plan for the synthesis of α(1→6)-linked octamannosyl thioglycoside 2
Following the synthesis of starting mannose synthons 5 and 6, the synthesis of 2 was carried out as illustrated in Scheme 1. In the first step of the glycosylation sequence, the p-thiotolyl disaccharide 9 was synthesized in high yield by selective anomeric activation of glycosyl fluoride 6 by AgClO4 and SnCl2 in CH2Cl2 and its coupling with the acceptor 5.12 The exclusive formation of α(1→6)-linked disaccharide 6 was supported by its analytical and spectral analysis.12
Scheme 1.
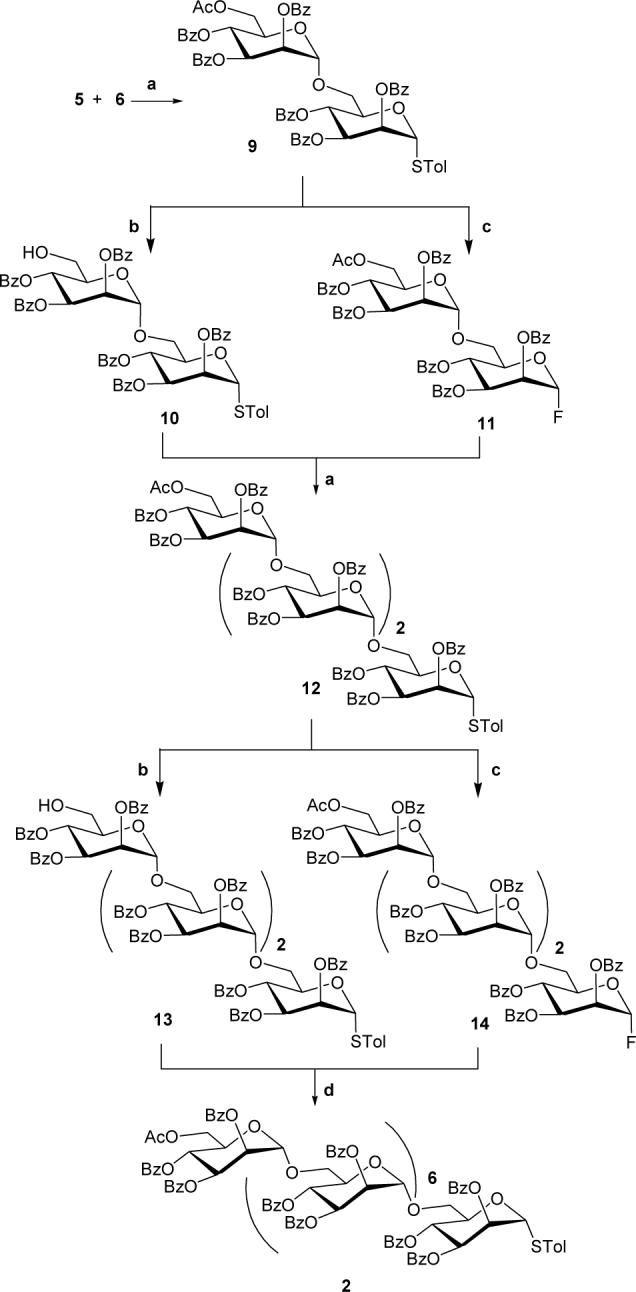
Reactions and Reagents. (a) AgClO4, SnCl2, dry CH2Cl2, 4Ǻ Mol sieves, rt, overnight, 9: 97%, 12: 80%. (c) AcCl/MeOH/CH2Cl2 (1:20:20 v/v), rt, overnight, 10: quantitative yield, 13: 86%. (b) DAST, NBS, dry CH2Cl2, −20 °C, 4 h, 11: 91%, 14: 84%. (d) Cp2HfCl2, AgOTf, dry CH2Cl2, 4A Mol sieves, rt, overnight, 61%.
Next, for the assembly of α(1→6)-linked tetramannosyl thioglycoside 12, the disaccharide 9 was converted to acceptor thiodisaccharide 10 and donor 1-fluorodisaccharide 11. Deacetylation of 9 by the overnight reaction with an AcCl/MeOH/CH2Cl2 mixture produced the desired acceptor disaccharide 10 in quantitative yield. In the 1H and 13C NMR spectra of compound 10, the absence of signals from the acetate group as seen in compound 9, confirmed the selective deacetylation. The disaccharide 9 was also converted to the donor fluoride 11 in 91% yield using DAST and NBS at −20 °C. The structure of 11 was supported by the 1H NMR spectra in which two characteristic peaks in thiotolyl glycosides [doublet at δ 7.20 ppm from two protons of tolyl ring and a singlet at δ 2.25 ppm from methyl] were not observed. The α-stereochemistry at the anomeric carbon was supported by a C-1 signal at δ 105.21 ppm (d, JC-1,F = 222.1 Hz) in the 13C NMR spectrum of 11, whereas, the signal at δ 98.11 ppm was attributable to the other anomeric carbon. In the 1H NMR spectrum of 11, the H-1 signal was found obscured with other protons in the multiplet at δ 5.92 ppm, however, the anomeric proton H-1′ was observed δ 5.15 ppm (J = 1.4 Hz).
With the acceptor disaccharide 10 and fluoride donor disaccharide 11 in hand, we next performed the [2+2] glycosylation reaction to access α(1→6)-linked tetramannosyl thioglycoside 12. The coupling reaction between glycosides 10 and 11 was carried out at room temperature with promotors AgClO4 and SnCl2 in CH2Cl2 over 4Ǻ molecular sieves and resulted in the desired glycoside 12 in 80% yield after purification by SiO2 column chromatography. The 1H NMR spectrum of 12 in CDCl3 showed four anomeric protons at δ 5.77, 5.25, 5.02 and 4.85 ppm as singlets suggesting α-glycosylation. In the 13C NMR spectrum of 12, the four anomeric carbons were observed at δ 98.37, 97.99, 97.79 and 86.91 ppm. The final structural confirmation was obtained by FABMS analysis of 12, which showed a peak at 2085.0 [M+Na]+, corresponding to the molecular formula C117H98O33SNa.
The α(1→6)-linked tetramannosyl thioglycoside 12 was further converted to thioglycosyl acceptor 13 and glycosyl fluoride donor 14 in 96% and 84% yields by similar reactions performed for the syntheses of glycosides 10 and 11 respectively. The characteristic acetate and thiotolyl groups were found absent in the 1H NMR and 13C NMR spectra of compounds 13 and 14 respectively. In the 13C NMR spectra of glycosyl fluoride 14, the four anomeric carbon signals were observed at δ 105.21 (d, JC-1,F = 223.0 Hz), 98.38, 97.92 and 97.56 ppm. Finally, the FABMS analyses of glycosides 13 and 14 confirmed the formation of these products.
Next, we pursued the [4+4] glycosylation reaction to access α(1→6)-linked octamannosyl p-tolylthioglycoside 2. The glycosyl donor 14 was treated with glycosyl acceptor 13 in the presence of promoters AgClO4 and SnCl2 in CH2Cl2. Even after 48 hrs at room temperature, only 20% yield of the desired octasaccharide 10 was achieved along with the recovery of unreacted acceptor glycoside 13. The glycosylation reaction between 13 and 14 was next performed with coupling reagents Cp2HfCl2 and AgOTf in CH2Cl2 overnight at room temperature. Octamannoside 2 was produced in 61% yield after purification by SiO2 column chromatography. The 1H NMR spectrum of 2 in CDCl3 showed the anomeric protons at δ 5.78, 5.24, 5.06, 5.01, 5.00, 4.99, 4.82 ppm as singlets and at δ 4.96 ppm (J1,2 = 1.4 Hz) as a doublet supporting the 1,2-trans glycosylation. In the 13C NMR spectrum of 2, the anomeric carbons were not well resolved and appeared as three signals at δ 98.41, 97.99 and 86.89 ppm. Lastly, the final structural confirmation was obtained by MALDI-TOF mass analysis of 2, which showed a peak at 3983.9 [M+Na]+ corresponding to the molecular formula C225H186O65SNa.
After the successful synthesis of 2, dansylated octamannan fluorescent probe 1 was synthesized in four steps (Scheme-2). In step one, α(1→6)-linked octamannosyl thioglycoside 2 was reacted with 1-azidoethanol 314 in presence of activator NIS and Lewis acid AgOTf at −4 °C for 45 minutes. Usual workup of the coupling reaction followed by purification of the reaction product by column chromatography gave the desired 1-azidoethyl α(1→6)-linked octamannosyl glycoside 15 in 89% yield. In the 1H NMR and 13C NMR spectra of 15, the signals from p-thiotolyl group were found absent. The 13C NMR spectrum of 15 clearly indicated the attributable to CH2N3 and OCH2 carbons respectively. The MALDI-TOF mass analysis of 15 showed a peak at 3947.1 [M+Na]+ corresponding to the molecular formula C220H183N3O66Na.
Scheme 2.
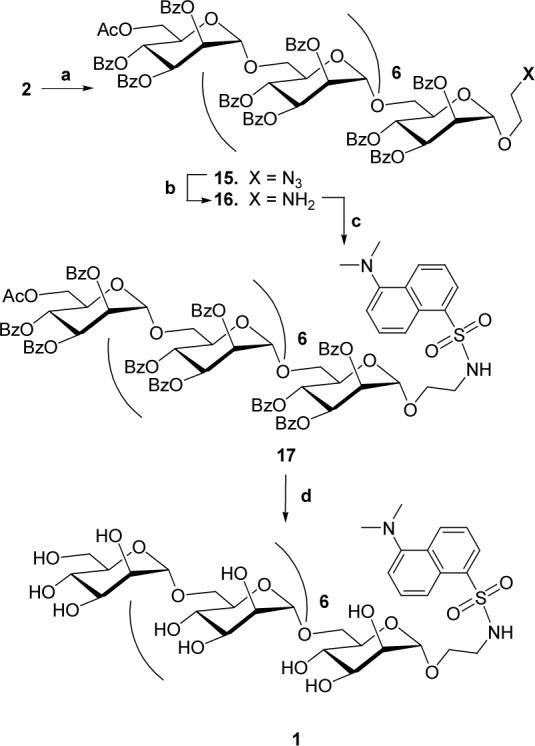
Reactions and Reagents. (a) HOCH2CH2N3, NIS, AgOTf, dry CH2Cl2, 4A Mol sieves, −4 °C, 45 min, 89%. (b) Pd/C, HCO2NH4, dry MeOH–CH2Cl2 (9:1), rt, 4 h, 82%. (c) Dansyl chloride, N-methylimidazole, dry CH2Cl2, 0 °C, overnight, 69%. (d) satd. NH3 in MeOH, dry CH2Cl2, rt, overnight, 71%.
In step two, the azido functionality in 15 was reduced using ammonium formate (HCO2NH4) over Pd/C in CH2Cl2-MeOH (9:1) and was monitored by TLC. Upon completion of the reaction, concentration and the usual workup produced 1-aminoethyl α(1→6)-linked octamannosyl glycoside 16. Without further purification, the crude 16 was reacted overnight at 0 °C in the dark with dansyl chloride 3 and N-methylimidazole. TLC showed a major fluorescent spot in long wave (365 nm) UV lamp and purification by column chromatography gave the desired blocked dansylated α(1→6)-linked octamannosyl glycoside 17 in 69% yield. The 1H NMR spectrum of 17 indicated formation of the desired product as it showed signals due to a dansyl group in the aromatic region as well as ppm [N(CH3)2]. In the APT 13C NMR spectrum of 17 in CDCl3, the carbons of CH2NH and N(CH3)2 were observed at δ 42.67 and 45.39 ppm respectively. In the MALDI-TOF mass analysis of 17 gave a peak at 4155.8 [M+Na]+ corresponding to the molecular formula C232H196N2O68SNa.
Finally, global de-protection of 17 was accomplished by overnight reaction with satd. NH3 solution in MeOH at room temperature. TLC in CHCl3:MeOH:H2O (65:35:10, lower layer) showed one fluorescent spot under a long wave UV lamp. The reaction mixture was concentrated to dryness and the syrup was washed several times with CH2Cl2 and finally with EtOAc. The syrup was purified by column chromatography on Sephadex LH-20 using MeOH as the mobile phase to furnish the desired fluorescent probe α(1→6)-linked octamannosyl glycoside 1 in 71% yield. The signals in the 1H NMR spectrum of 1 in D2O were not well resolved at 300 MHz, but showed protons in aromatic and sugar regions along with a singlet at δ 3.11 PPM (CH2NH) and δ 2.29 [N(CH3)2]. Similarly, in the 75 MHz 13C NMR spectrum of 1 in D2O, the presence of signals at δ 46.82 ppm [N(CH3)2] and δ 43.56 ppm (CH2NH) supported the structure. The final structural confirmation was obtained by FABMS analysis of 1, which showed a peak at 1613.1 [M+Na]+, corresponding to the molecular formula C62H98N2O43SNa.
In conclusion, we have reported a successful and efficient synthesis of a dansylated homolinear α(1→6)-linked octamannan fluorescent probe for its future biochemical potential use as a delivery vehicle for drugs via mannan receptors present in the infected macrophages.
Supplementary Material
Acknowledgments
Authors acknowledge College of Arts and Science, Western Illinois University for faculty start-up funds and NIH grant R21AI059270.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
Experimental procedures, analytical and spectral data of all new compounds can be found in the online version.
References
- 1.a Nakajima T, Ballou CE. J. Biol. Chem. 1974;249:7679–7684. [PubMed] [Google Scholar]; b Suzuki S, Shibata N, Kobayashi H. In: Fungal Cell Wall and Immune Responses, NATA ASI Ser. Latge JP, Boucias D, editors. H53. Speinger-Verlag; Berlin: 1991. pp. 111–121. [Google Scholar]; c Kobayashi H, Kojimahara T, Takahashi K, Takihawa M, Takahashi S, Shibata N, Okawa Y, Suzuki S. Carbohydr. Res. 1991;214:131–145. doi: 10.1016/s0008-6215(00)90536-6. [DOI] [PubMed] [Google Scholar]
- 2.a van Kooyk Y, Geijtenbeek TBH. Nat. Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]; b Geijtenbeek TBH, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GCF, Middel J, Cornelissen ILMHA, Nottet HSLM, Kewalramani VN, Littman DR, Figdor CG, van Kooyk Y. Cell. 2000;100:587–597. doi: 10.1016/s0092-8674(00)80694-7. [DOI] [PubMed] [Google Scholar]; c Kobata A. Acc. Chem. Res. 1993;26:319. [Google Scholar]
- 3.a Stratford M. Yeast. 1992;8:635–643. doi: 10.1002/yea.320080807. [DOI] [PubMed] [Google Scholar]; b Kanbe T, Culter JE. Infect. Immunol. 1994;62:1662–1669. doi: 10.1128/iai.62.5.1662-1668.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Irache JM, Salman HH, Gamazo C, Espuelas S. Expert Opin Drug Deliv. 2008;5:703–724. doi: 10.1517/17425247.5.6.703. [DOI] [PubMed] [Google Scholar]; b Diebold SS, Plank C, Cotton M, Wagner E, Zenke M. Somat Cell Mol Genet. 2002;27:65–74. doi: 10.1023/a:1022975705406. [DOI] [PubMed] [Google Scholar]; c Chakraborty P, Bhaduri AN, Das PK. Biochem. Biophys. Res. Commun. 1990;166:404–410. doi: 10.1016/0006-291x(90)91959-v. [DOI] [PubMed] [Google Scholar]; d Kaneo Y, Uemura T, Tanaka T, Kanoh S. Biol. Pharm. Bull. 1997;20:181–187. doi: 10.1248/bpb.20.181. [DOI] [PubMed] [Google Scholar]; e Jung YJ, Lee JS, Kim HH, Kim YT, Kim YM. Arch. Pharm. Res. 1998;21:179–186. doi: 10.1007/BF02974025. [DOI] [PubMed] [Google Scholar]
- 5.a Kodama T, Freeman M, Rohrer L, Zabrecky J, Matsudaira P, Krieger M. Nature. 1990;343:531–535. doi: 10.1038/343531a0. [DOI] [PubMed] [Google Scholar]; b Koren H, Becker S. CRC-Press. 1992:747–769. [Google Scholar]; c Suzuki H, Kurihara Y, Takeya M, et al. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz YS, Dushkin MI, Vavilin VA, Melnikova EV, Khoschenko OM, Kozlov VA, Agafonov AP, Alekseev A,Y, Rassadkin Y, Shestapalov AM, Azaev MS, Saraev DV, Filimonov PN, Kurunov Y, Svistelnik AV, Krasnov VA, Pathak AK, Derrick SC, Reynolds RC, Morris S, Blinov VM. Antimicrob. Agents Chemother. 2006;50:1982–1988. doi: 10.1128/AAC.00362-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Pathak AK, Pathak V, Bansal N, Maddry JA, Reynolds RC. Tetrahedron Letters. 2001;42:979–982. [Google Scholar]; b Pathak AK, Pathak V, Seitz L, Riordan JM, Gurcha SS, Besra GS, Reynolds RC. Bioorg. Med. Chem. 2007;15:5629–5650. doi: 10.1016/j.bmc.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Heng L, Ning J, Kong F. J. Carbohydr. Chem. 2001;20:285. [Google Scholar]; b Zhu Y, Chen L, Kong F. Carbohydr. Res. 2002;337:207–215. doi: 10.1016/s0008-6215(01)00307-x. [DOI] [PubMed] [Google Scholar]; c Ning J, Heng L, Kong F. Tet. Lett. 2002;43:673–675. [Google Scholar]; d Xing Y, Ning J. Tet. Assym. 2003;14:1275–1283. [Google Scholar]; e Ratner DM, Plante OJ, Seeberger PH. Eur. J. Org. Chem. 2002:826–833. [Google Scholar]; f Crich D, Banerjee A, Yao Q. J. Am. Chem. Soc. 2004;126:14930–14934. doi: 10.1021/ja047194t. [DOI] [PubMed] [Google Scholar]; g López JC, Agocs A, Uriel C, Gómeza AM, Fraser-Reid B. Chem. Commun. 2005:5088–5090. doi: 10.1039/b507468a. [DOI] [PubMed] [Google Scholar]; h Jayaprakash KN, Chaudhuri SR, Murty CVSR, Fraser-Reid B. J. Org. Chem. 2007;72:5534–5545. doi: 10.1021/jo070018t. [DOI] [PubMed] [Google Scholar]
- 9.Zhu Y, Kong F. Carbohydr. Res. 2001;332:1–21. doi: 10.1016/s0008-6215(01)00070-2. [DOI] [PubMed] [Google Scholar]
- 10.Toshima K. Carbohydr. Res. 2000;327:15–26. doi: 10.1016/s0008-6215(99)00325-0. [DOI] [PubMed] [Google Scholar]
- 11.a Nicolaou KC, Dolle RE, Papahatjis DP, Randall JL. J. Am. Chem Soc. 1984;106:4189. [Google Scholar]; b Nicolaou KC, Ueno H. In: Oligosaccharide Synthesis from Glycosyl Fluoride and Sulfides. In Preparative Carbohydrate Chemistry. Hanessian S, editor. Marcel Dekker; New York: 1996. pp. 313–338. [Google Scholar]
- 12.Pathak AK, Yerneni CK, Young Z, Pathak V. Org. Lett. 2008;10:145–148. doi: 10.1021/ol702743x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heng L, Ning J, Kong F. Carbohydr. Res. 2001;331:431–437. doi: 10.1016/s0008-6215(01)00059-3. [DOI] [PubMed] [Google Scholar]
- 14.Smith RH, Mehl AF, Shantz DL, Jr., Chmurny GN, Michejda CJ. J. Org. Chem. 1988;53:1467–1471. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.