

Abstract
The Ca2+-induced interaction between cardiac troponin I (cTnI) and actin plays a key role in the regulation of cardiac muscle contraction and relaxation. In this report we investigated changes of this interaction in response to strong crossbridge formation between myosin S1 and actin and PKA phosphorylation of cTnI within reconstituted thin filament. The interaction was monitored by measuring Förster resonance energy transfer (FRET) between the fluorescent donor 5-(iodoacetamidoethyl)aminonaphthalene-1-sulfonic acid (AEDANS) attached to the residues 131, 151, 160 167, 188 and 210 of cTnI and the nonfluorescent acceptor 4-dimethylaminophenylazophenyl-4′-maleimide (DABM) attached to cysteine 374 of actin. The FRET distance measurements showed that bound Ca2+ induced large increases in the distances from actin to the cTnI sites, indicating a Ca2+-triggered separation of actin from cTnI. Strongly bound myosin S1 induced additional increases in these distances in the presence of bound Ca2+. These two-step changes in the observed FRET distances provide a direct link of structural changes at the interface between cTnI and actin to the three-state model of thin filament regulation of muscle contraction and relaxation. When cTnC was inactivated through mutations of key residues within the 12-residue Ca2+-binding loop, strongly bound S1 alone induced increases in the distances in spite of the fact that the filaments no longer bound regulatory Ca2+. These results suggest bound Ca2+ or strongly bound S1 alone can partially activate thin filament, but full activation requires both bound Ca2+ and strongly bound S1. The distributions of the FRET distances revealed different structural dynamics associated with different regions of cTnI in different biochemical states. The second actin-binding region appears more rigid than the inhibitory/regulatory region. In the Mg2+ state, the regulatory region appears more flexible than the inhibitory region, and in the Ca2+ state, the inhibitory region becomes more flexible. PKA phosphorylation of cTnI increased the FRET distance from actin to cTnI residue 131 by 2.2 - 5.2 Å in different biochemical states and narrowed the distributions of the distances from actin to the inhibitory and regulatory regions of cTnI. The observed phosphorylation effects are likely due to an intramolecular interaction of the phosphorylated N-terminal segment and the inhibitory region of cTnI.
Keywords: Interactions between Cardiac Troponin I and Actin, Calcium activation of cardiac thin filament, Forster resonance energy transfer (FRET), Strong cross-bridge, PKA phosphorylation of cardiac troponin I
The Ca2+ dependent interaction between TnI and actin is one of the most important events in the regulation of striated muscle contraction and relaxation. We have investigated structural features of the interaction and how they are modulated by strong crossbridge and PKA phosphorylation of cTnI. Ca2+ regulation of myofilaments is linked to the thin filament, composed of the heterotrimeric troponin complex (Tn) and tropomyosin (Tm) bound to the double helical actin filament (1, 2). Tn is formed by troponin C (TnC), troponin I (TnI), and troponin T (TnT). Subunit TnC is the Ca2+ binding protein, TnI binds actin and inhibits actomyosin ATPase in relaxed muscle, and TnT anchors the Tn complex on the actin filament. In relaxed muscle, at resting concentration of Ca2+, TnI-Tm acting as a regulatory switch prevents crossbridge formation between actin and the myosin subfragment-1 (S1) through steric blocking of myosin-binding sites on actin (3-5). To activate actomyosin ATPase and force intracellular [Ca2+] rises to saturate the regulatory sites in cardiac TnC, triggering a series of conformational transitions among the thin filament proteins, including opening of the N-terminal domain of TnC, TnC-TnI interaction and TnI-actin interaction. These conformational changes ultimately result in strong force-generating interactions between myosin and actin (6). The strong actin-myosin interaction promotes additional changes in these thin filament structural transitions and enhances Ca2+ binding to the thin filament (7-9). This feedback activation of the thin filament is generally accepted to play a more prominent role in activation of cardiac muscle than skeletal muscle (6, 10-14). Yet, there is little information related to the molecular basis for the protein-protein interactions within the thin filament that underlie the feedback of strong crossbridges on myofilament activation.
In addition to direct activation of cardiac muscle by Ca2+ through its binding to cardiac TnC (cTnC) and the feedback activation of the strong actin-myosin interaction, muscle activation can be further modulated by cardiac TnI (cTnI) phosphorylation (15, 16). In comparison with skeletal TnI, cTnI has an additional 32-33 amino acids at its N-terminus containing two cAMP-dependent protein kinase A (PKA) phosphorylation sites at positions 23 and 24. There is evidence that phosphorylation of these two serine residues results in a reduction in the affinity of Ca2+ for cTnC without changing maximum MgATPase activity (17, 18), and an increase in relaxation rate (19) and cross bridge cycling rate (20) during β-adrenergic stimulation. The phosphorylation also modulates the Ca2+-dependent binding of myosin S1 to reconstituted thin filaments (21). This modulation mechanism through cTnI phosphorylation plays an important role in the functional adaptation of cardiac muscle to physiological or pathological stress. Despite extensive studies on functional effects of the phosphorylations within the thin filament (16), the molecular basis by which PKA phosphorylation of cTnI modulates the Ca2+-induced cascade of allosteric changes in the thin filament is still elusive. In our early studies, we showed that the rate of both Ca2+ binding and dissociation from cTnC were changed by cTnI phosphorylation (22). These changes appeared to be related to a global conformational change of phosphorylated cTnI (23-26) and phosphorylation-induced changes in protein-protein interactions within the cTn complex (27-31).
A main feature of regulation of cardiac muscle is the dynamics of the interaction among the thin filament proteins involving multiple structural transitions at the thin filament protein interfaces. It gives rise to cooperativity of structural interactions and kinetic coupling of signaling steps during muscle activation. An important challenge in the study of cardiac thin filament regulation is to understand the significance of alterations at the molecular level, i.e., the protein-protein interactions in response to signal transduction from Ca2+ binding to force generation. This information is essential to our understanding of how actin-myosin interaction is activated and how this activation is modulated by crossbridge formation and other mechanisms. In this report we focused our investigation on structural changes in the interaction between the C-terminal region of cTnI and actin in response to Ca2+ binding to troponin, strong crossbridge formation, and cTnI PKA phosphorylation. The C-terminal region of cTnI contains several functional domains including the inhibitory region (residues 130 -150), regulatory region (residues 151-167), and the second or “mobile” actin binding domain (residues 168-210). The interaction between the C-terminal region of cTnI and actin within the thin filament was monitored by measuring Förster resonance energy transfer (FRET) between the fluorescent 5-(iodoacetamidoethyl)aminonaphthalene-1-sulfonic acid (AEDANS) as donor attached to selected residues in the C-terminal region of cTnI and the nonfluorescent 4-dimethylaminophenylazophenyl-4′-maleimide (DABMI) as acceptor attached to cysteine 374 of actin. Our results provide quantitative information on a two-step movement of the inhibitory/regulatory region of cTnI (residue 130-167) from actin filament following Ca2+ activation and strong crossbridge formation between myosin S1 and actin. This two-step transition is well correlated with the three-state model of thin filament activation. An analysis of distribution of the measured FRET distances indicates the structural dynamics of the inhibitory/regulatory region of cTnI is significantly altered upon phosphorylation of the N-terminal segment of cTnI by PKA.
Materials and Methods
Protein Preparations
A recombinant wild-type cTnT mutant was generated from a full-length rat cTnT clone subcloned into a pET-17b vector, and a recombinant wild-type cTnC was generated from a cTnC cDNA clone from chicken slow skeletal muscle as previously reported (32). cTnI mutants containing a single cysteine at positions 131 (C81I/C98S/Q131C), 151 (C81I/C98S/S151C), 160 (C81I/C98S/L160C), 167 (C81I/C98S/S167C), 188 (C81I/C98S/E188C), and 210 (C81I/C98S/E210C) were generated from a mouse cDNA clone subcloned into a pET-3d vector as previously described (33, 34). All plasmids were transformed into BL21(DE3) cells (Invitrogen) and expressed under isopropyl-1-thio-D-galactopyranoside induction. Cells were harvested and suspended in a CM buffer containing 30 mM citric acid, pH 6.0, 1 mM EDTA, 1 mM DTT and protease inhibitors. After sonication and centrifugation, the supernatant was fractionated with ammonia sulfate. The pellets obtained from 30% to 60% of ammonia sulfate saturation were dissolved in a CM buffer and dialyzed against the buffer overnight. After centrifugation to remove any insoluble materials, the sample was loaded to CM column. Proteins were eluted from the column with a gradient from 0 to 0.6 M of NaCl. The purity of the protein was checked with SDS gel. Cardiac Tm (cTm) (35), actin (36), and myosin subfragment 1(S1) from chymotryptic digestion of myosin (37) were obtained from bovine cardiac tissue.
Protein Labeling
Modification of the single-cysteine residues of cTnI mutants with IAEDANS were performed according to previously described procedures (32, 34). Briefly, the lyophilized protein was re-suspended in a labeling buffer containing 50 mM Mops (pH 7.4), 3 M urea, 100 mM KCl, 1 mM EDTA, 1 mM DTT, then stepwise dialyzed against the labeling buffer to reduce [DTT] to ∼10 μM. Each single-cysteine cTnI (70–100 μM) was incubated with a 3 molar excess of IAEDANS under constant stirring overnight at 4 °C. Unreacted probes were reduced by adding DTT to a final concentration of 2 mM from a 1 M stock solution, then removed through dialysis. Modification of cysteine 374 of actin was carried out using a previously described procedure with modification (38, 39). The purified actin (2.5 mg/ml) was dialyzed against a buffer containing 50 mM Mops (pH 7.5), 100 mM KCl, 1 mM EDTA, 1 mM DTT. DTT was reduced to 10 μM by dialyzing against the same buffer containing no DTT. The sample was mixed with a 5-fold molar excess of DABMI and incubated at 4 °C for 20 hours. The reaction was terminated by adding 2 mM DTT. Unreacted probe was removed by centrifugation and dialysis. Label ratio was determined using ε325nm = 6,000 cm-1M-1 for AEDANS and ε460nm = 24,600 cm-1M-1 for DABM, respectively. The identities of all cTnI mutants and labeled proteins were verified using electrospray mass spectrometric analysis.
Reconstitution of the Troponin Complex and the Thin Filament
Reconstituted cardiac troponin (cTn) were obtained using a previous procedure (40) with modifications. cTnC, cTnI, and cTnT were separately dialyzed against a reconstitution buffer (50 mM Tris—HCl, pH 8.0, 6 M urea, 500 mM KCl , 5 mM CaCl2, 2 mM DTT), then mixed at a 1.0 : 1.1 : 1.1 molar ratio (10, 11, and 11 μM final concentrations). The mixture was gently shaken for two hours at room temperature, then stepwise dialyzed against a high salt buffer containing 1 M KCl, 20 mM Mops (pH 7.0), 5.0 mM MgCl2, 1.0 mM CaCl2, 1.5 mM DTT to successively reduce the urea concentration (6, 4, 2, and 0 M). The KCl concentration was subsequently reduced to 1, 0.7, 0.5, 0.3, and 0.15 M by stepwise dialysis against a working buffer containing 150 mM KCl, 50 mM Mops (pH 7.0), 5 mM MgCl2, 2 mM EGTA, and 1 mM DTT. The sample was centrifuged to remove insoluble unbound cTnI and cTnT. The formation and stoichiometry of troponin complex were verified by native gel and SDS page electrophoresis. cTn-cTm complexes were formed by mixing cTn and cTm 1:1.1 (4 μM cTn) in the same working buffer plus a sufficient volume of 3 M KCl to bring the final KCl concentration to 300 mM. Excessive salt was then reduced to 150 mM KCl in two dialysis steps. cTn-cTmA7 was prepared by mixing cTn-cTm, and polymerized actin 1:7.5 (2:15 μM) in the working buffer with 300 mM KCl, followed by reducing [KCl] to 150 mM as for the cTn-cTm complex. cTn-cTmA7-S1-ADP was prepared by adding S1 to the cTn-cTmA7 mixture at ratio of one cTn-cTm, seven actin and seven S1 after KCl had been decreased to 150 mM. ADP was added to the preparation immediately prior to measurements to a final concentration of 4 mM from a MgADP stock solution prepared from MgCl2 (200 mM) and Na2ADP (200 mM) in the working buffer.
PKA Phosphorylation of cTnI
cTnI was phosphorylated by the catalytic subunit of PKA, using a cTnC affinity column (28). Briefly, a sample of purified cTnI mutant was loaded on a cTnC affinity column equilibrated in 50 mM KH2PO4 at pH 7.0, 500 mM KCl, 10 mM MgCl2, and 0.5 mM DTT, and 125 U PKA/mg cTnI was added directly to the column. To initiate the reaction ATP was added to the column to a final concentration of 1.0 mM. After 30 min at 30 °C, the column was washed with a buffer containing 50 mM Mops at pH 7.0, 500 mM KCl, 2 mM CaCl2 and 0.5 mM DTT. Phosphorylated cTnI was eluted with a buffer containing 6 M urea, 10 mM EDTA, 0.5 mM DTT and 50 mM Mops at pH 7.0. The extent of phosphorylation was quantified by mass spectrometry and by treatment of the sample with alkaline phosphatase, followed by determination of inorganic phosphate using the EnzChek Phosphate Assay kit (41). Phosphorylation of the two PKA sites was >90%.
Actin-activated Acto-S1 ATPase Measurements
The biochemical activity of the labeled cTnI mutants was tested by Ca2+-dependent regulation of acto-S1 ATPase activity. Measurements were performed at 30 °C in 60 mM KCl, 5.6 mM MgCl2, 2 mM ATP, 30 mM imidazole (pH 7.0), 1 mM DTT and either 500 μM CaCl2 for the +Ca2+ state or 1 mM EGTA for the -Ca2+ state. The protein concentrations used were 4.2 μM F-actin, 0.6 μM cTm, 0.6 μM cTn and 0.5 μM S1. The amounts of inorganic phosphate released were determined colorimetrically as previously reported (42). Absorption was measured at 630 nm with a Beckman DU-640 spectrophotometer.
Fluorescence Measurements
AEDANS steady-state fluorescence measurements were carried out on an ISS PCI photon-counting spectrofluorometer, using a band-pass of 3 nm on both the excitation and the emission monochromators. Time-resolved fluorescence anisotropy decays were collected in the time domain using an IBH 5000U fluorescence lifetime system equipped with a 343 nm LED as the light source. Anisotropy decays of AEDANS attached to the selected residues of cTnI were determined by measuring the emission at 480 nm polarized in the vertical and horizontal directions with vertically polarized excitation. The polarized decay data were fitted to a biexponential function to recover the limiting anisotropy at zero time (43).
Total fluorescence intensity decays of AEDANS from the donor-alone and donor-acceptor samples were collected at 480 nm with a time-correlated single photon counting system associated with the IBH 5000U under identical experimental condition using a polarizer set to the magic angle. These decays were used to calculate the distribution of intersite distances between donor and acceptor as in previous work (40, 44) using the following equation:
| (1) |
where IDA (r,t) is the distance-dependent donor intensity decay of a donor-acceptor pair separated by a given distance r; αDi is the fractional amplitude associated with the lifetime τDi for the ith component; R0 is the Förster critical distance at which the energy transfer efficiency is 0.5 and can be experimentally derived for each specific FRET donor-acceptor pair. The experimentally determined Ro was in the range of 40-43 Å for different protein preparations and biochemical state. These values were used to calculate distance distributions
The observed donor intensity decay may be treated in the most general case as an ensemble of donor-acceptor pairs and is given by the average of the individual decays weighted by the distance probability distribution [P(r)] of the donor-acceptor pair (45),
| (2) |
The probability distribution is usually assumed to be a Gaussian with a mean distance (r) and half-width (hw) of the distribution. The standard deviation (σ) of the Gaussian is related to the half-width by hw = 2.354σ. The distributions of the distances between the selected residues of cTnI and Cys374 of actin were calculated using Global Curve, (46) a general purpose global non-linear least-squares program. Confidence estimates from time-resolved FRET data were obtained using a grid search of the reduced chi squares ratio (ϰR2) as described (46, 47). An analysis of the dependence of this ratio on the mean distance and the half-width were used to judge goodness of fit for the distribution and determine the upper and lower estimates of the mean distance and the half-width at the 68% confidence level (45, 48).
Results
Characterization of cTnI mutants
To examine whether labeled cTnI mutants have the same regulatory function as wild-type protein, Ca2+ regulation of the actin-activated S1-ATPase activity assay was carried out. The results are summarized in Table 1. The ATPase activity of S1 in the presence of actin, but in the absence of troponin and Tm was taken as 100%. The Ca2+ sensitivity was 0.785 for the control preparation containing wild-type cTn. The Ca2+ sensitivities for all other preparations containing labeled mutants of cTnI were similar to that of the control, suggesting that the effects of the mutations and modifications of cTnI on the Ca2+ regulatory activity were negligible.
Table 1.
Effects of modified cTnI mutants on reconstituted actomyosin ATPase activity
| Troponin Complex | ATPase Activity | Ca2+ Sensitivity | |
|---|---|---|---|
| EGTA | Ca2+ | ||
| cTnC(wt)-cTnT(wt)-cTnI(wt) | 0.017 | 0.079 | 0.785 |
| cTnC(wt)-cTnT(wt)-cTnI(Q131C) | 0.015 | 0.074 | 0.797 |
| cTnC(wt)-cTnT(wt)-cTnI(S151C) | 0.019 | 0.084 | 0.773 |
| cTnC(wt)-cTnT(wt)-cTnI(L160C) | 0.016 | 0.079 | 0.797 |
| cTnC(wt)-cTnT(wt)-cTnI(S167C) | 0.018 | 0.085 | 0.788 |
| cTnC(wt)-cTnT(wt)-cTnI(S188C) | 0.017 | 0.077 | 0.779 |
| cTnC(wt)-cTnT(wt)-cTnI(S210C) | 0.016 | 0.072 | 0.778 |
Ca2+-dependent acto-S1 ATPase activity was measured at 30 °C in 60 mM KCl, 5.6 mM MgCl2, 2 mM ATP, 30 mM imidazole (pH 7.0), 1 mM DTT and either 500 μM CaCl2 for the Ca2+ state or 1 mM EGTA for the EGTA state. The protein concentrations used were 4.2 μM F-actin, 0.6 μM cTm, 0.6 μM cTn and 0.5 μM S1. The amounts of inorganic phosphate released were determined colorimetrically and expressed in μmol Pi/sec/g 42. Ca2+ sensitivity was defined as {1-(ActivityEGTA/ ActivityCa)}×10042. cTnC(wt), cTnI(wt) and cTnT(wt) are wild-type cTnC, cTnI, and cTnT, respectively. The cTnI mutants were labeled at the single cysteine with AEDANS.
SDS-PAGE and native gels were used to examine the stability and stoichiometry of the troponin complex reconstituted with mutant proteins and labeled cTnI. The complexes were reconstituted by incubating labeled cTnI mutants, cTnC and cTnT in a molar ratio of 1.0 : 1.1 : 1.1 on ice for 30 min, and then dialyzing against the working buffer containing 50 mM Mops (pH 7.0), 1 mM DTT, and 0.1M KCl in the presence of 5 mM Mg2+. The samples were centrifuged at 10,000 x g for 10 min to remove all insoluble cTnI and cTnT before gel electrophoresis. Gel analysis showed that both wild-type and mutant troponin complexes existed as single complexes with correct stoichiometry (Fig. 1). Multiple experiments were performed on samples prepared within three weeks and gels showed no evidence of protein degradation.
Figure 1.
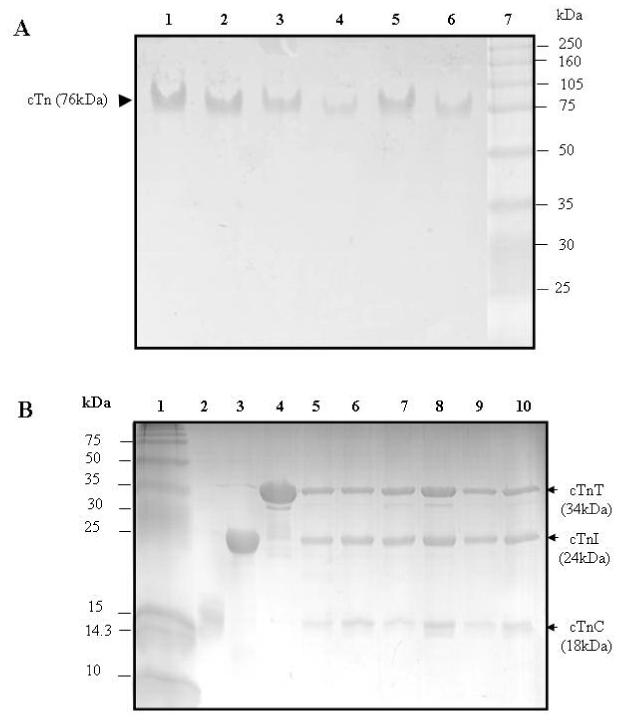
Electrophoresis analysis of reconstituted cardiac troponin
Panel A: Native PAGE gels (8% resolving and 4% stacking). Lanes 1-6, troponin reconstituted from wild-type cTnC, wild-type cTnT, and a cTnI mutant in which the single-cysteine was labeled with AEDANS. Lane 7, HMW protein standards (Amersham Biosciences). (1) cTnI(131C), (2) cTnI(151C), (3) cTnI(160C), (4) cTnI(167C), (5) cTnI(188C), and (6) cTnI(210C). These gels showed all six reconstituted samples ran as a single component with a mass of ∼ 76 kDa.
Panel B: SDS-PAGE gels (18% resolving and 4% stacking). Lane 1, LMW protein standards (Amersham Bisciences). (2) wild-type cTnC, (3) wild-type cTnI, and (4) wild-type cTnT. Lanes 5-10, troponin reconstituted from wild-type cTnC, wild-type cTnT, and a cTnI mutant in which its single cysteine was labeled with AEDANS. (5) cTnI(131C), (6) cTnI(151C), (7) cTnI(160C), (8) cTnI(167C), (9) cTnI(188C), and (10) cTnI(210C). The six reconstituted troponin samples were each resolved into three bands. Densitometry analysis of the bands was done with Bio-Rad Quantity One Software, and the results showed that the three resolved bands corresponded to cTnC, cTnI, and cTnT with a molar ration of 1:1:1. No significant degradation products were found on the gels.
Conformational changes in cTnI at the actin-cTnI interface
Fluorescence of the donor AEDANS attached to the six single-Cys residues in the C-terminal region of cTnI in reconstituted thin filaments was quenched in the presence of the acceptor DABM attached to actin C374 to different extents, depending upon the locations of the donor in cTnI (data not shown). Differences in the quenching reflected differences in the efficiency of energy transfer (FRET) and suggested different intersite distances between actin C374 and the cTnI sites. Donor fluorescence was sensitive to the presence of Ca2+ in the presence of acceptor, indicating a Ca2+-induced alteration of the intersite distances. Figure 2 shows a series of donor intensity decay plots with the donor attached to cTnI residue 167 and the acceptor attached to actin C374 in different ionic environments and biochemical states. These decays were analyzed in terms of a distribution of the distances between donor and acceptor (eq. 2). Figure 3 shows is a typical analysis of one of the tracings from Fig. 2 for the intensity decay of donor attached to cTnI(167C). The distributions of this intersite distance between cTnI(167C) and actin(C374) determined in four biochemical states are shown in Fig. 4. Summarized in Table 2 are distance results for the six distances between actin(C374) and the cTnI sites. In the absence of Ca2+ (Mg2+ alone), the mean distance between actin and cTnI(167C) was 40.8 Å and the half-width of the distribution was 19.0 Å. In the presence of Ca2+ (Mg2+ + Ca2+), the distance increased by 12.3 Å to 53.1 Å and the half-width decreased by 3.8 Å to 15.2 Å. This effect of Ca2+ on the distance is consistent with that previously reported for the equivalent intersite distance in skeletal muscle thin filaments (39). Similarly to the effect of Ca2+ on the actin-cTnI(C167) distance, bound Ca2+ induced an increase in the actin-cTnI(C160) distance by 10.2 Å from 47.1 Å to 57.3 Å and a decrease in the half-width of the distance distribution by 4.9 Å from 19.5 Å to 14.6 Å. Myosin S1 strongly bound to thin filaments lengthened these two intersite distances regardless of whether Ca2+ was absent (Mg2+ alone) or present (Mg2+ + Ca2+), and in each ionic condition. Bound S1 increased the half-widths of the distribution (0.6-4.2 Å). However, bound Ca2+reduced the half-width of the distributions of both distances determined with fully regulated thin filaments containing bound S1. Taken together, these results suggested that Ca2+ binding to cTnC in thin filaments induced a separation between the regulatory region of cTnI and the C-terminus of actin. This separation was further increased by the strong binding of myosin S1 to the thin filaments. The decrease in the half-width of the distance distributions induced by Ca2+ binding to thin filaments with or without bound S1 suggested a loss of conformational flexibility in the region between the actin C-terminus and the regulatory region of cTnI.
Figure 2.

Fluorescence intensity decays of AEDANS (donor) attached to residue 167 of cTnI in reconstituted cardiac thin filaments. Dotted curves, donor-only sample, solid curve, donor-acceptor sample in which actin was labeled with acceptor DABM at C374. Black, no Ca2+ (Mg2+ only); red, Ca2+ (+ Mg2+); Pink, myosin S1 is strongly bound to the filaments and in the absence of Ca2+, and blue, myosin is strongly bound to the filaments and in the presence of Ca2+. The decays were measured with excitation at 343 nm, and the emission collected at 500 nm. Sample were in 50 mM Mops at pH 7.0, 1mM DTT, 1 mM EGTA, 5 mM MgCl2, 0.2 M KCl. When Ca2+ was present, it was 2 mM CaCl2. The initial fast decays of these tracings are shown in the inset.
Figure 3.

An analysis of energy transfer from AEDANS attached to cTnI(167C) to DABM attached to actin(C374) in reconstituted cardiac thin filaments. The analysis shown here is for the corresponding decay curve shown in Fig. 2. The sharp peak on the left is the excitation light pulse. The donor intensity decay in the presence of acceptor (broad peak on the right) was fit to eq. 2 with a single Gaussian as P(r) and a sum of two exponential terms for IDA(r,t) (eq. 1). The residual plot of the fit is displayed in the lower panel across the figure, and the autocorrelation plot is shown as an inset at the upper right-hand corner. The best-fit data from this analysis were used to calculate the distribution of the inter-site distances between cTnI C167 and actin C374.
Figure 4.

Area-normalized distributions of intersite distances between actin C374 and cTnI(167C) in cardiac thin filaments in different biochemical states. 1, in the absence of Ca2+; 2, in the presence of Ca2+, 3, in the presence of strongly bound myosin S1 (-Ca2+), and 4, in the presence of strongly bound S1 (+Ca2+).
Table 2.
FRET distances between actin(C374) and cTnI in regulated cardiac thin filaments
| Thin Filament | Thin Filament + S1-ADP | ||||
|---|---|---|---|---|---|
| Distance | Conditions | Mean Distance (Å) |
Half-Width (Å) |
Mean Distance (Å) |
Half-Width (Å) |
| cTnI(131C)- actin(C374) |
Mg2+ | 37.3 (36.3, 38.5) |
12.8 (11.2, 13,7) |
41.4 (40.0, 42.9) |
15.0 (13.9, 16.3) |
| Mg2+ + Ca2+ |
39.0 (38.1, 40.3) |
15.1 (14.3, 16.4) |
44.0 (42.8, 45.1) |
21.5 (20.3, 22.9) |
|
| cTnI(151C) - actin(C374) |
Mg2+ | 43.5 (42.8, 44.2) |
13.0 (12.1, 13.7) |
53.9 (52.6, 55.1) |
18.0 (16.9, 19.6) |
| Mg2+ + Ca2+ |
54.6 (53.2, 55.8) |
17.1 (16.3, 17.8) |
56.8 (55.6, 57.8) |
22.7 (20.9, 23.3) |
|
| cTnI(160C)- actin(C374) |
Mg2+ | 47.1 (45.9, 48.0) |
19.5 (18.2, 20.8) |
56.3 (54.0, 58.1) |
21.6 (20.2, 22.8) |
| Mg2+ + Ca2+ |
57.3 (56.3, 58.6) |
14.6 (12.4, 16.3) |
59.8 (58.3, 61.7) |
15.2 (13.6, 17.1) |
|
| cTnI(188C)- actin(C374) |
Mg2+ | 40.8 (39.3, 41.7) |
19.0 (18.1, 20.3) |
54.3 (53.8, 55.9) |
23.2 (21.9, 24.1) |
| Mg2+ + Ca2+ |
53.1 (52.2, 54.3) |
15.2 (14.3, 16.6) |
60.7 (59.2, 62.2) |
17.3 (15.9, 18.4) |
|
| cTnI(167C)- actin(C374) |
Mg2+ | 46.7 (45.8, 47.7) |
6.6 (6.1, 7.3) | 53.7 (52.1, 55.3) |
6.8 (6.2, 7.3) |
| Mg2+ + Ca2+ |
51.3 (50.2, 52.5) |
8.6 (7.8, 10.2) |
58.4 (57.2, 59.6) |
9.6 (8.7, 11.2) |
|
| cTnI(210C)- actin(C374) |
Mg2+ | 47.3 (46.1, 48.4) |
5.4 (4.9, 5.8) | 54.2 (53.2, 55.2) |
4.9 (4.3, 5.6) |
| Mg2+ + Ca2+ |
53.1 (52.0, 54.3) |
7.2 (6.4, 8.3) | 57.5 (56.4, 59.0) |
8.4 (6.9, 9.8) | |
• Numbers in parentheses are the lower and upper bounds of the best fitted parameters at the 68% confidence level (one standard deviation).
• Thin Filament + S1-ADP was prepared by mixing each troponin component at a ratio of one cTn, one cTm, 7.5 polymerized actin, and seven S1-ADP (see Materials and Methods).
Within the inhibitory region of cTnI, Ca2+ binding induced a small increase (> 2 Å) in the distance between actin and cTnI(131C) and a larger increase (11.1 Å) in the other distance between actin and cTnI(151C). The half-widths of these two distance distributions also increased from the Mg2+ state to the Ca2+ state. Similar Ca2+-induced increases were also observed in the presence of strongly bound S1. cTnI residue 131 is located at the N-terminal end of the inhibitory region and adjacent to the cTnI-cTnC interface and residue 151 is located at the junction between the inhibitory and regulatory regions. These two regions are contiguous and expected to move together by Ca2+ and S1 binding. The inhibitory region of cTnI is expected to be released from actin by Ca2+ binding, and this release conferred a more flexible inhibitory region as reflected by increases in the half-widths of the distance distributions.
In the second actin binding region or mobile domain of cTnI (residues 188 and 210), small increases in inter-site distances (4.6-5.8 Å) between actin and these two residues were induced by Ca2+. The half-widths of the distance distributions were more than a factor of two smaller than those for the other distance distributions regardless of whether Ca2+ was absent or present (5.4-8.6 Å). These narrower distributions suggested that the second actin-binding region in cTnI is relatively rigid in comparison with the other regions of cTnI in the thin filament, which is consistent with the rigid helical structure of the second actin binding region demonstrated in the Ca2+-saturated core domain of cardiac troponin by X-ray crystallography (49), and the organized structure of the second actin binding domain in the absence of Ca2+ demonstrated by NMR (50), and by electron microscopy reconstruction (51). Strongly bound S1 lengthened these two inters-site distances, but its effect on the half-widths of the two distance distributions was small (< 1 Å).
The thin filament is organized around a central filament of actin which is a symmetric right-handed double helix. Each regulatory unit within the filament consists of one cTn, one cTm and seven actin monomers. This structure potentially can give rise to multiple FRET from a donor attached to a cTnI site to acceptors on neighboring actin monomers. In a previous modeling study (52), an analysis method was developed that incorporated multiple transfer from skeletal muscle TnI (sTnI) residue 133 to each of several actin residues. The results showed that the contribution of multiple transfer to the observed transfer efficiency was negligible for transfer to actin 374, but appreciable for transfer to other actin sites. Since sTnI(133) is equivalent to cTnI(167), the present observed FRET efficiency between cTnI(167) and actin 374 likely contained little of no multiple transfer. However, the contribution of multiple transfer to the observed transfer efficiency was not known for the other five FRET distances. Since we were interested in changes of the distributions of distances resulting from ligand binding to the thin filament, the apparent distributions enabled an analysis of these changes in the absence of correction for multiple transfer. Also in our distance calculations a value of ⅔ was used for the orientation factor κ2, assuming that both donor and acceptor probes tumbled rapidly and isotropically. If ligand binding to thin filaments substantially modified probe mobility, a different value would need to be used for κ2 to calculate distance parameters for systems containing bound Ca2+ and S1. Since the acceptor probe was non-fluorescent, it was not feasible to evaluate with some certainty the possible range of κ2 that would be applicable to the systems with and without bound ligands. We measured the anisotropy decay of the donor probe attached to each of the six sites in cTnI, and determined the anisotropies associated with the attached probe. The results (not shown) showed very similar donor mobility in all systems, suggesting that ligand binding to the thin filaments did not affect the mobility of the donor in all cTnI sites. The observed changes in both inter-site distance and the half-width of the distance distribution induced by bound Ca2+ and S1 reflected a conformational effect and not a change in donor mobility.
To further investigate the effect of strongly bound S1 on the six inter-site distances, we used a second preparation of thin filaments reconstituted with a cTnC mutant (D65V/D67A) to repeat distance measurements. This mutant cannot chelate Ca2+ because the first and third acidic residues within the 12-residue Ca2+-binding loop were replaced by hydrophobic side chains (53). The results are summarized in Table 3. As expected, Ca2+ had no effect on all six inter-site distances and negligible effects (within experimental errors) on the half-widths of the distributions. Bound S1 conferred increases in the distances in the range of 4-10 Å. This range was comparable to that observed (4-14 Å) with the preparation reconstituted with functionally competent cTnC between the Mg2+ state and the (Mg2+ + S1) state (Table 2). With the exception of the actin-cTnI(210C) distance, bound S1 induced small increases in the half-width (2.8-4.7 Å) of the other five distributions. These increases were similar to those (0.2-2.5 Å) elicited by bound S1 in Ca2+-competent thin filaments. The distribution of the distance actin-cTnI(210C) showed a decrease (-4.6 Å) in the half-width with thin filaments complexed with S1. A marginal decrease (-0.5 Å) in the half-width of the distribution of the same actin-cTnI(160C) distance was also observed with thin filaments containing functionally competent cTnC. Taken together these results showed that bound S1 alone was capable of inducing a separation of the C374 region of actin from several regions of cTnI.
Table 3.
FRET distances from actin C374 to cTnI in cardiac thin filaments reconstituted with cTnC mutant5 D65V/D67A which does not bind regulatory Ca2+*
| Thin Filament | Thin Filament + S1-ADP | ||||
|---|---|---|---|---|---|
| Distance | Conditions | Mean Distance (Å) | Half-Width (Å) | Mean Distance (Å) | Half-Width (Å) |
| cTnI(131C)-actin(C374) | Mg2+ | 38.3 (37.1, 40.0) | 15.8 (14.2, 16,7) | 42.6 (41.3, 43.9) | 20.5 (19.3, 21.9) |
| Mg2+ + Ca2+ | 37.7 (36.5, 38.9) | 15.7 (14.3, 16.8) | 44.9 (43.8, 46.1) | 19.0 (17.9, 19.9) | |
| cTnI(151C)-actin(C374) | Mg2+ | 46.4 (45.8, 47.2) | 10.5 (9.1, 11.7) | 51.7 (50.9, 52.8) | 15.3 (13.4, 16.0) |
| Mg2+ + Ca2+ | 46.2 (45.0, 47.1) | 14.2 (13.4, 15.1) | 52.1 (51.5, 52.9) | 18.4 (16.7, 19.9) | |
| cTnI(160C)-actin(C374) | Mg2+ | 47.3 (46.5, 48.3) | 16.3 (15.8, 16.7) | 54.6 (53.7, 55.9) | 20.6 (19.7, 21.6) |
| Mg2+ + Ca2+ | 47.9 (46.8, 48.6) | 17.2 (16.8, 17.7) | 56.6 (55.3, 57.5) | 20.7 (19.6, 21.5) | |
| cTnI(167C)-actin(C374) | Mg2+ | 43.1 (42.0, 43.9) | 19.4 (18.9, 19.8) | 52.4 (51.7, 53.7) | 22.7 (21.9, 23.5) |
| Mg2+ + Ca2+ | 42.4 (41.8, 43.7) | 21.1 (20.7, 21.9) | 53.5 (52.2, 54.7) | 23.4 (21.7, 26.0) | |
| cTnI(188C)-actin(C374) | Mg2+ | 46.1 (45.3, 47.5) | 4.3 (3.9, 4.6) | 56.1 (55.2, 57.8) | 7.1 (6.6, 7.7) |
| Mg2+ + Ca2+ | 46.8 (45.3, 47.9) | 6.0 (5.7, 6.6) | 56.0 (55.1, 57.4) | 10.8 (9.9, 11.1) | |
| cTnI(210C)-actin(C374) | Mg2+ | 48.8 (47.8, 49.6) | 11.6 (10.8, 12.4) | 54.3 (53.2, 55.7) | 7.0 (6.6, 7.4) |
| Mg2+ + Ca2+ | 49.0 (48.1, 50.4) | 10.7 (9.9, 11.3) | 54.6 (53.7, 55.7) | 3.6 (3.3, 3.8) | |
• Numbers in parentheses are the lower and upper bounds of the best fitted parameters at the 68% confidence level (one standard deviation)
The results of inter-site distances shown in Table 2 and Table 3 were obtained with thin filaments preparations to which cations were first added, followed by addition of (S1 + ADP). Each addition resulted in an increase in the distance. It was of interest to establish whether the increase elicited by bound S1 was independent of the cation state. Additional experiments were performed in which S1 was first added, followed by addition of Ca2+. The results for the distance between actin and cTnI(C167) are shown in Fig. 5 in a closed pathway. Starting from the initial state of the thin filament in the presence of Mg2+(TF•Mg2+), the clockwise pathway showed an increase of 9 Å in the distance induced by bound Ca2+ in the intermediate state, and an additional increase of 10.6 Å by the subsequent addition of S1-ADP to form the final state, TF•S1•Mg2+•Ca2+. The counter clockwise pathway showed an increase of 13.5Å in the initial step in which S1-ADP was added, and a second increase of 6.4 Å induced by the addition of Ca2+ to reach the same final state. The sum of the two increases was the same for the two pathways. This additive effect was also observed in the other five actin-cTnI distances, with differences between the two pathways in each case being < ± 0.2 Å. The experiments with thin filaments reconstituted from a non-functional cTnC mutant (Table 3) were repeated by reversing the order of addition of Ca2+ and S1-ADP. The total distance increase from actin to each cTnI site between the initial and final states were within 0.1-0.3 Å of both clockwise pathway and counter clockwise pathway (results not shown). These results suggested that the conformational effect of strongly bound S1 on the disposition of the C-terminus of actin from functionally important regions of cTnI was not influenced by the presence of functional cTnC.
Figure 5.
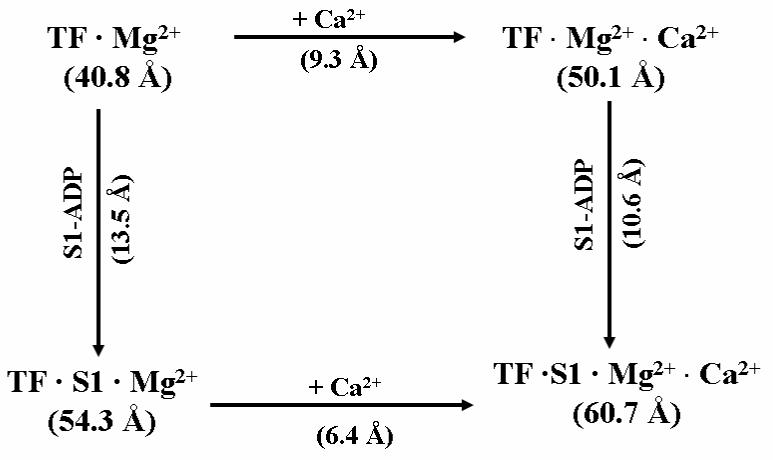
A cyclic representation of the FRET distance between actin C374 and cTnI C167C in cardiac thin filaments (TF) determined in three states. The initial complex is TF•Mg2+ in the Mg2+ state in which the distance is 40.8 Å. In the clockwise direction, Ca2+ binding to TF•Mg2+ to form the second complex TF•Mg2+•Ca2+ increases the distance by 9.3 Å, and the subsequent binding of S1 to form the final complex TF•S1•Mg2+•Ca2+ (right vertical pathway) lengthens the distance by an additional 10.6 Å. The sum of the two increases in these two clockwise steps is 9.3 + 10.6 = 19.9 Å. In the counter clockwise pathway, S1 first binds to TF•Mg2+ to form TF•S1•Mg2+, increasing the distance by 13.5 Å. Ca2+ binding to form the final complex TF•S1•Mg2+•Ca2+ (lower pathway) lengthens the distance by another 6.4 Å. The sum of these two increases in the counter clockwise direction is 13.5 + 6.4 = 19.9 Å. The difference in the distances between TF•Mg2+ (initial state) and TF•S1•Mg2+•Ca2+(final state) is 19.9 Å (60.7 – 40.8), independent of how the final state is reached, whether along the clockwise or the counter clockwise direction.
The unique N-terminal extension of cTnI has two PKA sites (Ser 23 and Ser 24). In a previous study we reported that bisphosphorylation of these two sites resulted in a bending of this segment toward the C-terminus (24). We investigated the effects of bisphosphorylation of cTnI on the proximity relationship between actin Cys374 and four of the six sites in cTnI (residues 131, 151, 160 and 167) in regulated thin filaments. These results are summarized in Table 4. Bisphosphorylation increased the distance from actin to cTnI(131) by 7 Å in the presence of Mg2+, and an increase of 5 Å in the presence of both Mg2+ and Ca2+. The difference of 1.7 Å was observed with non-phosphorylated thin filaments between the two ionic environments. This distance in the bisphosphorylated preparation was not sensitive to Ca2+ (Table 4). Bound S1 induced a small separation (2.1 Å) in the presence of Mg2+ and a larger separation (5.2 Å) in the presence of both Mg2+ and Ca2+ between the two sites in which cTnI was bisphosphorylated. The effects of bisphosphorylation on the separations from actin to the other three cTnI sites were negligibly smaller (± 1 Å), except that a change of 2.2 Å between actin and cTnI(151) was observed in the presence of Ca2+ and strongly bound S1. The distributions of the four distances determined with bisphosphorylated cTnI were all narrower than the corresponding distributions from thin filaments containing non-phosphorylated cTnI. The half-widths of these narrower distributions were smaller than those from the corresponding non-phosphorylated samples by 1-10 Å. This may suggest that phosphorylation of the N-terminus of cTnI can affect structural dynamics of the regulatory and inhibitory regions of cTnI at the interface between cTnI and actin.
Table 4.
FRET distances from actin C374 to cTnI in cardiac thin filaments reconstituted with bisphosphorylated cTnI
| Thin Filament | Thin Filament + S1-ADP | ||||
|---|---|---|---|---|---|
| Distance | Conditions | Mean Distance (Å) |
Half-Width (Å) | Mean Distance (Å) |
Half-Width (Å) |
| p-cTnI(131C)-actin(C374) | Mg2+ | 44.3 (43.1, 45.6) | 11.5 (10.2, 13.6) | 46.4 (44.3, 58.3) | 12.4 (11.9, 15.9) |
| Mg2+ + Ca2+ | 44.3 (43.4, 45.4) | 12.0 (10.8, 13.3) | 49.5 (48.4, 50.3) | 14.4 (12.7, 16.9) | |
| p-cTnI(151C)-actin(C374) | Mg2+ | 44.9 (43.6, 55.7) | 9.5 (6.9, 11.6) | 53.5 (52.1, 54.5) | 11.7 (11.0, 12.4)) |
| Mg2+ + Ca2+ | 53.4 (52.0, 55.2) | 7.0 (6.2, 7.7) | 59.0 (48.0, 50.3) | 10.1 (9.3, 11.1) | |
| p-cTnI(160C)-actin(C374) | Mg2+ | 48.3 (47.1, 49.6) | 10.3 (9.4, 11.1) | 56.2 (55.1, 57.7) | 9.9 (8.3, 11.4)) |
| Mg2+ + Ca2+ | 58.0 (56.8, 59.3) | 11.1 (9.7, 12.8) | 60.1 (58.9, 61.6) | 12.8 (11.1, 14.3)) | |
| p-cTnI(167C)-actin(C374) | Mg2+ | 42.1 (41.0, 43.4) | 15.1 (14.2, 15.9) | 55.4 (54.2, 56.8) | 16.2 (15.6, 16.9) |
| Mg2+ + Ca2+ | 54.0 (52.7, 55.4) | 12.7 (10.9, 14.3) | 60.0 (59.0, 61.4) | 11.7 (11.1, 12.7) | |
• Numbers in parentheses are the lower and upper bounds of the best fitted parameters at the 68% confidence level (one standard deviation.
Discussion
Regulation of cardiac thin filaments is triggered by Ca2+ binding to cTnC which results in a series of conformational changes within the filament, including changes in the actin-cTnI interaction. In the present study, we have determined FRET between a donor attached to several residues in the C-terminal region of cTnI and an acceptor attached to actin C374 in its C-terminus. Results from these measurements enable us to investigate conformational changes that occur at the interface between actin and the C-terminal region of cTnI resulting from the binding of regulatory Ca2+ and myosin, and bisphosphorylation of cTnI in its N-terminal extension.
The current three-state model for activation of thin filaments in striated muscle includes a blocked state, a closed state, and an open state (11, 54). The equilibrium between the blocked and closed states is Ca2+-sensitive in which the actin binding sites for myosin are sterically blocked by tropomyosin, and this blocking is removed by bound Ca2+ in the closed state. The Ca2+-induced closed state enables crossbridges to weakly bind to the thin filament, and the open state is regulated and stabilized by crossbridges strongly binding to actin. The open state involves movement of tropomyosin on the helical actin surface to expose binding sites for crossbridges to bind strongly to actin, thus generating force. A reversible shift between the blocked and closed states involves several Ca2+- induced structural transitions within the thin filament, which include a closed-open conformational change of the N-terminal domain of TnC, alternate interactions of the regulatory/inhibitory regions of TnI with TnC and with actin. These structural transitions can be modulated by strong crossbridge formation between actin and play a role in thin filament regulation.
The recent structures of the ternary core domain of cardiac troponin (49) have provided valuable insights into the structures of isolated troponin in the Ca2+-bound state. Previous FRET studies have shown the magnitude of Ca2+-induced opening of the N-domain of cTnC (32), and the kinetics of this opening and the closing of the N-domain associated with Ca2+ dissociation (55). The present results on inter-site separations from the penultimate residue of actin to several sites in the C-terminal region of cTnI suggest large Ca2+-induced movements of the regulatory region of cTnI away from actin C-terminus and rigid conformation of the second or “mobile” actin binding domain of cTnI. These movements are in accord with the idea of a release of cTnI inhibitory region from actin upon Ca2+ activation. The movements of the regulatory region provide a mechanism for lateral movement of tropomyosin on the actin surface to expose sites for interactions to occur between actin and myosin to form crossbridges. A Ca2+-triggered large separation of actin C-terminus from the regulatory region of cTnI (residues 151-167) is demonstrated by the observed increases of 10-12 Å in the inter-site distances between actin C374 and the cTnI residues. These large Ca2+-induced separations are accompanied by smaller separations of actin C-terminus from the N-terminus of the inhibitory region (< 3 Å, residue 131) and from the second actin-binding region (< 6 Å, residues 188 and 210) of cTnI. The large increases in the inter-site distances are a consequence of the movement of the regulatory region away from actin toward the Ca2+-saturated N-domain of cTnC in which a hydrophobic segment of the N-domain interacts with hydrophobic residues of the regulatory region. This sliding of the regulatory region drags along the contiguous inhibitory region thus converting its helix-loop-helix motif into an extended segment (34, 56) and releasing the inhibitory region from actin. Such a “drag and release” mechanism (40) is consistent with the observed large increases of the separations from actin to the cTnI regulatory region and the C-terminus of the inhibitory region and a smaller increase in the separation from actin to the N-terminus of the cTnI inhibitory region (residue 131, < 3 Å). These Ca2+-induced separations in the cTnI-actin interface would allow movements of tropomyosin on the actin surface to expose sites for interaction with crossbridges. These separations between actin C-terminus and cTnI sites are further enhanced by the binding of myosin S1+ADP (2-7 Å). These additional increases are elicited by strong interactions between S1 and actin, equivalent to strong crossbridge formations in muscle. These two-step separations between cTnI and actin revealed by our FRET are consistent with the three-state model for activation of thin filaments (11, 54).
FRET distance measurements has also shown that bound S1 alone can also lengthen the inter-site distances in the absence of bound regulatory Ca2+, or in the presence of an inactivated Ca2+ binding site II in cTnC. If thin filament activation requires structural transitions resulting from separations of regions of actin and cTnI to allow movements of tropomyosin, the present results would suggest that either bound Ca2+ or bound myosin alone could elicit partial activation since the binding of each ligand independently results in increases in inter-site distances. Full activation, however, would require both bound Ca2+ and strongly bound myosin allow complete movements of tropomyosin. An early study showed that activation of thin filaments by strongly bound myosin was independent of bound Ca2+(57). Other studies (58, 59) have suggested that strongly bound crossbridges are also required to expose enough myosin binding sites on the actin filament to accommodate optimal movements of tropomyosin for full activation. While optimal movements of tropomyosin are essential for thin filament activation, the structural basis that promotes the movements is separations of the actin-cTnI interface. These separations are elicited by the binding of regulatory Ca2+ and the binding of myosin in which the effects of binding of the two ligands are independent and additive.
The distribution of inter-site distances provides a qualitative assessment of the dynamics of the interaction between the two sites and conformational flexibility between two regions within a molecular complex. The half-widths of the distributions of the two distances from actin to the residues (131 and 151) in the inhibitory region of cTnI are very similar and suggest a moderate conformational flexibility between actin C-terminus and this region of cTnI in the absence of bound Ca2+. Residue 131 is within the N-terminal helical segment of the inhibitory region that is followed by a segment (residues 134-145) predicted to be in a coil conformation (60). Residue 151 is located in the helical segment that follows the flexible coil. In the absence of bound Ca2+, the flexible coil segment is thought to be bound to actin. The two flanking helical segments appear to have moderate conformational flexibility within the actin-cTnI complex. Bound Ca2+ releases the flexible segment of cTnI from actin, which results in only small increases (2-4 Å) in the half-widths suggesting no large gain in conformational flexibility of the both end segments of the cTnI inhibitory region. Strong binding of S1 to the thin filament increases the half-width of the two distributions by 5-6 Å, suggesting that bound S1 further enhances the conformational flexibility. On the other hand, the half-widths of the distributions for the two distances from actin to the regulatory region (cTnI residues 160, 167) are 6 Å larger than those for the inhibitory region in the absence of bound Ca2+. This larger flexibility is reasonable since the regulatory region is a distance away from the loop segment of the inhibitory region. In the presence of bound Ca2+, these larger half-widths are reduced by 4-5 Å. This loss of conformational flexibility occurs because the regulatory region has moved into the Ca2+-saturated open N-domain of cTnC in which much of the regulatory region is restricted because of a hydrophobic interaction between cTnC and the regulatory region.
The second actin binding domain in the C-terminal segment of cTnI has no contact with cTnC or cTnT within the core structure of troponin. The distributions of the distances from actin C-terminus to cTnI residues 188 and 210 are narrow with half-widths being 5-7 Å in the absence of Ca2+. Bound Ca2+and S1 confer only marginal broadening of the distributions, indicative of restricted conformational flexibility of the residues relative to actin C-terminus. An α-helix is formed between cTnI residues 165 and 189 in the crystal structure (49), and the C-terminus (residues 170-211) is the putative second actin-binding site in the absence of Ca2+(61). A recent NMR study shows that the mobile domain or the second actin-binding domain of skeletal TnI containing residues 131-182, corresponding to the residues 164-210 of cTnI, forms a helix (residues 132-140), a mini-globular subdomain (residues143-167) and a helix (residues 170-177) (50). The structural model constructed by docking of the NMR derived mobile domain structure into the cryo-electron microscopy density map of the thin filament suggests an interaction between the domain of TnI and the outer domain of actin in the absence of Ca. The narrow distributions of the two FRET distances are consistent with this model. The electron density of several residues associated the C-terminus of cTnI including the last 19 residues (193-211) was undefined in the crystal structure with bound regulatory Ca2+. A latching mechanism proposed for the regulatory role of cTnI in a recent study suggests that the second or “mobile” actin binding domain of cTnI is highly dynamic in the presence of Ca2+ and becomes more structural upon interaction with actin in the absence of Ca2+(62). However, the present results suggest that regions around two of these residues have limited flexibilities in either the presence or the absence of bound Ca2+.
Phosphorylation of the two serines at residues 23 and 24 of cTnI by PKA plays important roles in modulation myofilament Ca2+ sensitivity and crossbridge kinetics (20, 21, 24, 27, 63). Evidences have suggested that in the absence of phosphorylation the N-terminal segment of cTnI interacts with the N-domain of cTnC and stabilizes the open conformation of the domain (27, 28, 64). Bisphosphorylation of cTnI induces a significant conformational change in the N-terminal segment producing a reduction in the axial ratio of cTnI and a more compact structure (23, 24, 65). These conformational changes weaken the interactions between the N-terminal segment of cTnI and the N-domain of cTnC. A recent NMR and modeling study has suggested that the bisphosphorylated N-terminal segment of cTnI interacts with its inhibitory region through electrostatic interactions (26). This suggestion supports the notion that the bisphosphorylation modulates the interactions between the inhibitory region of cTnI and actin-tropomyosin. The observed increases in the FRET distances between cTnI(131) and actin induced by bisphosphorylation are in accord with the NMR and modeling results. The effect of bisphosphorylation on the distances from actin to the other three cTnI sites is not on the magnitude of the distances, but on the half-width of the distance distributions. The reduced half-widths are correlated with decreases in structural dynamics of cTnI in the interface between troponin and the actin filament. These results suggest that PKA phosphorylation of cTnI has minimal structural effects, but they may have significant structural dynamic and kinetic effects on cTnI-actin interaction to modulate myofilament Ca2+ sensitivity and crossbridge kinetics.
In summary, we investigated the effects of bound regulatory Ca2+, strongly bound myosin, and PKA bisphosphorylation of cTnI on the separations from actin Cys374 to several actin-binding sites within the inhibitory region, regulatory region and the second actin binding region of cTnI. The results show that bound Ca2+ induces large increases in the distances from actin to the cTnI sites, indicating a Ca2+ triggered separation of actin from cTnI. Bound myosin S1 (S1.MgADP) induces additional increases in these distances. These two-step changes in distances between cTnI and actin provide direct evidence to link structural changes at the interface between cTnI and actin to the three-state model of thin filament regulation of muscle contraction and relaxation. In the absence of bound Ca2+, bound S1 also lengthens these distances. These results suggest that either bound Ca2+ or bound S1 alone can partially activate the thin filament, but full activation requires both bound Ca2+ and strongly bound myosin. The FRET distance distribution analysis suggests that the second actin binding region of cTnI within the thin filament is rigid comparing with the inhibitory/regulatory region. The inhibitory region is more flexible in the Ca2+ bound state than in the Mg2+ state. PKA phosphorylation of cTnI changes the interaction between actin and the inhibitory region of cTnI. The bisphosphorylation also alters the flexibility of both the inhibitory and regulatory regions of cTnI. These alterations may be related to changes in the electrostatic interactions between the N-terminal segment and the inhibitory region of cTnI.
Abbreviations
- Tn
troponin
- TnC
troponin C
- TnI
troponin I
- TnT
troponin T
- Tm
tropomyosin
- c
cardiac muscle
- PKA
Protein Kinase A
- FRET
Förster resonance energy transfer
- DTT
dithiothreitol
- Mops
3-(N-mopholino)propanesulfonic acid
- EGTA
ethylene glycol-bis-(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid
- IAEDANS
5-(iodoacetamidoethyl)aminonaphthelene-1-sulfonic acid
- DABM
4-dimethylaminophenylazophenyl-4’-maleimide
Footnotes
This work was supported in part by National Institutes of Health Grant HL80186 (W.-J. D.) and HL52508 (H. C. C.).
References
- (1).Ebashi S, Endo M, Otsuki I. Control of muscle contraction. Q Rev Biophys. 1969;2:351–84. doi: 10.1017/s0033583500001190. [DOI] [PubMed] [Google Scholar]
- (2).Farah CS, Reinach FC. The troponin complex and regulation of muscle contraction. FASEB J. 1995;9:755–767. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- (3).Haselgrove JC. X-ray evidence for a conformational change in the actin-containing filaments of vertebrate striated muscle. Cold Spring Harbor Symp. Qant. Biol. 1972;37:341–352. [Google Scholar]
- (4).Huxley HE. Structural changes in the actin-and myosin-containing filaments during contraction. Cold Spring Harbor Symp. Qant. Biol. 1972;37:361–376. [Google Scholar]
- (5).Parry DA, Squire JM. Structural role of tropomyosin in muscle regulation: analysis of the x-ray diffraction patterns from relaxed and contracting muscles. J. Mol. Biol. 1973;75:33–55. doi: 10.1016/0022-2836(73)90527-5. [DOI] [PubMed] [Google Scholar]
- (6).Gordon AM, Homsher E, Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- (7).Gordon AM, Ridgway EB. Extra calcium on shortening in barnacle muscle. Is the decrease in calcium binding related to decreased cross-bridge attachment, force, or length? J Gen Physiol. 1987;90:321–40. doi: 10.1085/jgp.90.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Guth K, Potter JD. Effect of rigor and cycling cross-bridges on the structure of troponin C and on the Ca2+ affinity of the Ca2+-specific regulatory sites in skinned rabbit psoas fibers. J. Biol. Chem. 1987;262:13627–13635. [PubMed] [Google Scholar]
- (9).Hannon JD, Martyn DA, Gordon AM. Effects of cycling and rigor crossbridges on the conformation of cardiac troponin C. Circ. Res. 1992;71:984–991. doi: 10.1161/01.res.71.4.984. [DOI] [PubMed] [Google Scholar]
- (10).Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- (11).McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- (13).Moss RL, Nwoye LO, Greaser ML. Substitution of cardiac troponin C into rabbit muscle does not alter the length dependence of Ca2+ sensitivity of tension. J Physiol. 1991;440:273–89. doi: 10.1113/jphysiol.1991.sp018708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wolff MR, McDonald KS, Moss RL. Rate of tension development in cardiac muscle varies with level of activator calcium. Circ Res. 1995;76:154–60. doi: 10.1161/01.res.76.1.154. [DOI] [PubMed] [Google Scholar]
- (15).Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ Res. 2004;94:146–58. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- (16).Solaro RJ. Modulation of cardiac myofilament activity by protein phosphorylation. In: Page EF, Harry A, Solaro RJ, editors. Handbook of physiology. Section 2: The Cardiovascular System. Volume I: The Heart. Oxford University Press; Oxford: 2002. pp. 264–300. [Google Scholar]
- (17).Moir AJ, Solaro RJ, Perry SV. The site of phosphorylation of troponin I in the perfused rabbit heart. The effect of adrenaline. Biochem J. 1980;185:505–13. doi: 10.1042/bj1850505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ray KP, England PJ. The identification and properties of phosphatases in skeletal muscle with activity towards the inhibitory subunit of troponin, and their relationship to other phosphoprotein phosphatases. Biochemical Journal. 1976;70:11–16. doi: 10.1042/bj1570369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Solaro RJ. Modulation of activation of cardiac myofilamens by beta-adrenergic agonists. In: Allen DG, Lee JA, editors. Modulation of Cardiac Calcium Sensitivity. Oxford University Press; Oxford: 1993. pp. 160–177. [Google Scholar]
- (20).Kentish JC, McCloskey DT, Layland J, Palmer S, Leiden JM, Martin AF, Solaro RJ. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ Res. 2001;88:1059–65. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- (21).Reiffert SU, Jaquet K, Heilmeyer LM, Jr., Ritchie MD, Geeves MA. Bisphosphorylation of cardiac troponin I modulates the Ca(2+)-dependent binding of myosin subfragment S1 to reconstituted thin filaments. FEBS Lett. 1996;384:43–7. doi: 10.1016/0014-5793(96)00274-8. [DOI] [PubMed] [Google Scholar]
- (22).Dong WJ, Wang CK, Gordon AM, Rosenfeld SS, Cheung HC. A kinetic model for the binding of Ca2+ to the regulatory site of troponin from cardiac muscle. J Biol Chem. 1997;272:19229–35. doi: 10.1074/jbc.272.31.19229. [DOI] [PubMed] [Google Scholar]
- (23).Liao R, Wang CK, Cheung HC. Coupling of calcium to the interaction of troponin I with troponin C from cardiac muscle. Biochemistry. 1994;33:12729–34. doi: 10.1021/bi00208a026. [DOI] [PubMed] [Google Scholar]
- (24).Dong WJ, Chandra M, Xing J, She M, Solaro RJ, Cheung HC. Phosphorylation-induced distance change in a cardiac muscle troponin I mutant. Biochemistry. 1997;36:6754–61. doi: 10.1021/bi9622276. [DOI] [PubMed] [Google Scholar]
- (25).Chandra M, Dong WJ, Pan BS, Cheung HC, Solaro RJ. Effects of protein kinase A phosphorylation on signaling between cardiac troponin I and the N-terminal domain of cardiac troponin C. Biochemistry. 1997;36:13305–11. doi: 10.1021/bi9710129. [DOI] [PubMed] [Google Scholar]
- (26).Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J Mol Biol. 2007;373:706–22. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- (27).Abbott MB, Dong WJ, Dvoretsky A, DaGue B, Caprioli RM, Cheung HC, Rosevear PR. Modulation of cardiac troponin C-cardiac troponin I regulatory interactions by the amino-terminus of cardiac troponin I. Biochemistry. 2001;40:5992–6001. doi: 10.1021/bi0100642. [DOI] [PubMed] [Google Scholar]
- (28).Finley N, Abbott MB, Abusamhadneh E, Gaponenko V, Dong W, Gasmi-Seabrook G, Howarth JW, Rance M, Solaro RJ, Cheung HC, Rosevear PR. NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett. 1999;453:107–12. doi: 10.1016/s0014-5793(99)00693-6. [DOI] [PubMed] [Google Scholar]
- (29).Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA, Robbins J. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem. 2005;280:703–14. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]
- (30).Schmidtmann A, Lohmann K, Jaquet K. The interaction of the bisphosphorylated N-terminal arm of cardiac troponin I-A 31P-NMR study. FEBS Lett. 2002;513:289–93. doi: 10.1016/s0014-5793(02)02340-2. [DOI] [PubMed] [Google Scholar]
- (31).Ward DG, Brewer SM, Gallon CE, Gao Y, Levine BA, Trayer IP. NMR and mutagenesis studies on the phosphorylation region of human cardiac troponin I. Biochemistry. 2004;43:5772–81. doi: 10.1021/bi036310m. [DOI] [PubMed] [Google Scholar]
- (32).Dong WJ, Xing J, Villain M, Hellinger M, Robinson JM, Chandra M, Solaro RJ, Umeda PK, Cheung HC. Conformation of the regulatory domain of cardiac muscle troponin C in its complex with cardiac troponin I. J. Biol. Chem. 1999;274:31382–31390. doi: 10.1074/jbc.274.44.31382. [DOI] [PubMed] [Google Scholar]
- (33).Dong WJ, Chandra M, Xing J, Solaro RJ, Cheung HC. Conformation of the N-terminal segment of a monocysteine mutant of troponin I from cardiac muscle. Biochemistry. 1997;36:6745–53. doi: 10.1021/bi962226d. [DOI] [PubMed] [Google Scholar]
- (34).Dong WJ, Xing J, Robinson JM, Cheung HC. Ca2+ induces an extended conformation of the inhibitory region of troponin I in cardiac muscle troponin. J. Mol. Biol. 2001;314:51–61. doi: 10.1006/jmbi.2001.5118. [DOI] [PubMed] [Google Scholar]
- (35).Smillie LB. Preperation and identification of a- and b-Tropomyosins. Methods in Enzymology. 1982;85:234–241. doi: 10.1016/0076-6879(82)85023-4. [DOI] [PubMed] [Google Scholar]
- (36).Pardee JD, Spudich JA. Purificatin of muscle actin. Methods in Enzymology. 1982;85:164–181. doi: 10.1016/0076-6879(82)85020-9. [DOI] [PubMed] [Google Scholar]
- (37).Xing J, Cheung HC. Vanadate-induced changes in myosin subfragment-1 from cardiac muscle. Arch. Biochem. Biophys. 1994;313:229–234. doi: 10.1006/abbi.1994.1381. [DOI] [PubMed] [Google Scholar]
- (38).Kobayashi T, Kobayashi M, Collins JH. Ca2+-dependent, myosin subfragment 1-induced proximity changes between actin and the inhibitory region of troponin I. Biochim. Biophys. Acta. 2001;1549:148–154. doi: 10.1016/s0167-4838(01)00254-0. [DOI] [PubMed] [Google Scholar]
- (39).Tao T, Gong BJ, Leavis PC. Calcium-induced movement of troponin-I relative to actin in skeletal muscle thin filaments. Science. 1990;247:1339–1341. doi: 10.1126/science.2138356. [DOI] [PubMed] [Google Scholar]
- (40).Robinson JM, Dong WJ, Xing J, Cheung HC. Switching of troponin I: Ca(2+) and myosin-induced activation of heart muscle. J Mol Biol. 2004;340:295–305. doi: 10.1016/j.jmb.2004.04.046. [DOI] [PubMed] [Google Scholar]
- (41).Dong WJ, Xing J, Chandra M, Solaro J, Cheung HC. Structural mapping of single cysteine mutants of cardiac troponin I. Proteins. 2000;41:438–47. [PubMed] [Google Scholar]
- (42).Wang CK, Cheung HC. Proximity relationship in the binary complex formed between troponin I and troponin C. J Mol Biol. 1986;191:509–21. doi: 10.1016/0022-2836(86)90145-2. [DOI] [PubMed] [Google Scholar]
- (43).She M, Dong WJ, Umeda PK, Cheung HC. Time-resolved fluorescence study of the single tryptophans of engineered skeletal muscle troponin C. Biophys J. 1997;73:1042–55. doi: 10.1016/S0006-3495(97)78137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).She M, Xing J, Dong WJ, Umeda PK, Cheung HC. Calcium binding to the regulatory domain of skeletal muscle troponin C induces a highly constrained open conformation. J. Mol. Biol. 1998;281:445–452. doi: 10.1006/jmbi.1998.1933. [DOI] [PubMed] [Google Scholar]
- (45).Lakowicz JR, Gryczynski I, Cheung HC, Wang CK, Johnson ML, Joshi N. Distance distributions in proteins recovered by using frequency-domain fluorometry. Applications to troponin I and its complex with troponin C. Biochemistry. 1988;27:9149–60. doi: 10.1021/bi00426a012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Robinson JM, Wang Y, Kerrick WG, Kawai R, Cheung HC. Activation of Striated Muscle: Nearest-neighbor Regulatory-unit and Cross-bridge Influence on Myofilament Kinetics. J. Mol. Biol. 2002b;322:1065–1088. doi: 10.1016/s0022-2836(02)00855-0. [DOI] [PubMed] [Google Scholar]
- (47).Bevington PR, Robinson DK. Data Reduction and Error Analysis for the Physical Sciences. 2 McGraw Hill; Boston: 1992. [Google Scholar]
- (48).Cheung HC, Wang CK, Gryczynski I, Wiczk W, Laczko G, Johnson ML, Lakowicz JR. Distance distributions and anisotropy decays of troponin C and its complex with troponin I. Biochemistry. 1991;30:5238–5247. doi: 10.1021/bi00235a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- (50).Murakami K, Yumoto F, Ohki SY, Yasunaga T, Tanokura M, Wakabayashi T. Structural basis for Ca2+-regulated muscle relaxation at interaction sites of troponin with actin and tropomyosin. J Mol Biol. 2005;352:178–201. doi: 10.1016/j.jmb.2005.06.067. [DOI] [PubMed] [Google Scholar]
- (51).Galinska-Rakoczy A, Engel P, Xu C, Jung H, Craig R, Tobacman LS, Lehman W. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. J Mol Biol. 2008;379:929–35. doi: 10.1016/j.jmb.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Robinson JM, Dong WJ, Cheung HC. Can Forster resonance energy transfer measurements uniquely position troponin residues on the actin filament? A case study in multiple-acceptor FRET. J Mol Biol. 2003;329:371–80. doi: 10.1016/s0022-2836(03)00424-8. [DOI] [PubMed] [Google Scholar]
- (53).Sweeney HL, Brito RM, Rosevear PR, Putkey JA. The low-affinity Ca2(+)-binding sites in cardiac/slow skeletal muscle troponin C perform distinct functions: site I alone cannot trigger contraction. Proc Natl Acad Sci U S A. 1990;87:9538–42. doi: 10.1073/pnas.87.24.9538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Maytum R, Lehrer SS, Geeves MA. Cooperativity and switching within the three-state model of muscle regulation. Biochemistry. 1999;38:1102–1110. doi: 10.1021/bi981603e. [DOI] [PubMed] [Google Scholar]
- (55).Dong WJ, Robinson JM, Xing J, Cheung HC. Kinetics of conformational transitions in cardiac troponin induced by Ca2+ dissociation determined by Forster resonance energy transfer. J Biol Chem. 2003;278:42394–402. doi: 10.1074/jbc.M304858200. [DOI] [PubMed] [Google Scholar]
- (56).Dong WJ, Robinson JM, Stagg S, Xing J, Cheung HC. Ca2+-induced conformational transition in the inhibitory and regulatory regions of cardiac troponin I. J Biol Chem. 2003;278:8686–92. doi: 10.1074/jbc.M212886200. [DOI] [PubMed] [Google Scholar]
- (57).Bremel RD, Weber A. Cooperation within actin filament in vertebrate skeletal muscle. Nature New Biol. 1972;238:97–101. doi: 10.1038/newbio238097a0. [DOI] [PubMed] [Google Scholar]
- (58).Butters CA, Tobacman JB, Tobacman LS. Cooperative effect of calcium binding to adjacent troponin molecules on the thin filament-myosin subfragment 1 MgATPase rate. J Biol Chem. 1997;272:13196–202. doi: 10.1074/jbc.272.20.13196. [DOI] [PubMed] [Google Scholar]
- (59).Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J. Mol. Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- (60).Tung CS, Wall ME, Gallagher SC, Trewhella J. A model of troponin-I in complex with troponin-C using hybrid experimental data: the inhibitory region is a beta-hairpin. Protein Sci. 2000;9:1312–1326. doi: 10.1110/ps.9.7.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Takeda S, Kobayashi T, Taniguchi H, Hayashi H, Maeda Y. Structural and functional domains of the troponin complex revealed by limited digestion. Eur J Biochem. 1997;246:611–7. doi: 10.1111/j.1432-1033.1997.00611.x. [DOI] [PubMed] [Google Scholar]
- (62).Hoffman RM, Blumenschein TM, Sykes BD. An interplay between protein disorder and structure confers the Ca2+ regulation of striated muscle. J Mol Biol. 2006;361:625–33. doi: 10.1016/j.jmb.2006.06.031. [DOI] [PubMed] [Google Scholar]
- (63).Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76:1028–35. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- (64).Gaponenko V, Abusamhadneh E, Abbott MB, Finley N, Gasmi-Seabrook G, Solaro RJ, Rance M, Rosevear PR. Effects of troponin I phosphorylation on conformational exchange in the regulatory domain of cardiac troponin C. J Biol Chem. 1999;274:16681–4. doi: 10.1074/jbc.274.24.16681. [DOI] [PubMed] [Google Scholar]
- (65).Heller WT, Finley NL, Dong WJ, Timmins P, Cheung HC, Rosevear PR, Trewhella J. Small-angle neutron scattering with contrast variation reveals spatial relationships between the three subunits in the ternary cardiac troponin complex and the effects of troponin I phosphorylation. Biochemistry. 2003;42:7790–800. doi: 10.1021/bi0341509. [DOI] [PubMed] [Google Scholar]