

Abstract
Solid-state hydrogen/deuterium exchange (ssHDX) with electrospray ionization mass spectrometry (ESI-MS) and Fourier transform infrared (FTIR) spectroscopy were used to assess protein conformation in amorphous solids. Myoglobin, lysozyme, β-lactoglobulin, ribonuclease A, E-cadherin 5, and concanavalin A were co-lyophilized with carbohydrates (trehalose, raffinose, and dextran 5000), linear polymers (polyvinyl alcohol and polyvinyl pyrrolidone) or guanidine hydrochloride (negative control). For ssHDX, samples were exposed to D2O vapor at 33% relative humidity and room temperature, and then reconstituted at low temperature (4°C) and pH 2.5 and analyzed by ESI-MS. Peptic digestion of selected proteins was used to provide region-specific information on exchange. FTIR spectra were acquired using attenuated total reflectance. FTIR and ssHDX of intact proteins showed preservation of structure by raffinose and trehalose, as indicated by FTIR band intensity and protection from exchange. ssHDX of peptic digests further indicated that these protective effects were not exerted uniformly along the protein sequence but were observed primarily in α-helical regions, a level of structural resolution not afforded by FTIR. The results thus demonstrate the utility of HDX with ESI-MS for analyzing protein conformation in amorphous solid samples.
INTRODUCTION
Proteins and other biotech drugs are among the fastest-growing sectors of the pharmaceutical industry. To protect these labile molecules from chemical and physical degradation, protein drugs are often marketed as solids. The properties of proteins and formulation additives (“excipients”) together with the processing methods used (e.g., lyophilization) typically produce solids that are amorphous rather than crystalline. Though amorphous solids are lower in energy than solutions, there is ample evidence that proteins undergo a variety of degradation processes in the amorphous solid state (1–4). Understanding and controlling these processes is central to the effective development of solid protein drug products.
Although the mechanisms of protein degradation in amorphous solids are far from clear, maintaining native conformation is generally considered critical for preventing degradation. Various methods have been used to assess protein structure in amorphous solids, though far fewer methods are available for solid proteins than for proteins in solution. The most commonly used technique is Fourier transform infrared (FTIR) spectroscopy. FTIR offers the advantages of applicability to both solid and solution samples, ease of sample preparation, and rapid analysis. However, although FTIR can detect gross changes in protein secondary structure, the method lacks sufficient resolution to detect more subtle structural changes. Other spectroscopic methods, such as near-infrared circular dichroism and Raman spectroscopy, and thermal methods such as differential scanning calorimetry (DSC) also have been used to acquire information on protein structure in amorphous solids but share the limited resolution of FTIR. Solid-state nuclear magnetic resonance spectroscopy (ssNMR) has made it possible to solve the structures of membrane proteins at atomic resolution. Current ssNMR methods allow the complete assignment of backbone and side-chain signals for solid proteins in the 5–10 kD range, but generally require that the sample possess a degree of microscopic order (e.g., crystallinity) and/or isotopic labeling. Since protein drugs are often much larger than 10 kD, lack microscopic order in the amorphous solid state, and are not routinely expressed in isotopically enriched forms, the routine application of current ssNMR methods to determine protein drug conformation in amorphous solids is impractical.
Hydrogen/deuterium exchange (HDX) has emerged as a new method for studying protein conformation and excipient interactions in the solid state. In solution, HDX has been used for more than 50 years to study protein conformation, folding, and ligand binding (5,6). Recently, efforts have been made to extend HDX to proteins in the solid state. Generally, lyophilized formulations are exposed to D2O vapor for variable lengths of time at a predetermined relative humidity (RH) value before being analyzed by a suitable technique. French et al. (7) used HDX with FTIR analysis to characterize human granulocyte colony stimulating factor (rhG-CSF) and recombinant consensus interferon-α (rConIFN) in spray-dried powders containing trehalose, using isotopic shifts in the amide II/II′ bands. Desai et al. (8) employed HDX with 1H NMR analysis to study the unfolding of bovine pancreatic trypsin inhibitor (BPTI) on lyophilization. In previous studies by our group (9–11), HDX with tandem liquid chromatography/electrospray ionization mass spectrometry (LC/+ESI-MS) and peptic mapping were used to provide site-specific information on HDX in solid samples of calmodulin. The results demonstrated that low-molecular-weight sugars (i.e., trehalose and sucrose) provided significant protection from exchange relative to excipient-free controls, and that this protection was exerted preferentially in the α-helical fragments. The studies presented here extend this method to other model proteins and compare the results with those obtained by FTIR. The work also tests the hypothesis that protection from HDX by excipients in amorphous solids is exerted nonuniformly along the protein sequence and depends on excipient type and protein structure.
MATERIALS AND METHODS
Materials
Myoglobin, lysozyme, ribonuclease A, β-lactoglobulin, concanavalin A (see Table 1), raffinose, trehalose, polyvinyl alcohol (PVA; average molecular weight of 30,000), polyvinyl pyrrolidone (PVP; average molecular weight of 10,000), tris[2-carboxyethyl] phosphine (TCEP), urea-d4, and guanidine hydrochloride (Gdn·HCl) were obtained from Sigma-Aldrich (St. Louis, MO). Dextran 5000 was obtained from Fluka (Milwaukee, WI). Isopropyl-β-D-thiogalactopyranoside (IPTG) for protein expression was purchased from Amresco (Solon, OH). Pepsin was obtained from Worthington Biochemical Corp. (Lakewood, NJ) and formic acid was obtained from Acros Organics (Morris Plains, NJ). All materials were of reagent grade or higher and used without further purification.
TABLE 1.
Molecular mass and secondary structure of model proteins
| Protein | Molecular mass (kD) | Number of S-S bonds | % α-Helix | % β-Sheet | % unordered |
|---|---|---|---|---|---|
| Myoglobin | 16.95 | 0 | 83 | 0 | 17 |
| Lysozyme | 14.3 | 4 | 40 | 6 | 54 |
| Ribonuclease A | 13.7 | 4 | 27 | 38 | 35 |
| β-Lactoglobulin | 18.3 | 2 | 12 | 35 | 53 |
| E-Cadherin | 12.6 | 2 | 7 | 30 | 60 |
| Concanavalin A | 25.6 | 0 | 7 | 79 | 14 |
Source—RCSB PDB: myoglobin (1WLA), lysozyme (1CXV), RNase A (1RBX), β-lactoglobulin (1CJ5), concanavalin A (1GKB). EC5 information from reports in literature (12).
Expression and purification of E-cadherin 5
E-cadherin 5 (EC5, Table 1) was expressed in Escherichia coli by transforming the recombinant plasmid into BL21 (DE3)-T1R competent cells according to a previously reported protocol (12). Briefly, cells were incubated in self-made LB agar plates with kanamycin as the inhibiting antibiotic. A colony was selected and IPTG added to induce overexpression. After further incubation, the cells were transferred to 4°C to stop growth, pelleted, reconstituted, and French-pressed to lyse the cells. The supernatant was centrifuged and loaded onto a Q-Sepharose column connected to a fast protein liquid chromatography (FPLC) system (Amersham Biosciences, Piscataway, NJ). EC5 fractions were collected and concentrated using Amicon Ultra-15 tubes with a 5000-mol wt cutoff (Millipore Corp., Billerica, MA). The concentrated protein was then further purified using a Superdex 200 column (GE Healthcare, Piscataway, NJ). EC5 concentration was determined by UV absorption at 280 nm.
Sample preparation
Solid samples were prepared by lyophilization from aqueous solution. Then 100 μL samples of protein in solution (4 mg/mL) were directly lyophilized or colyophilized with one of the excipients (i.e., trehalose, raffinose, dextran, PVA, or PVP) in a ratio of 1:1 (w/w). For samples containing Gdn·HCl, 100 μL of a 3 M Gdn·HCl solution were added to the 100 μL protein solution before lyophilization. All samples were lyophilized by first freezing at −35°C for 2 h. Drying was then performed under a vacuum of 15 mT at a shelf temperature of −35°C for 2 h, −5°C for 8h, 5°C for 6 h, and 25°C for 10 h.
Solids characterization
Lyophilized solids were analyzed to determine moisture content, glass transition temperature (Tg), crystallinity, and protein secondary structure.
Thermogravimetric analysis
Thermogravimetric analysis (TGA) was used to measure the water content in the lyophilized samples after exposure to 33% RH conditions for 72 h. Samples were analyzed by a Q50 TGA (TA Instruments, New Castle, DE) with a thermal scan from ambient to 200°C at a scan rate of 10°C/min in an open platinum pan with nitrogen purge. Universal Analysis software (version 4.1, TA Instruments) was used to determine water content from the measured mass loss.
DSC
DSC was used to determine the Tg of the lyophilized formulations after exposure to 33% RH for 72 h. Modulated-temperature DSC (MTDSC) was used to distinguish glass transition events from other kinetic thermal events, such as dehydration and degradation, using a Q100 DSC (TA Instruments). Samples were held isothermally at 25°C for 5 min before increasing the temperature at a ramp rate of 1°C/min with modulation amplitudes of ±0.32°C and a modulation period of 60 s. Universal Analysis software (version 4.1, TA Instruments) software was used for analysis.
Powder x-ray diffraction
Powder x-ray diffraction (PXRD) was performed to ascertain whether the lyophilized samples were in the crystalline or the amorphous state after exposure to 33% RH for 72 h. The lyophilized samples were packed into the shallow cell of a plastic sample holder and covered with a glass slide. A Bruker D8 Discover powder diffractometer with a solid-state detector (Bruker AXS, Madison, WI) was used with Cu-Kα at a scan rate of 1.2° 2θ/min from 10° to 40°. Crystallinity was assessed visually from the PXRD patterns.
FTIR spectroscopy
FTIR spectroscopy was performed to assess protein secondary structure in lyophilized solids. A PerkinElmer FTIR One spectrometer (PerkinElmer Life and Analytical Sciences, Waltham, MA) with a universal attenuated total reflectance accessory (UATR) was used to acquire the spectra. The solid sample was placed on a diamond crystal surface and covered with a stainless-steel slide, and pressure (>100 Torr) was applied to ensure good contact between the protein and the crystal. GRAM AI software (Thermo Electron Corp., Waltham, MA) was used to analyze the spectra. The raw absorption spectra (amide I band) were derivatized followed by area normalization using the GRAMS software to enable direct qualitative and quantitative comparisons between spectra. Quantitation was performed using the method of Kendrick et al. (13). Bands at ∼1660–1650 cm−1 were assigned to α-helical structures, and those at ∼1640–1625 cm−1 were assigned to β-sheet structures. Because the pyrrolidone ring of PVP absorbs in the amide I region, masking the protein signal, FTIR measurements were not performed on samples containing PVP. FTIR spectra were not acquired for samples Gdn·HCl due to partial crystallization.
Solid-state HDX
Solid-state HDX (ssHDX) experiments were performed according to the protocol previously reported by our group (9,10). Lyophilized samples were placed in sealed desiccators at room temperature and 33% RH over D2O, achieved by storing the samples over a saturated solution of MgCl2 in D2O. Samples were collected in triplicate at designated times for immediate analysis or stored at –80°C for later analysis. ssHDX experiments were performed for intact protein and for peptic digests, as described below.
ssHDX for intact protein
ssHDX studies of intact protein were performed to determine the total deuterium uptake by the protein upon exposure to D2O vapor at 33% RH for 72 h. Preliminary data (not shown) and our previous studies (9,10) showed that proteins generally reach a plateau in deuterium uptake after 72 h at 33% RH; this standard storage time was applied to all proteins. Lyophilized formulations were reconstituted with solvent A (94.5% H2O, 5% acetonitrile, and 0.5% formic acid, pH 2.3) to a protein concentration of 4 mg/mL. A 2 μL aliquot was then removed and diluted with an additional 48 μL of solvent A. The sample was then injected into a short C18 trap column (Upchurch Scientific, Oak Harbor, WA) and washed with the aqueous phase for 1.3 min before being eluted with solvent B (19.5% H2O, 80% acetonitrile, and 0.5% formic acid) into the mass spectrometer. A Micromass Q-Tof II mass spectrometer (Waters Corp., Milford, MA) was used in +ESI mode for analysis. The intact protein was detected within 3 min after reconstitution. MassLynx software (version 4.0, Waters Corp.) was used to deconvolute the mass spectrum for the intact molecules, and the mass was taken as the centroid of each deconvoluted peak. Back exchange was corrected using the method of Zhang and Smith (14), and total deuterium incorporation (D) was calculated as
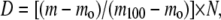 |
(1) |
where m is the mass of the sample protein at any time, mo is the mass of the native protio form of the protein, m100 is the mass of the fully deuterated protein, and N is the total number of exchangeable amides on the protein backbone. The protection (P) from exchange provided by a particular excipient is defined as the difference between the percent deuterium incorporation for an excipient-free control (Dn) and the percent deuterium incorporation for the excipient-containing sample (De), (i.e., P = Dn − De).
ssHDX of peptic digests
Solid samples of selected proteins (i.e., EC5, β-lactoglobulin, and myoglobin) were subjected to proteolytic digestion with pepsin to determine the distribution of deuterium incorporation along the protein sequence. Lyophilized pepsin was dissolved in 10 mM sodium acetate to a final concentration of 15 mg/mL. For each digestion, lyophilized proteins without disulfide bonds (i.e., myoglobin) were reconstituted with 40 μL solvent A followed by the addition of 12 μL pepsin (1:3, protein/pepsin w/w). Proteins with disulfide bonds (i.e., EC5 and β-lactoglobulin) were reconstituted with 35 μL 1M TCEP/4M urea-d4 solution (15) and placed in an ice bath for 4 min to denature the protein and reduce the disulfide bonds before the addition of 12 μL of pepsin solution. In either case (i.e., proteins with or without disulfide bonds), the mixture was introduced into the injection loop (on ice) and digestion was allowed to occur online for 3 min before LC+ESI-MS. The peptides were separated on a C4 reverse phase column (Grace Vydac, Hesperia, CA) with a gradient from 5% to 50% B in 7 min followed by a wash and reequilibration step. Data analysis was performed using the MS scan with the highest ion count; the cluster was then smoothed and centralized to calculate the deuterium uptake after correcting for back exchange by the method of Zhang and Smith (14). The injection port, column, and tubing were kept on ice and low pH solvents were used to minimize back exchange. Deuterium incorporation for each peptic fragment was calculated relative to the deuterated control (see Eq. 1). Back exchange is assumed to be independent of fragment secondary structure or corrected by the use of deuterated controls, as is typical in solution HDX studies (16,17) and as observed in our previous ssHDX studies on calmodulin (9).
RESULTS
Physical characterization of solids
Solid samples were subjected to thermal analysis (DSC and TGA) and x-ray diffraction (PXRD) to establish the physical state (i.e., amorphous versus crystalline) and measure key physical properties (i.e., Tg and water content). PXRD patterns for all samples were consistent with amorphous material, with the exception of samples treated with Gdn·HCl, which showed partial crystallinity (not shown). TGA and DSC were performed for samples containing various excipients and either myoglobin (a representative α-helical protein) or β-lactoglobulin (a representative β-sheet protein). Regardless of the excipient used, water content did not exceed 8% (Table 2), and was highest in dextran and PVP formulations. DSC studies were performed on amorphous formulations to determine Tg. For myoglobin, Tg values increased in the order PVA < trehalose < raffinose < dextran < PVP, whereas for β-lactoglobulin the order was trehalose < PVA < raffinose < dextran ∼ PVP (Table 2). Tg values were >40°C in all cases, indicating that the solids are in the glassy state at the temperature of the FTIR and ssHDX studies (room temp. ∼25°C).
TABLE 2.
Moisture content and Tg values for myoglobin and β-lactoglobulin formulations*
| Additive
|
|||||||
|---|---|---|---|---|---|---|---|
| Protein | Property | Trehalose | Raffinose | Dextran | PVA | PVP | None |
| Myoglobin | Moisture content (%) | 4.53 ± 0.25 | 4.42 ± 0.09 | 6.11 ± 0.79 | 5.12 ± 0.2 | 7.00 ± 0.85 | 3.94 ± 0.64 |
| Tg (°C) | 52.41 ± 0.98 | 56.18 ± 0.52 | 95.06 ± 0.95 | 41.72 ± 0.49 | 96.3 ± 2.09 | NA | |
| β-Lactoglobulin | Moisture content (%) | 6.18 ± 0.38 | 5.67 ± 0.24 | 7.23 ± 0.23 | 4.68 ± 0.59 | 6.32 ± 0.78 | 6.85 ± 0.44 |
| Tg (°C) | 43.53 ± 0.58 | 47.34 ± 2.17 | 94.93 ± 0.58 | 44.41 ± 0.06 | 94.69 ± 0.50 | NA | |
After exposure to 33% RH, 72 h. Protein/excipient = 1:1 (w/w).
The solids-characterization studies thus establish that, with the exception of formulations containing Gdn·HCl, the myoglobin and β-lactoglobulin samples are glassy amorphous solids with a moisture content of ∼4–8%. Samples containing Gdn·HCl are partially crystalline with a moisture content of 4–8%. The properties of samples containing other proteins are assumed to be similar.
Protein conformation in amorphous solids by FTIR and ssHDX
Amide I FTIR spectra and ssHDX data were acquired for six proteins with different secondary structures. Data are grouped by protein and ordered from proteins with the greatest α-helix content to those with the greatest β-sheet content. Within each protein, FTIR data are presented first followed by ssHDX data for the intact (i.e., undigested) protein. When available, ssHDX data for peptic digests are then presented.
Myoglobin
Myoglobin is a globular protein with high (83%) α-helix content (Table 1). The α-helical structure is retained in samples lyophilized in the absence of excipients, as reflected in the strong FTIR bands at 1660 and 1650 cm−1 (Fig. 1 A). The band intensity is greater for solid samples containing trehalose or raffinose, consistent with an increase in α-helix content and greater retention of structure. FTIR spectra for samples containing dextran were similar to the excipient-free samples, whereas those containing PVA showed a loss in α-helix band intensity relative to controls, consistent with a loss in structure. Changes in intensity of the unstructured band (∼1640 cm−1) mirror changes in the α-helix band. Peak assignments are in agreement with previous FTIR reports for myoglobin (18–20).
FIGURE 1.

Myoglobin conformation in amorphous solids containing various additives (1:1 w/w). (A) Second derivative FTIR spectra. (B) ssHDX of intact protein. (C) ssHDX of peptic digests. Additives were trehalose (Tre), raffinose (Raf), dextran (dex), polyvinyl alcohol (PVA), polyvinylpyrrolidone (PVP), and guanidine hydrochloride (Gdn). In C, peptic fragments and approximate secondary structures were: 1 (G1-W7; 42% α-helix), 2 (Q8-L11; 100% α-helix), 3 (N12-A19; 67% α-helix, 33% turn), 4 (I21-L29, 100% α-helix), 5 (I30-L40, 100% α-helix), 6 (G35-L40, 100% α-helix), 7 (M55-L69, 100% α-helix), 8 (T70-L76, 100% α-helix), 9 (G80-L86, 71% α-helix), 10 (K87-Y103, 47% α-helix), 11 (A110-D126, 52% α-helix, 17% turn), 12 (I142-K147, 100% α-helix), and 13 (L149-G153, 20% α-helix). In B, asterisk indicates significant difference from the value for the “None” sample (α = 0.05); n = 3 ± SD.
ssHDX results for intact myoglobin (i.e., not subjected to peptic digestion) show a pattern of exchange consistent with the FTIR results (Fig. 1 B). The greatest protection from exchange was observed for samples containing trehalose and raffinose, and these samples also showed the strongest α-helix band intensity on FTIR (Fig. 1 A). Exchange was greater for samples containing PVA or excipient-free samples, in agreement with weak FTIR α-helix band intensities. The loss of FTIR band intensity at 1650 cm−1 for samples containing PVA relative to excipient-free controls is not reflected in higher exchange, however. Samples containing dextrose showed intermediate α-helix band intensity and extent of exchange. LC/+ESI-MS spectra of myoglobin showed evidence of dimer formation (∼4%) after storage at 33% RH for 72 h.
ssHDX was also performed for myoglobin after peptic digestion. Digestion produced 28 fragments detectable by LC/+ESI-MS, 13 of which were selected to provide maximum sequence coverage (74%; Fig. 1 C). Raffinose and trehalose showed the greatest protection from exchange for all fragments, consistent with ssHDX results for the intact protein. As in our previous studies of calmodulin (9–11), the greatest protective effect of these sugars was observed for the α-helical fragments (fragments 2–9 and 12; Fig. 1 C). Most of the unstructured fragments also showed some protective effect (fragments 1, 10, 11, and 13; Fig. 1 C). Although these fragments were assigned to the “unstructured” group, all but fragment 13 had some α-helix content (Fig. 1 C). Dextran exhibited a moderate protective effect primarily in the α-helical fragments, whereas PVA and PVP showed no protective effects in any fragment.
As expected, the greatest deuterium incorporation occurred in myoglobin samples exposed to Gdn·HCl. For some fragments (fragments 4–8, 11, and 12; Fig. 1 C) deuterium incorporation was >100% in solid samples treated with Gdn·HCl. Since the percent deuterium incorporation is calculated relative to the fully folded and fully deuterated protein in solution (see Eq. 1), values >100% suggest that the protein was denatured in Gdn·HCl solutions, remained unfolded upon lyophilization, and in that form showed greater deuterium uptake from D2O(g) in the solid protein than in the native protein in D2O solution. Of the proteins subjected to digestion, this effect was only observed for myoglobin.
Lysozyme
Lysozyme's mixed α-helix and β-sheet structure (Table 1) is reflected in the FTIR spectra of solid samples. FTIR spectra for samples without excipients showed moderately strong α-helix bands at 1658 and 1648 cm−1 together with strong β-sheet bands at 1638 cm−1 (Fig. 2 A), consistent with previous reports (19,21). In samples containing excipients, the α-helix bands show modest increases in intensity (versus “none”) indicative of increased secondary structure (22). Samples containing trehalose or PVA also showed modest increases in β-sheet band intensity (1638 cm−1). Lysozyme has been reported to undergo structural perturbation on drying, leading to loss of secondary structure (23).
FIGURE 2.

Lysozyme conformation in amorphous solids containing various additives (1:1 w/w). (A) Second derivative FTIR spectra. (B) ssHDX of intact protein. Additives were Tre, Raf, dex, PVA, PVP, and Gdn. In B, asterisk indicates significant difference from the value for the “None” sample (α = 0.05); n = 3 ± SD.
ssHDX studies of intact lysozyme (Fig. 2 B) show moderate protection from exchange by trehalose, raffinose, and dextran with little protection by PVA and PVP. Lysozyme and other proteins with disulfide bonds showed lower percentage deuterium uptake after Gdn·HCl exposure (i.e., lysozyme, ribonuclease A, β-lactoglobulin; Figs. 2 B, 3 B, and 4 B, Table 1). This suggests that the disulfide bonds, which were not reduced here, provided some protection from exchange after Gdn·HCl treatment. Since lysozyme could not be digested quickly with pepsin, ssHDX studies of peptic digests were not performed.
FIGURE 3.

RNase conformation in amorphous solids containing various additives (1:1 w/w). (A) Second derivative FTIR spectra. (B) ssHDX of intact protein. Additives were Tre, Raf, dex, PVA, PVP, and Gdn. In B, asterisk indicates significant difference from the value for the “None” sample (α = 0.05); n = 3 ± SD.
FIGURE 4.

β-Lactoglobulin conformation in amorphous solids containing various additives (1:1 w/w). (A) Second derivative FTIR spectra. (B) ssHDX of intact protein. (C) ssHDX of peptic digests. Additives were Tre, Raf, dex, PVA, PVP, and Gdn. In C, peptic fragments and approximate secondary structures were: 1 (V3-D11, 11% α-helix, 27% turn), 2 (I12-W19, 50% α-helix), 3 (Y20-M24, 100% β-sheet), 4 (I29-V32, 75% turn), 5 (D33-V41, 22% turn), 6 (Y42-L54, 61.5% β-sheet), 7 (L46-L54, 44% β-sheet), 8 (K75-F82, 25% β-sheet, 25% turn), 9 (N90-L95, 83% β-sheet), 10 (D96-L104, 22% β-sheet), 11 (N109-L117, 33% α-helix), 12 (P113-V123, 27% α-helix, 54% β-sheet), 13 (V123-L133, 27% α-helix, 9% β-sheet, 18% turn), 14 (E134-L149, 25% α-helix, 19% β-sheet), 15 (L143-L149, 42% β-sheet), and 16 (S150-L156, 57% α-helix). In B, asterisk (*) indicates significant difference from the value for the “None” sample (α = 0.05); n = 3 ± SD.
RNase A
RNase A is an endonuclease with mixed α-helix and β-sheet structure (Table 1). The dominant feature of the FTIR spectra for solid samples is a strong β-sheet band (∼1638 cm−1) that increases in intensity when excipients are included (Fig. 3 A). Weak α-helix bands are also observed at 1658 and 1650 cm−1; the latter shows minor increases in intensity for samples containing PVA or dextran. Peak assignments are in agreement with previous FTIR studies of RNase A (18,19,24,25).
ssHDX studies of intact RNase A show moderate protection from exchange by trehalose and raffinose, with little protection by the other excipients (Fig. 3 B). This pattern is not consistent with the effects of the excipients on the β-sheet and α-helix bands of the FTIR spectrum (see Discussion). As was the case with lysozyme, RNase A could not be digested quickly with pepsin, so ssHDX studies of peptic digests were not performed.
β-lactoglobulin
β-lactoglobulin is a lipocalin with mixed α-helix and β-sheet structure (Table 1). FTIR spectra of solid samples showed a strong band at 1626 cm−1 assigned to β-sheet (Fig. 4 A). The position and intensity of this band are relatively unaffected by the carbohydrate excipients, whereas inclusion of PVA is associated with a decrease in its intensity. Although β-lactoglobulin has some α-helix content (Table 1), these bands are not prominent in the FTIR spectra of the solid samples. The spectra are in good agreement with those previously reported for β-lactoglobulin (24).
As for RNase A, ssHDX results for the intact β-lactoglobulin show significant protection from exchange by trehalose and raffinose, more limited protection by dextran, and little to no protection by PVA and PVP (Fig. 4 B). Although trehalose and raffinose show protection from exchange, inclusion of these excipients had little effect on the FTIR spectum.
ssHDX analysis of peptic digests of β-lactoglobulin was also conducted. Sixteen of the 32 peptic fragments detected by LC/+ESI-MS were used to map the protein (Fig. 4 C), providing 76% sequence coverage. The β-sheet regions of β-lactoglobulin generally showed lower deuterium incorporation than other types of secondary structure, and values were less sensitive to excipient type (Fig. 4 C). Protection from exchange in the α-helical fragments followed the pattern observed for myoglobin, with the greatest protection provided by trehalose and raffinose. Exchange in fragments assigned to the “mixed” or “loops and other” categories was variable. In some fragments, inclusion of PVA or PVP resulted in greater deuterium incorporation than in the excipient-free control (e.g., fragments 1, 12, 13, and 16; Fig. 4 C), suggesting that PVA and PVP promote exposure to D2O(g) in these fragments.
EC5
EC5 is a domain of the adhesion protein E-cadherin with high β-sheet content. FTIR spectra for EC5 show a strong β-sheet band at 1644 cm−1, suggesting that the β-sheet structure is retained in the solid samples (Fig. 5 A). The spectrum is relatively unaffected by the inclusion of excipients. ssHDX studies of intact EC5 showed high deuterium incorporation that was insensitive to excipient type (Fig. 5 B), consistent with the FTIR results.
FIGURE 5.

EC5 conformation in amorphous solids containing various additives (1:1 w/w). (A) Second derivative FTIR spectra. (B) ssHDX of intact protein. (C) ssHDX of peptic digests. Additives were Tre, Raf, dex, PVA, PVP, and Gdn. In C, peptic fragments and approximate secondary structures were: 1 (P2-F10, 70% β-sheet), 2 (F11-D25, 87% β-sheet), 3 (A26-E38, 38% β-sheet), 4 (L39-W58, 55% β-sheet), 5 (S58-L67, unstructured), 6 (I60-A66, unstructured), 7 (E68-L76, 55% β-sheet), 8 (Y72-L76, 100% β-sheet), 9 (L78-D96, 79% β-sheet), 10 (D96-E111, 6% β-sheet). In B, asterisk indicates significant difference from the value for the “None” sample (α = 0.05); n = 3 ± SD.
ssHDX was performed on peptic digests of EC5 (Fig. 5 C). Eleven peptic fragments were selected for analysis, giving a total sequence coverage of 97%, and assigned to either β-sheet or unstructured regions (Fig. 5 C). Fragments with partial β-sheet character (i.e., fragments 1, 3, and 4; Fig. 5 C) were included in the unstructured group on the basis of their exchange behavior. Peptic fragments of EC5 generally showed greater percent deuterium incorporation than observed for other proteins, suggesting a less compact structure in the solid state. The β-sheet fragments (fragments 2 and 5–9; Fig. 5 C) showed some protection from exchange by the various excipients, to a degree roughly comparable to the β-sheet regions of β-lactoglobulin (Fig. 5 C). The unstructured fragments of EC5 were insensitive to excipient type, and exposure to Gdn·HCl did not increase deuterium incorporation relative to the other excipients (fragments 1, 3, 4, 10, and 11; Fig. 5 C), again suggesting a loose structure.
Con A
Con A is a lectin with a predominantly β-sheet structure (Table 1). FTIR spectra for solid samples of Con A show an intense β-sheet band at 1633 cm−1, consistent with retention of secondary structure (Fig. 6 A). Relative to the excipient-free control, the intensity of the band increases in solids containing trehalose or raffinose with a corresponding decrease in the shoulder at ∼1625 cm−1, suggesting increased secondary structure. The spectrum is relatively insensitive to other excipients. Peak assignments agree with previous FTIR reports for Con A (18).
FIGURE 6.

Con A conformation in amorphous solids containing various additives (1:1 w/w). (A) Second derivative FTIR spectra. (B) ssHDX of intact protein. Additives were Tre, Raf, dex, PVA, PVP, and Gdn. In B, asterisk indicates significant difference from the value for the “None” sample (α = 0.05); n = 3 ± SD.
ssHDX studies of intact Con A showed a pattern of exchange consistent with the FTIR results (Fig. 6 B). The greatest protection from exchange was observed for trehalose and raffinose, whereas other excipients showed more modest effects. Protection from deuterium exchange was generally more limited for Con A than for other proteins. Of interest, Con A showed relatively low deuterium incorporation on ssHDX after exposure to Gdn·HCl, although the protein contains no disulfide bonds. Con A forms dimers, tetramers, and aggregates in aqueous solution (26), which may be retained during lyophilization and may protect the Gdn·HCl-treated protein from deuterium exchange in the solid state. Unlike the other proteins, solids containing Con A were hazy upon reconstitution, further supporting the presence of aggregation and/or oligomerization. LC/+ESI-MS spectra showed peaks corresponding to dimeric and trimeric forms of Con A after exposure to 33% RH for 72 h. The areas of these deconvoluted MS peaks were ∼8% and 4%, respectively, of the area of intact Con A. Con A could not be digested quickly with pepsin, so ssHDX studies of peptic digests were not performed.
Statistical analysis
For each protein, FTIR spectra for samples containing excipients were compared quantitatively with those for the excipient-free control using the “area of overlap” method of Kendrick et al. (13) (Table 3), which compares spectra on the basis of the fraction of overlapping area in area-normalized second derivative spectra. All matrix entries were ≥0.90, indicating that the spectra for samples containing excipients are very similar to those of the excipient-free controls (13).
TABLE 3.
Comparison of FTIR spectra for formulations containing excipients to the excipient-free control, using the “area of overlap” method of Kendrick et al. (13); see text
| Excipient
|
||||
|---|---|---|---|---|
| Protein | +Tre | +Raf | +Dex | +PVA |
| Myoglobin | 0.94 | 0.95 | 0.97 | 0.92 |
| Lysozyme | 0.95 | 0.97 | 0.94 | 0.94 |
| RNase A | 0.97 | 0.96 | 0.92 | 0.90 |
| β-Lactoglobulin | 0.97 | 0.98 | 0.98 | 0.91 |
| EC5 | 0.99 | 0.99 | 0.99 | 0.99 |
| Con A | 0.97 | 0.98 | 0.98 | 0.98 |
Table entries are the ratio of the areas of the area-normalized second derivative spectra; a value of one indicates identical spectra.
In ssHDX studies of intact protein, deuterium incorporation in samples containing excipients was compared with that in excipient-free controls using one-way analysis of variance, with Tukey's test for post hoc comparison of the means (Origin 7.0, OriginLab Corp., Northampton, MA). Differences in percent exchange of 2–3% were generally significant at 95% confidence (α = 0.05; see part B in Figs. 1–6).
DISCUSSION
In these studies, protein conformation in amorphous solids was assessed with the use of FTIR and ssHDX. A comparison of the results of these two methods requires an appreciation of the fundamental bases of the measurements. FTIR measures the absorption of infrared light that occurs when the frequency of light and the frequency of a vibrational mode of the protein coincide (27–29). In the amide I region that is often employed for protein structural analysis (1600–1700 cm−1), the spectrum is dominated by C=O stretching vibrations of the protein backbone (27,29). Theoretical analysis indicates that the C=O stretching vibrations are most strongly influenced by the interactions of amide dipoles that occur through space (i.e., transition dipole coupling (TDC)) and by hydrogen bonding, although other factors such as through-bond coupling and the dielectric constant of the medium may also contribute (27). In computations for an infinite α-helix, the strongest TDC interactions are with direct neighbors in the chain and with groups that are hydrogen bonded to the amide of interest (27). For an antiparallel β-sheet, the strongest TDC interactions are between peptide groups that are hydrogen bonded to one another, or are on different chains, but in close proximity (27). The C=O stretching vibrations determined by these interactions are observable because they are delocalized over large regions of the protein, so that infrared light of particular frequencies is absorbed (28). Thus, FTIR in the amide 1 region measures the absorption of infrared light at frequencies corresponding to C=O stretching vibrations of the protein, which in turn are determined primarily by through-space interactions (i.e., TDC) and by hydrogen bonding.
In solution, an HDX experiment measures the rate and/or extent of a hydrogen transfer reaction. A typical solution HDX study involves the exposure of protein in the native form (“protio”) to D2O. Hydrogen atoms (1H) throughout the molecule are replaced with deuterium (2H) in a reaction catalyzed by acid, base, and/or water. The exchange of a particular amide hydrogen for deuterium requires that 1), the amide hydrogen be accessible to solvent; and 2), the N-H bond be broken (6,30,31). The reaction is sensitive to protein structure because amide hydrogen atoms participating in protein secondary structure (e.g., α-helix and β-sheet) or buried in the protein core exchange much more slowly than those that are solvent-exposed (31,32); intrinsic exchange rate constants (ki) for exposed amide hydrogen atoms are of the order 10 s−1, whereas those for hydrogen-bonded or core hydrogen atoms may be reduced by eight orders of magnitude or more (31–33). At selected times the reaction may be quenched by low pH (∼2.5) and low temperature (∼0°C), which reduces ki by ∼105 (31), and the samples can be analyzed for deuterium incorporation (e.g., by LC/MS or NMR). During analysis, deuterium incorporated into the amino acid side chains undergoes rapid back exchange to the protio form, whereas deuterium incorporated into backbone amides is generally retained. Kinetic analysis of HDX in solution has been used to provide information on protein dynamics (6).
For HDX in solid samples, it is reasonable to assume that exchange of a particular amide hydrogen also requires that 1), the amide hydrogen atom be accessible to D2O; and 2), the N-H bond be broken. The factors that affect accessibility of the amide hydrogen in the solid state are likely to differ from those for solution, however. For example, accessibility of amide hydrogen atoms in solids may involve D2O vapor sorption and diffusion of D2O in the solid—processes that are not relevant in solution. In solution, protein structure is determined by intramolecular hydrogen bonds (e.g., in α-helix or β-sheet) and by intermolecular hydrogen bonds to water. Intermolecular hydrogen bonds certainly contribute to structure and accessibility in solids, but intramolecular hydrogen bonds to water may be “replaced” by hydrogen bonds to other components of the dry solid (e.g., sugars) (1,34). The time course of HDX in solution provides information about protein dynamics and the relative rates of unfolding and exchange. In glassy solids, however, the matrix may restrict the dynamic motions of the protein; thus, the time required for large-scale motions (e.g., global unfolding) may be far longer than that for solution, and in fact may be far longer than the ssHDX experiment. For example, α (i.e., global) relaxation times on the order of 105 h have been reported for proteins in glassy solids (35). In addition, there is growing evidence that amorphous solids are spatially and dynamically heterogeneous (36,37), and thus exchange may occur in an ensemble of physical environments. Sequestration of D2O by excipients is unlikely to be a factor in ssHDX studies, however, since D2O vapor is in equilibrium with a relatively large volume D2O solution (see Materials and Methods).
In this context, we interpret the 72-h percent exchange values of our ssHDX studies as a pseudo-equilibrium measure of the accessibility of backbone amide hydrogen atoms in solid samples to D2O, a value determined primarily by intra- and intermolecular hydrogen bonds. This is a “pseudo-equilibrium” measurement, since at “true” equilibrium all backbone amide hydrogen atoms will be exchanged for deuterium, a process that may take years to complete. ssHDX is similar to FTIR in that both report on backbone amide groups whose interactions determine protein secondary structure. The methods differ in that amide I FTIR measures C=O stretching vibrations, whereas ssHDX measures the extent of a reaction. The C=O stretching vibrations of FTIR are determined primarily by through-space interactions, hydrogen bonding, and other factors, as noted above. In contrast, the extent of ssHDX is likely to be determined primarily by the intra- and intermolecular hydrogen bonds of the protein in the solid matrix.
The effects of excipients on the amide I FTIR spectra and on the extent of ssHDX measured in intact proteins are in reasonably good agreement for many of the proteins studied here. For example, the α-helical protein myoglobin shows protection from exchange by excipients in the order of trehalose, raffinose > dextran > PVA, PVP, none (Fig. 1 B). With the exception of PVA (discussed below), this order is consistent with observed decreases in the intensity of the α-helix band in the FTIR spectrum (1650 cm−1, Fig. 1 A). Proteins with high β-sheet content (i.e., EC-5 and Con A) also show agreement between excipient effects measured by the two techniques, although the effects are not as large (Figs. 5 and 6). The poorest agreement between the two methods is observed for RNase A, a protein with mixed α-helix and β-sheet content (Table 1). The FTIR spectrum for RNase is dominated by the strong β-sheet band (1640 cm−1); the intensity of this band is increased similarly by all the excipients relative to the excipient-free control (Fig. 3 A). The weaker α-helix bands (1650, 1660 cm−1) show some differences among the excipients, with PVA and dextran showing the greatest effects. In contrast, the ssHDX results show that the greatest protection from exchange for RNase is provided by trehalose and raffinose, with minimal effects by dextran and PVA (Fig. 3 B). These differences may be due to the propensity of RNase to form oligomers, which have been routinely produced by lyophilization from acidic solution (38,39). The RNase FTIR band at 1640 cm−1 was assigned to the β-sheet here and elsewhere (40), but has also been attributed to cross-β structures in RNase oligomers (40). The dominant FTIR β-sheet band for RNase (Fig. 3 A) may reflect oligomeric species that are relatively insensitive to excipient selection; weak α-helix bands of the FTIR spectrum provide limited information on excipient effects in those regions. The ssHDX results may primarily reflect the behavior of the α-helical domains, which tend to be more excipient-sensitive (Fig. 3 B) and may be relatively unaffected by oligomer formation. Oligomeric species were not detected on LC/MS analysis of RNase samples, suggesting that any oligomers formed were dissociated during sample preparation and analysis. For several of the proteins, differences between FTIR and ssHDX results were observed when PVA was included as an excipient (e.g., myoglobin and lysozyme). Interpretation of FTIR spectra for PVA may be complicated by low levels of residual acetate groups that absorb in the amide I region; PVA is manufactured by hydrolysis of poly(vinyl acetate). These impurities are not expected to contribute significantly to the ssHDX results.
Analysis of the peptic digests of ssHDX samples provides a level of structural resolution that cannot be achieved by FTIR. ssHDX data for digests of myglobin, β-lactoglobulin, and EC-5 (Figs. 1 C, 4 C, and 5 C) show that protection from exchange is not conferred uniformly along the protein backbone, but instead is greatest in fragments corresponding to structured regions of the proteins. The α-helical fragments of myoglobin (Fig. 1 C) and β-lactoglobulin (Fig. 4 C) show the greatest protection from exchange by many of the excipients, a finding that is consistent with our previous studies of the α-helical protein calmodulin (9–11). The β-sheet fragments of β-lactoglobulin and EC-5 show more limited protection (Figs. 4 C and 5 C). Although the origins of these site-specific effects are unclear, the data suggest that hydrogen-bonding interactions between the excipients and the protein backbone are not involved, since these would be expected to be greatest in unstructured fragments. As with solution HDX, the use of immobilized pepsin columns could be used to improve the ability to digest resistant proteins (41,42); however, that approach was not attempted here.
In summary, the results demonstrate that ssHDX with ESI/MS analysis provides information on the structure and intermolecular interactions of proteins in amorphous solids. The technique is complementary to amide I FTIR in that both methods report on backbone amide groups. The application of peptic digestion to ssHDX samples provides peptide-level resolution and has been used to demonstrate that protein-excipient interactions in the solid state are preferentially exhibited in structured fragments. ssHDX with ESI/MS analysis thus shows promise for characterizing proteins in amorphous solids, with potential applications in the pharmaceutical, biotechnology, and food industries.
Acknowledgments
Access to the PerkinElmer ATR FTIR instrument was provided Dr. Paulette Spencer, director of the Bioengineering Research Center (BERC) of the University of Kansas. We gratefully acknowledge this access and the assistance provided by Drs. Charles Ye and Jonggu Park of the BERC. We also acknowledge the assistance provided by members of the Mass Spectrometry Service Laboratory of the University of Kansas. The Q-TOF-2 was purchased with support from KSTAR (the Kansas-administered National Science Foundation Experimental Program to Stimulate Competitive Research (T. D. Williams)) and the University of Kansas.
Sandipan Sinha's present address is Pfizer Inc., St. Louis, MO.
Editor: Heinrich Roder.
References
- 1.Carpenter, J. F., B. S. Chang, W. Garzon-Rodriguez, and T. W. Randolph. 2002. Rational design of stable lyophilized protein formulations: theory and practice. In Rational Design of Stable Protein Formulations. J. F. Carpenter and M. C. Manning, editors. Kluwer Academic/Plenum Publishers, New York. 109–133. [DOI] [PubMed]
- 2.Song, Y., A. D. Wilson, R. Li, M. J. Hageman, R. L. Schowen, and E. M. Topp. 2000. Solid-state chemical stability of peptides and proteins: application to controlled release formulations. In Handbook of Pharmaceutical Controlled Release Technology. D. L. Wise, editor. Marcel Dekker, New York. 693–724.
- 3.Stotz, C. E., S. L. Winslow, M. L. Houchin, A. J. D'Souza, and J. T. Ji. 2003. Degradation pathways for lyophilized peptides and proteins. In Lyophilization of Biopharmaceuticals. M. J. Pikal, and H. R. Costantino, editors. AAPS Press, Arlington, VA, 433–480.
- 4.Yu, L. 2001. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 48:27–42. [DOI] [PubMed] [Google Scholar]
- 5.Krishna, M. M. G., L. Hoang, Y. Lin, and S. W. Englander. 2004. Hydrogen exchange methods to study protein folding. Methods. 34:51–64. [DOI] [PubMed] [Google Scholar]
- 6.Wales, T. E., and J. R. Engen. 2006. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 25:158–170. [DOI] [PubMed] [Google Scholar]
- 7.French, D. L., T. Arakawa, and T. Li. 2004. Fourier transform infared spectroscopy investigation of protein conformation in spray-dried protein/trehalose powders. Biopolymers. 73:524–531. [DOI] [PubMed] [Google Scholar]
- 8.Desai, U. R, O. J. Osterhout, and A. M. Klibanov. 1994. Protein structure in the lyophilized state: a hydrogen isotope exchange/NMR study with bovine pancreatic trypsin inhibitor. J. Am. Chem. Soc. 116:9420–9422. [Google Scholar]
- 9.Li, Y., T. D. Williams, R. L. Schowen, and E. M. Topp. 2007. Characterizing protein structure in amorphous solids using hydrogen/deuterium exchange with mass spectrometry. Anal. Biochem. 366:18–28. [DOI] [PubMed] [Google Scholar]
- 10.Li, Y., T. D. Williams, R. L. Schowen, and E. M. Topp. 2007. Trehalose and calcium exert site-specific effects on calmodulin conformation in amorphous solids. Biotechnol. Bioeng. 97:1650–1653. [DOI] [PubMed] [Google Scholar]
- 11.Li, Y., T. D. Williams, and E. M. Topp. 2008. Effects of excipients on protein conformation in lyophilized solids by hydrogen/deuterium exchange mass spectrometry. Pharm. Res. 25:259–267. [DOI] [PubMed] [Google Scholar]
- 12.Zheng, K., C. R. Middaugh, and T. J. Siahaan. 2008. Evaluation of the physical stability of the EC5 domain of E-cadherin: effects of pH, temperature, ionic strength, and disulfide bonds. J. Pharm. Sci. In press. [DOI] [PMC free article] [PubMed]
- 13.Kendrick, B. S., A. Dong, S. D. Allison, M. C. Manning, and J. F. Carpenter. 1996. Quantitation of the area of overlap between second-derivative amide I infrared spectra to determine the structural similarity of a protein in different states. J. Pharm. Sci. 85:155–158. [DOI] [PubMed] [Google Scholar]
- 14.Zhang, Z., and D. L. Smith. 1993. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2:522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan, X., H. Zhang, J. Watson, M. I. Schimerlik, and M. L. Deinzer. 2002. Hydrogen/deuterium exchange and mass spectrometric analysis of a protein containing multiple disulfide bonds: solution structure of recombinant macrophage colony stimulating factor-β(rhM-CSFβ). Protein Sci. 11:2113–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsutsui, Y., and P. L. Wintrobe. 2007. Hydrogen/deuterium exchange-mass spectrometry: a powerful tool for probing protein structure, dynamics and interactions. Curr. Med. Chem. 14:2344–2358. [DOI] [PubMed] [Google Scholar]
- 17.Hotchko, M., G. S. Anand, E. A. Komives, and L. F. Ten Eyck. 2006. Automated extraction of backbone deuteration levels from amide H/2H mass spectrometry experiments. Protein Sci. 15:583–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong, A., P. Huang, and W. S. Caughey. 1990. Protein secondary structures in water from second-derivative amide I infrared spectra. Biochemistry. 29:3303–3308. [DOI] [PubMed] [Google Scholar]
- 19.Smith, B. M., and S. Franzen. 2002. Single-pass attenuated total reflection Fourier transform infrared spectroscopy for the analysis of proteins in H2O solution. Anal. Chem. 74:4076–4080. [DOI] [PubMed] [Google Scholar]
- 20.Meersman, F., L. Smeller, and K. Heremans. 2002. Comparative Fourier transform infrared spectroscopy study of cold-, pressure-, and heat-induced unfolding and aggregation of myoglobin. Biophys. J. 82:2635–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Allison, S. D., B. Chang, T. W. Randolph, and J. F. Carpenter. 1999. Hydrogen bonding between sugar and protein is responsible for inhibition of dehydration-induced protein unfolding. Arch. Biochem. Biophys. 365:289–298. [DOI] [PubMed] [Google Scholar]
- 22.Luthra, S., D. S. Kalonia, and M. J. Pikal. 2007. Effect of hydration on the secondary structure of lyophilized proteins as measured by Fourier transform infrared (FTIR) spectroscopy. J. Pharm. Sci. 96:2910–2921. [DOI] [PubMed] [Google Scholar]
- 23.Carpenter, J. F., and J. H. Crowe. 1989. An infrared spectroscopic study of the interactions of carbohydrates with dried proteins. Biochemistry. 28:3916–3922. [DOI] [PubMed] [Google Scholar]
- 24.Ishida, K. P., and P. R. Griffiths. 1993. Comparison of the amide I/II intensity ratio of solution and solid-state proteins sampled by transmission, attenuated total reflectance, and diffuse reflectance spectrometry. Appl. Spectrosc. 47:584–589. [Google Scholar]
- 25.Bentaleb, A., A. Abele, P. Haikel, P. Schaaf, and J. C. Voegel. 1998. FTIR-ATR and radiolabeling study of the adsorption of ribonuclease A onto hydrophilic surfaces: correlation between the exchange rate and the interfacial denaturation. Langmuir. 14:6493–6500. [Google Scholar]
- 26.Xu, Q., and T. A. Keiderling. 2005. Trifluoroethanol-induced unfolding of concanavalin A: equilibrium and time-resolved optical spectroscopic studies. Biochemistry. 44:7976–7987. [DOI] [PubMed] [Google Scholar]
- 27.Barth, A., and C. Zscherp. 2002. What vibrations tell us about proteins. Q. Rev. Biophys. 35:369–430. [DOI] [PubMed] [Google Scholar]
- 28.Ganim, Z., H. S. Chung, A. W. Smith, L. P. DeFlores, K. C. Jones, and A. Tokmakoff. 2008. Amide I two-dimensional infrared spectroscopy of proteins. Acc. Chem. Res. 41:432–441. [DOI] [PubMed] [Google Scholar]
- 29.Moran, A., and S. Mukamel. 2004. The origin of the vibrational model couplings in various secondary structural motifs of polypeptides. Proc. Natl. Acad. Sci. USA. 101:506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanman, J., J. Prevelige, and E. Peter. 2004. High-sensitivity mass spectrometry for imaging subunit interactions; hydrogen/deuterium exchange. Curr. Opin. Struct. Biol. 14:181–188. [DOI] [PubMed] [Google Scholar]
- 31.Busenlehner, L. S., and R. N. Armstrong. 2005. Insights into enzyme structure and dynamics elucidated by amide H/D exchange mass spectrometry. Arch. Biochem. Biophys. 433:34–46. [DOI] [PubMed] [Google Scholar]
- 32.Yan, X., J. Watson, P. S. Ho, and M. L. Deinzer. 2004. Mass spectrometric approaches using electrospray ionization exchange for determining protein structures and their conformational changes. Mol. Cell. Proteomics. 3:10–23. [DOI] [PubMed] [Google Scholar]
- 33.Hoofnagle, A. N., K. A. Resing, and N. G. Ahn. 2003. Protein analysis by hydrogen exchange mass spectrometry. Ann. Rev. Biophys. Biomol. Struct. 32:1–25. [DOI] [PubMed] [Google Scholar]
- 34.Hill, J. J., E. Y. Shalaev, and G. Zografi. 2005. Thermodynamic and dynamic factors involved in the stability of native protein structure in amorphous solids in relation to levels of hydration. J. Pharm. Sci. 94:1636–1667. [DOI] [PubMed] [Google Scholar]
- 35.Chang, L. L., D. Shepherd, J. Sun, X. C. Tang, and M. J. Pikal. 2005. Effect of sorbitol and residual moisture on the stability of lyophilized antibodies: implications for the mechanism of protein stabilization in the solid state. J. Pharm. Sci. 94:1445–1455. [DOI] [PubMed] [Google Scholar]
- 36.Thurau, C. T., and M. D. Ediger. 2002. Spatially heterogeneous dynamics during physical aging far below the glass transition temperature. J. Polym. Sci. Part Polym. Phys. 40:2463–2472. [Google Scholar]
- 37.Ediger, M. D. 2000. Spatially heterogeneous dynamics in supercooled liquids. Annu. Rev. Phys. Chem. 51:99–128. [DOI] [PubMed] [Google Scholar]
- 38.Gotte, G., M. Donadelli, D. V. Laurents, F. Vottariello, M. Morbio, and M. Libonati. 2006. Increase of RNase A N-terminus polarity or C-terminus apolarity changes the two domains' propensity to swap and form the two dimeric conformers of the protein. Biochemistry. 45:10795–10806. [DOI] [PubMed] [Google Scholar]
- 39.Gotte, G., D. V. Laurents, and M. Libonati. 2006. Three-dimensional domain-swapped oligomers of ribonuclease A: identification of a fifth tetramer, pentamers and hexamers, and detection of trace heptameric, octameric and nonameric species. Biochim. Biophys. Acta. 1764:44–54. [DOI] [PubMed] [Google Scholar]
- 40.Yan, Y.-B., J. Zhang, H.-W. He, and H.-M. Zhou. 2006. Oligomerization and aggregation of bovine pancreatic ribonuclease A: characteristic events observed by FTIR spectroscopy. Biophys. J. 90:2525–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu, Y., S. Kaveti, and J. R. Engen. 2006. Extensive deuterium back-exchange in certain immobilized pepsin columns used for H/D exchange mass spectrometry. Anal. Chem. 78:1719–1723. [DOI] [PubMed] [Google Scholar]
- 42.Hamuro, Y., S. J. Coales, K. S. Molnar, S. J. Tuske, and J. A. Morrow. 2008. Specificity of immobilized porcine pepsin in H/D exchange compatible columns. Rapid Commun. Mass Spectrom. 22:1041–1046. [DOI] [PubMed] [Google Scholar]