

Abstract
Using x-ray diffraction, solid-state 2H-NMR, differential scanning calorimetry, and dilatometry, we have observed a perturbation of saturated acyl chain phosphatidylglycerol bilayers by the antimicrobial peptide peptidyl-glycylleucine-carboxyamide (PGLa) that is dependent on the length of the hydrocarbon chain. In the gel phase, PGLa induces a quasi-interdigitated phase, previously reported also for other peptides, which is most pronounced for C18 phosphatidylglycerol. In the fluid phase, we found an increase of the membrane thickness and NMR order parameter for C14 and C16 phosphatidylglycerol bilayers, though not for C18. The data is best understood in terms of a close hydrophobic match between the C18 bilayer core and the peptide length when PGLa is inserted with its helical axis normal to the bilayer surface. The C16 acyl chains appear to stretch to accommodate PGLa, whereas tilting within the bilayer seems to be energetically favorable for the peptide when inserted into bilayers of C14 phosphatidylglycerol. In contrast to the commonly accepted membrane thinning effect of antimicrobial peptides, the data demonstrate that pore formation does not necessarily relate to changes in the overall bilayer structure.
INTRODUCTION
Naturally occurring antimicrobial peptides (AMPs) are small amphipathic and mostly cationic molecules that are considered to be promising candidates to fight microbial infections (1). Although it is not known how AMPs are able to kill bacteria so effectively, it is generally accepted that AMPs encounter the bacterial plasma membrane at some stage. Besides the common hypothesis that AMPs affect the physical properties of the lipid bilayers of Gram-positive and Gram-negative bacteria, several different modes of interaction with membranes have been proposed. These mechanisms involve the formation of nonlamellar phases, membrane micellization, lipid segregation, or pore formation (for recent reviews see, e.g., (2,3)). The latter mechanism has been especially strongly supported experimentally by Huang and co-workers (4,5), who believe that it involves the concerted formation of either toroidal or barrel-stave pores upon peptide insertion from a surface adsorbed state above a certain AMP threshold concentration. However, the available data imply that the mode of interaction strongly depends on the physicochemical properties of both the lipid bilayer and the respective AMP.
We recently demonstrated how profoundly the outcome of experimental results can be influenced by the choice of the lipid model system (6,7). In those studies, the human multifunctional peptide LL-37, the frog-skin AMP peptidyl-glycylleucine-carboxyamide (PGLa), and melittin from the venom of the European honey bee were found to induce disklike micelles in short-chain phosphatidylcholines (PC), whereas a quasi-interdigitated phase was formed in phosphatidylglycerol (PG) and long-chain PC bilayers. In turn, it was conjectured that LL-37 would bind to the surface in the presence of phosphatidylserine bilayers, whereas no interaction was observed for phosphatidylethanolamine bilayers. Furthermore, in binary mixtures of these lipids, the resulting mode of interaction was dominated by one of the two lipids, though not necessarily by the charged one, demonstrating that net charge is not the only important parameter to be considered. These effects, and especially the formation of either disklike micelles or quasi-interdigitated phases, are pronounced mainly below the chain-melting transition, Tm, of the lipid bilayers. The question, however, is how, in the case of PGLa, these effects translate into the biologically more relevant fluid phase, as our previous experiments showed that LL-37 leads to a thinning of PC and PG membranes (7), as observed also in other lipid/peptide interaction studies (4,8). In another recent study, we observed furthermore that PGLa undergoes a temperature-dependent realignment in dimyristoyl phosphatidylcholine (DMPC)/dimyristoyl phosphatidylglycerol (DMPG) bilayers, from an inserted transmembrane state in the gel phase, via a tilted-helix alignment near the lipid chain-melting transition, to a surface-bound state at the elevated temperatures in the fluid phase (9).
This work focuses on the effects of PGLa on the gel and fluid phases of PG, the predominant lipid of Gram-positive bacterial membranes (3), and in addition addresses the influence of hydrocarbon chain length. In the relevant buffer conditions (20 mM Na-phosphate and 130 mM NaCl, pH 7.4), PG bilayers are negatively charged (10). PGLa (21 amino acids: GMASKAGAIAGKIAKVALKAL-NH2) belongs to the magainin family of peptides and was originally isolated from the skin of the African clawed frog Xenopus laevis (11,12). The peptide exhibits a broad spectrum of antimicrobial activity against Gram-positive and Gram-negative bacteria, fungi, and protozoa, whereas it has minimal toxicity toward eukaryotic cells (13,14). Previous biophysical studies indicate that PGLa is unstructured in aqueous solution and does not adopt a specific conformation in the presence of uncharged lipid bilayers if the peptide is added exogenously (15–17). However, if the peptide is codispersed from a dry lipid/peptide film, and there is no excess of water present in the sample into which the cationic peptide can escape, it will form an α-helix even in neutral phosphatidylcholine (18) and phosphatidylethanolamine bilayers (19). This observation indicates the important step of electrostatic attraction of the peptide to the membrane surface, which is further supported by the general observation of a predominantly α-helical structure of PGLa in the presence of negatively charged bilayers or micelles (15–17). Discrimination between neutral and negatively charged lipid species is further supported by monolayer studies (20), where PGLa was found to mix with negatively charged lipids, but to form separate islands in zwitterionic PC monolayers. PGLa was also reported to adopt different orientations in the membrane depending on peptide concentration and synergistic interactions with magainin-2, ranging from a surface-aligned monomeric state, via tilted dimers, to an upright inserted state (21,22).
Combining x-ray scattering, 2H-NMR spectroscopy, differential scanning calorimetry and dilatometry, we found in this study that PGLa assumes a surface alignment in the gel state of PG membranes, inducing a quasi-interdigitated phase, as reported previously (6,7). Above the lipid chain-melting transition, in the fluid phase, the PGLa helix inserts into the membrane above a certain threshold concentration, most likely with a tilted angle for DMPG and parallel to the bilayer normal for dipalmitoyl phosphatidylglycerol (DPPG) and distearoyl phosphatidylglycerol (DSPG). This interplay between membrane and peptide properties leads to an increase of the membrane thickness for DMPG and DPPG, whereas DSPG bilayers remain unaffected. Thus, our study demonstrates that contrary to what was previously accepted, peptide pore formation is not necessarily related to membrane thinning, and may lead to no detectable changes at all in membrane structure or even to membrane thickening.
MATERIALS AND METHODS
Sample preparation
Lipids
DMPG, DPPG, DSPG, and their chain perdeuterated analogs, of purity >99%, were purchased as sodium salts from Avanti Polar Lipids (Alabaster, AL) and used without further purification. Stock solutions were prepared in chloroform/methanol (9:1 v/v). Before and after experiments, the purity of the phospholipids was checked by thin-layer chromatography, which showed only a single spot using CHCl3/CH3OH/NH3,conc. (65:35:5 v/v) as a solvent. Ammonia, chloroform, methanol, sodium hydrogen phosphate, and sodium dihydrogen phosphate were all purchased in pro analysis quality from Merck (Darmstadt, Germany).
Peptide
PGLa was synthesized as the C-terminal amide using the solid phase method by NeoMPS (San Diego, CA). The peptide was purified to homogeneity by reverse-phase high-performance liquid chromatography to >98%. The crystalline powder was stored at −18°C and rehydrated in 20 mM potassium phosphate buffer with 130 mM NaCl, pH 7.4, before the experiments.
Liposomes
Appropriate amounts (see below for details) of the phospholipid stock solutions were dried under a stream of nitrogen and stored in vacuum overnight to remove the organic solvent totally. The dried lipid films with the respective amounts of the PGLa stock solution were dispersed by vigorous vortex-mixing in double-distilled water containing 20 mM potassium phosphate and 130 mM NaCl, pH 7.4. DMPG samples were then thermostated to 50°C,and DPPG and DSPG samples to 65°C for 90 min, with intermittent, vigorous vortex mixing for 1 min. After cooling to room temperature for 20 min, the samples were used immediately for differential scanning calorimetry (DSC), dilatometry, and x-ray measurements. For NMR measurements, the lipid suspension was centrifuged and the concentrated lipid pellet was transferred into the NMR sample tubes.
Differential scanning calorimetry
Calorimetric experiments were performed on a high-sensitivity VP-DSC (Microcal, Northampton, MA). Before scanning, the samples were degassed for 10 min and equilibrated for 30 min at low temperatures. A scan rate of 0.5°C/min was used throughout. The lipid concentration was 1 mg ml−1. The scans were repeated at least twice to ensure reproducibility. Data acquisition and analysis was performed using Microcal's Origin software. The enthalpy change of the phase transition, ΔH, was obtained from the area under the peak and the mass of phospholipid in each sample. The phase transition temperature, Tm, was defined as the temperature at the peak maximum.
Dilatometry
Similar to previous reports from our laboratory (8,23–25), the molecular lipid volume, V, was determined from 30-mg ml−1 dispersions using the DSA 5000 (Anton Paar, Graz, Austria). The molecular volume is directly related to the specific partial volume (26)
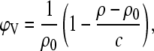 |
(1) |
where c is the lipid concentration, and ρ and ρ0 are the measured densities of the lipid dispersion and the buffer, respectively.
Nuclear magnetic resonance spectroscopy
2H-NMR experiments were performed on perdeuterated lipid dispersions using a Varian (Palo Alto, CA) Inova spectrometer operating at 107.4 MHz 2H resonance frequency (700 MHz for 1H), and a Bruker Avance spectrometer (Bruker Biospin, Karlsruhe, Germany) operating at 76.7 MHz 2H resonance frequency (500 MHz for 1H). Deuterium NMR spectra were acquired employing a quadrupolar echo experiment with a 3- to 4-μs pulse width, 24 μs pulse spacing, and a recycle delay of 0.5 s, on a single-channel home-built probe (107.4 MHz) or on a triple-channel Bruker probe (76.7 MHz). The sample was equilibrated for 15–20 min at each temperature. To extract the quadrupolar splittings, the 2H-NMR Pake doublets were deconvoluted using a Fourier transform based “de-Paking” algorithm (27) implemented with our own computer software.
Small- and wide-angle x-ray scattering (SWAXS)
SWAXS diffraction experiments were performed on a modified Kratky compact camera (Hecus X-Ray Systems, Graz, Austria). Ni-filtered CuKα radiation (λ = 1.54 Å) originating from a sealed tube x-ray generator (Seifert, Ahrensburg, Germany) operating at 50 kV and 40 mA was selected in combination with a pulse-height discriminator. The x-ray beam size was 0.5 mm × 3.5 mm (V × H). The camera was equipped with a Peltier-controlled, programmable cuvette (temperature precision ±0.1°C) and linear, one-dimensional, position-sensitive detectors (PSD 50-M, Hecus X-Ray Systems). Calibration of the small-angle region was performed with silver stearate, and p-bromo-benzoic acid was used to calibrate the wide-angle region.
The lipid dispersions (50 mg ml−1), were filled into thin-walled 1-mm-thick quartz-glass capillaries, sealed, and equilibrated for 10 min at each temperature before the x-ray diffraction experiments. Exposure times of 1800 s and 3600 s were used for the SAXS and WAXS regimes, respectively.
Background-corrected SAXS data were analyzed using the previously described program GAP (global analysis program) (28–30), which in the present case allowed us to retrieve the membrane thickness,
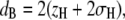 |
(2) |
and the hydrocarbon chain length,
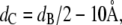 |
(3) |
from a full q-range analysis of the SAXS patterns. The parameters zH and σH are the position and width of the Gaussian, respectively, used to describe the electron-dense headgroup region within the electron density model. In combination with the molecular lipid volume determined from dilatometry, we further calculated the lateral area/lipid using
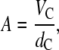 |
(4) |
where VC = V – VH gives the volume of the hydrocarbon chains, and VH = 257 Å3 (24) is the volume of the PG headgroup. Those gel-phase SAXS patterns that show the coexistence of a quasi-interdigitated and a noninterdigitated phase were analyzed as previously (6,7,24,31) by using a linear combination of the scattering intensities:
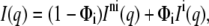 |
(5) |
where Φi is the fraction of the interdigitated phase and the superscripts “ni” and “i” refer to the noninterdigitated and interdigitated phases, respectively.
RESULTS
Fig. 1 reports the DSC results of the relevant PGs at different lipid/peptide (L/P) molar ratios. The complete thermodynamic data is listed in Table 1, including also the DSC data on the perdeuterated lipid analogs. Our results in the absence of the peptide are in good agreement with previously published data (31,32). In the case of DMPG, the enthalpy of the pretransition at Tp ∼ 10°C decreased with PGLa concentration (Fig. 1 A) and an additional phase transition peak, ∼2°C higher than the original Tm, appeared at L/P = 100. The position of this peak did not shift significantly upon the addition of further peptide, but its enthalpy increased on account of ΔHm. Similar findings have been reported previously (6,33) and indicate the formation of a peptide-rich lipid phase, which melts at a different temperature than the pure DMPG bilayer. Both phases coexist, and their relative amount depends on the peptide concentration. Comparable, although more pronounced, effects were observed via DSC for DPPG/PGLa interactions (Fig. 1 B). The pretransition had disappeared already at L/P = 50:1, and a new peak with a phase transition temperature of 41.5°C (i.e., 1.4°C higher than the Tm for pure DPPG) accounted for ∼40% of the total enthalpy at L/P = 100:1. Upon increasing the PGLa concentration to 50:1 the original main-phase transition peak of DPPG disappeared, and at 25:1 the peak at higher temperature started to broaden, in agreement with early studies at low ionic strength (17). The influence of PGLa was most pronounced for DSPG (Fig. 1 C). The pretransition disappeared at L/P = 100:1, and the peak, at a temperature 0.9°C higher, was already predominant at this low PGLa concentration. Increasing the peptide concentration further broadened the main transition peak. The DSC studies were repeated for perdeuterated PG analogs, where the results were comparable to the nondeuterated lipids but shifted to lower temperatures (Table 1).
FIGURE 1.

Excess heat capacity curves of DMPG (A), DPPG (B), and DSPG (C), at different PGLa concentrations. Selected cooling scans are shown as dash-dotted lines. Numbers indicate the lipid/peptide molar ratio.
TABLE 1.
Thermodynamic data from DSC measurements of various PGs in the presence of PGLa
| Lipid† | L/P | Tp (°C) | ΔHp (kcal/mol) | Tm (°C) | ΔHm (kcal/mol) | Tm†‡ (°C) |
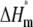 (kcal/mol) (kcal/mol) |
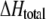 § (kcal/mol) § (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| DMPG | 10.3 | 0.6 | 22.4 | 7.2 | 7.2 | |||
| DMPG | 100:1 | 10.4 | 0.4 | 22.4 | 4.8 | 24.0 | 2.2 | 7.0 |
| DMPG | 50:1 | 11.6 | 0.1 | 22.7 | 3.1 | 24.5 | 5.1 | 8.2 |
| DMPG | 25:1 | 10.7 | 0.1 | 22.5 | 2.3 | 24.3 | 4.5 | 6.8 |
| DMPG-d | 8.9 | 0.6 | 18.8 | 7.2 | 7.2 | |||
| DMPG-d | 25:1 | 19.0 | 1.7 | 20.7 | 6.6 | 8.3 | ||
| DPPG | 33.3 | 1.5 | 40.2 | 10.8 | 10.8 | |||
| DPPG | 100:1 | 32.3 | 0.6 | 40.1 | 4.9 | 41.5 | 3.9 | 8.7 |
| DPPG | 50:1 | 41.5 | 9.3 | 9.3 | ||||
| DPPG | 25:1 | 41.5 | 8.5 | 8.5 | ||||
| DPPG-d | 27.8 | 35.7 | 9.7 | 9.7 | ||||
| DPPG-d | 100:1 | 35.7 | 9.2 | 36.9 | 3.9 | 13.1 | ||
| DPPG-d | 50:1 | 35.7 | 6.8 | 37.0 | 6.2 | 13.0 | ||
| DPPG-d | 25:1 | 35.7 | 2.7 | 37.0 | 10.3 | 13.0 | ||
| DSPG | 50.4 | 1.0 | 53.6 | 13.0 | 13.0 | |||
| DSPG | 100:1 | 53.8 | 5.8 | 54.6 | 6.1 | 11.9 | ||
| DSPG | 50:1 | 54.7 | 11.2 | 11.2 | ||||
| DSPG | 25:1 | 54.6 | 11.8 | 11.8 | ||||
| DSPG-d | 48.8 | 14.1 | 14.1 | |||||
| DSPG-d | 100:1 | |||||||
| DSPG-d | 50:1 | |||||||
| DSPG-d | 25:1 | 49.8 | 13.3 | 13.3 |
Lipid name followed by d indicates a chain-perdeuterated lipid.
Transition temperature of the peptide-induced phase-transition peak.
ΔHtotal = ΔHm + 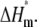
Deuterium NMR was used to monitor the impact of PGLa on the dynamics and structure of the surrounding lipid environment. To obtain a detailed picture of the changes in local mobility in the hydrophobic core of the membrane, 2H-NMR spectra of DPPG and DSPG, 2H-labeled in their acyl chains, were measured at different temperatures and lipid/peptide ratios. The results below the Tm are shown in Fig. 2, where in both cases a slight change in the shoulders of the spectra was observed upon varying L/P. A more pronounced change occurred in the center of the lineshapes of the DPPG/PGLa samples, as resonances with reduced splittings emerged and flattened the central Pake doublet. As this central signal originates from the methyl groups, this observation at higher peptide concentration suggests increased disorder at the terminus of the DPPG hydrocarbon chains. In contrast, there was no additional intensity observed in the spectra of DSPG in the presence of peptide.
FIGURE 2.

2H-NMR gel-phase spectra of chain-deuterated DPPG at 25°C (A) and DSPG at 40°C (B), containing PGLa at different lipid/peptide ratios.
Fig. 3 shows representative 2H-NMR spectra of the DPPG and DSPG samples in the fluid phase (45°C and 60°C, respectively), recorded at 76.7 MHz. Deuterium spectra at 107.4 MHz resulted in a similar picture, but were less well resolved (data not shown). Pronounced changes in the 2H-NMR spectra with increasing peptide concentration were observed in the case of DPPG (Fig. 3 A). The quadrupolar splittings, each originating from different labeled positions along the acyl chain, increased with an increasing amount of PGLa. In addition to this change of the splitting sizes, a broadening of the individual doublets and an overall change in the lineshape was observed with increasing peptide concentration. Furthermore, the doublet intensities in the spectrum of pure DPPG are seen to increase with increasing splitting, but they are more evenly distributed in the presence of peptide. In contrast, for DSPG samples above Tm, no differences in the 2H-NMR spectra were observed in the presence and absence of PGLa (Fig. 3 B).
FIGURE 3.

2H-NMR fluid-phase spectra of chain-deuterated DPPG vesicles at 45°C (A) and DSPG at 60°C (B), containing PGLa at different lipid:peptide ratios.
Because individual doublets for each 2H-labeled site were resolved for carbon positions C7–C16 in DPPG and C7–C18 in DSPG, respectively, we were able to analyze the 2H-NMR spectra in the form of an order parameter profile, SCD. This allowed us to address the degree of motional averaging along the acyl chain, and how it is affected by PGLa. The profiles (Fig. 4) were determined from “de-Paked” spectra. In accordance with the raw data presented in Fig. 3, the overall order of the DPPG membranes increased with increasing peptide concentration. It is interesting to note that the variation of SCD along the acyl chain remains almost unaffected by PGLa in the case of DPPG (Fig. 4 A). SCD changed only by an overall constant upshift above L/P = 100:1. Considering a previous interpretation of the order parameter in terms of chain length (34,35), the increase of SCD indicates a modest increase in membrane thickness upon the addition of peptide. In contrast, the presence of PGLa had no effect on the order parameter profile of DSPG (Fig. 4 B).
FIGURE 4.

NMR order parameter profiles (SCD) obtained from deconvoluted (de-Paked) 2H-NMR powder spectra of chain-deuterated DPPG (A) and DSPG (B) in the absence of PGLa (○) and at L/P = 100:1 (⋄) and 25:1 (•). Errors are within the size of the symbols.
Finally, SWAXS experiments were performed to address the effect of PGLa on the overall structural properties of DMPG, DPPG, and DSPG. Fig. 5 shows the scattering patterns of all three lipids in the lamellar gel phase, Lβ′, at selected temperatures and in the presence of PGLa (L/P = 25:1). The SAXS patterns of all lipids display a pure diffuse modulation originating from positionally uncorrelated bilayers as a result of electrostatic repulsion of the anionic lipids (Fig. 5 A). A detailed analysis of the patterns in terms of a global model (29) showed that the pure DMPG and DPPG dispersions consist of a single phase. The DSPG sample, however, shows a coexistence of a Lβ′ and interdigitated LβI phases, as previously reported in detail (24). A similar phase coexistence was recently found in lysyl-DPPG bilayers (31). The LβI phase shows up as additional diffuse scattering around q ∼ 0.2 Å−1 and precludes a global data analysis in terms of a single phase, as evidenced by the disagreement of such a model with this scattering regime (Fig. 5 A, dash-dotted lines). It is of interest that DPPG and DSPG bilayers displayed a similar coexistence of interdigitated and noninterdigitated phases in the presence of PGLa, which is again most pronounced for DSPG. This has been reported before also for LL-37 and melittin in PG bilayers (6,7) and can be understood in terms of the formation of a quasi-interdigitated phase LβIq, where the hydrophobic lipid tails are shielded by the peptide (see Fig. 6 in Sevcsik et al. (6)). The quality of the data presented here precludes us from detecting the LβIq phase in the case of DMPG. The WAXS patterns, which report on the chain lattice, are characteristic of orthorhombic packing with tilted hydrocarbon chains for pure DMPG and DPPG (Fig. 5 B). The WAXS pattern of pure DSPG can in turn be described by a linear superposition of tilted hydrocarbon chains in an orthorhombic lattice and nontilted hexagonally packed acyl chains (24). The occurrence of the latter chain lattice is due to the LβI phase. The introduction of PGLa into DMPG and DPPG bilayers led to significant disorder, as evidenced by the diffuse chain-packing peak in the WAXS regime. This is in agreement with the NMR data, which suggested increased methyl mobility in the presence of the peptide below the Tm (see above). Also in agreement with the NMR data (Fig. 2 B), DSPG displayed a well-defined WAXS peak in the presence of PGLa (Fig. 5), showing nontilted hexagonally packed chains, most likely originating from quasi-interdigitated bilayers.
FIGURE 5.

Small- and wide-angle scattering of DMPG (i), DPPG (ii), and DSPG (iii) in the gel phase in the absence and presence of PGLa (P/L = 1:25). (A) SAXS data on a logarithmic scale with an increment of 10. (B) WAXS data on a linear scale with an increment of 1000. Solid lines show the model fits to the data in the absence of PGLa, and dashed lines to the data in the presence of PGLa. The resulting uncertainties of the model parameters are presented in Table 2 and Fig. 7. Dash-dotted lines correspond to fits of a single bilayer phase. Fits to the WAXS patterns were performed using a combination of Gaussian distribution and are presented here for illustration purposes only. The pattern of pure DSPG shows a coexistence of orthorhombically and hexagonally packed hydrocarbon chains corresponding to the Lβ′ and LβI phases, respectively (dash-dotted lines). A detailed WAXS data analysis has been presented previously (24). The selected temperatures are 5°C for DMPG, 30°C for DPPG, and 45°C for DSPG. Data have been scaled for a better view.
FIGURE 6.

Small-angle scattering of DMPG (i), DPPG (ii), and DSPG (iii) in the fluid phase in the absence and presence of PGLa (L/P = 25:1). Solid lines show the model fits to the data in the absence of PGLa, and dash-dotted lines to the data in the presence of PGLa. The selected temperatures are 30°C for DMPG, 45°C for DPPG, and 60°C for DSPG. Data have been scaled for a better view.
Above the chain-melting transition, in the fluid lamellar Lα phase, none of the sample SAXS patterns exhibited any contribution from a coexisting phase, but could all be described in terms of a single-phase model, even in the presence of PGLa (L/P = 25:1) (Fig. 6). The fits yielded χ2 values close to 1, demonstrating the significance of the derived structural parameters. Table 2 lists the results in the absence and presence of PGLa and also contains the molecular volume, hydrocarbon volume, lateral area, and hydrocarbon length of the lipids. The molecular volumes were determined using dilatometry on liposomal dispersions. The other structural parameters were derived as described in the previous section. The area/lipid values in the presence of PGLa were calculated assuming that the partial molecular lipid volume is unaffected by the peptide, in agreement with a recent report (8). It is most interesting that both DMPG and DPPG show an increase of the membrane thickness in the Lα phase by 1–2 Å (Table 2). In contrast, dB remains within the error of the measurement constant for DSPG upon the addition of the peptide. The increase of membrane thickness goes hand in hand with a 2 Å2 decrease of A for DMPG, and 4 Å2 in the case of DPPG (Table 2), indicating increased lipid-packing density in the presence of PGLa.
TABLE 2.
Structural parameters of the studied PGs in the fluid phase in the absence and presence of peptide
| Lipid* | T (°C) | dB (Å) | dC (Å) | A (Å2) | V (Å3) | VC (Å3) |
|---|---|---|---|---|---|---|
| DMPG | 30 | 48.4 ± 0.4 | 14.2 ± 0.4 | 55.1 ± 2 | 1039 ± 10 | 782 ± 10 |
| + PGLa | 49.3 ± 0.4 | 14.7 ± 0.4 | 53.3 ± 2 | |||
| DPPG | 45 | 50.2 ± 0.4 | 15.1 ± 0.4 | 61.0 ± 2 | 1178 ± 10 | 921 ± 10 |
| + PGLa | 52.2 ± 0.4 | 16.1 ± 0.4 | 57.2 ± 2 | |||
| DSPG | 60 | 52.8 ± 0.4 | 16.4 ± 0.4 | 63.8 ± 2 | 1303 ± 10 | 1046 ± 10 |
| + PGLa | 52.5 ± 0.4 | 16.3 ± 0.4 | 64.4 ± 2 |
When peptide was present, the lipid/peptide ratio was L/P = 25:1.
In Fig. 7, the calculated membrane thicknesses of the pure PG bilayers are compared to those in the presence of PGLa (L/P = 25:1) both above and below the Tm. The general increase of dB with chain length, observed for all phases, can be attributed to the addition of CH2 groups. The coexisting noninterdigitated phase remains unaffected, with the exception of DPPG, which displayed a slightly smaller dB value. The reduction of the membrane thickness of DPPG could be due to an increase in the molecular tilt angle or simply to increased molecular disorder, as evidenced also in the corresponding WAXS patterns (Fig. 5 B) and NMR data (Fig. 2 A).
FIGURE 7.

Chain length dependence of the membrane thickness in the Lβ′ (squares), LβI (triangles), and Lα phases (circles). Solid symbols correspond to the results in the presence of PGLa (L/P = 25:1), and open symbols to those in the absence of peptide.
DISCUSSION
Using DSC, 2H-NMR, and x-ray scattering, we have provided experimental evidence that PGLa interacts with PG bilayers in a chain-length- and concentration-dependent manner. Moreover, the effects of the peptide vary strongly with the phase state of the lipid. Below the chain-melting transition, PGLa induces a quasi-interdigitated phase that coexists in various proportions with the original lamellar gel phase. The presence of the LβIq phase was most prominent for DSPG, which exhibits a co-existence of interdigitated and noninterdigitated phases even in the absence of the peptide (24). We have previously reported the occurrence of LβIq phases for PGs and long-chain PCs in the presence of the peptides PGLa, melittin, and LL-37 (6,7). Their formation can be understood assuming that the peptide gets embedded into the gel phase bilayer at the water/lipid interface, with the helical axis aligned parallel to the membrane surface. This creates a void below the peptide, which is compensated for by moving the methyl termini of the opposing lipid monolayer close to the peptide, thereby generating the LβIq phase (Fig. 8). A compensation of the void by an elastic monolayer deformation is not possible due to the high bending rigidity (∼100 kBT) in the gel phase (36). The increased acyl chain disorder observed in DPPG from both WAXS and NMR data (Figs. 2 and 5) is most likely due to defect zones between the coexisting gel phases. It is interesting that no such disorder was observed for DSPG. This indicates that PGLa is more effective in inducing a LβIq phase due to the larger void induced and the DSPG propensity to form an interdigitated phase even in the absence of the peptide (24).
FIGURE 8.

Schematic representation of PGLa-associated structural changes in PG bilayers. Below the main phase transition (T < Tm), all three studied lipids exhibit a quasi-interdigitated phase in the presence of PGLa. The peptide aligns parallel to the membrane surface and its hydrophobic moieties are shielded by the hydrocarbon chains of the opposing lipid monolayer. In the fluid phase (T > Tm), above a critical threshold concentration, PGLa inserts into the PG membranes and most likely forms toroidal pores. In DMPG membranes, PGLa inserts at an angle and leads to a small increase in membrane thickness. The membrane thickening effect is most pronounced for DPPG membranes, where PGLa inserts vertically into the bilayer, and least pronounced for DSPG bilayers, in which no membrane deformation is necessary due to a match of the hydrophobic lipid core with the hydrophobic peptide length.
In the fluid phase, the bending rigidity of lipid bilayers is ∼1 order of magnitude lower than in the gel phase. Thus, the bilayer may respond elastically to a peptide embedded in the bilayer interface, leading to a local dimple deformation (4). Averaged globally over the lipid bilayer, this shows up as membrane thinning, as previously observed in many x-ray diffraction studies on lipid/peptide interactions (4,8,37). At sufficiently high concentrations, the peptides may change their orientation by inserting vertically or at an oblique angle into the lipid bilayer, forming pores of either barrel stave or toroidal type. Recent experiments suggest that the process of pore formation is driven by the entropy of the membrane-adsorbed peptide ensemble (8). Matching of the hydrophobic peptide length with the hydrocarbon core of the lipid bilayer may also lead to a local membrane deformation and thinning on the global average over the membrane (4). Thus, the observation of membrane thinning in connection with lipid/peptide interactions has been hitherto generally taken as “the” fingerprint for pore formation.
It is striking that our x-ray and NMR experiments, in excellent agreement, show an overall increase of membrane-order DPPG in the fluid phase (Table 2 and Fig. 4 A). Furthermore, in the case of DSPG, the order-parameter profile and membrane thickness were not affected at all by PGLa, even at L/P = 25:1 (Fig. 4 B). This is difficult to reconcile in terms of the currently accepted views on lipid/peptide interactions. The absence of any effect on DSPG membranes in the fluid phase could be simply attributed to an expulsion of the peptide from the bilayer. A possible scenario would be that the hydrophobic interactions between hydrocarbon chains, which increase with acyl chain length, finally overrule the electrostatic attraction, rendering the bilayer practically impenetrable. Indeed, if one compares the area/lipid values for the three studied PGs in the absence of PGLa (Table 2) to the reported data for the chain analogous PCs (DMPC, A = 59.6 Å2 (38); DPPC, A = 64.0 Å2 (38); and DSPC, A = 66.7 Å2 (39)), one finds significantly smaller values for the PGs. This signifies increased packing density for PG bilayers compared to PC membranes, which is unexpected in view of the negative charges carried by the PG headgroups. A similar effect was recently reported for phosphatidylserine bilayers in the absence of salts (40), which might be due to hydrogen-bond formation, as suggested previously for PG membranes (32). As an alternative, however, screening of the headgroup charges by the cations present in the used buffer could also account for this effect. In any case, the increased molecular packing would seem to support the above picture of peptide exclusion. However, it is then difficult to explain why DPPG, whose area/lipid differs by about the same amount from DPPC as from DSPG and DSPC, is affected by the peptide and DSPG not. Moreover, the cooling DSC scans are comparable to the heating scans (Fig. 1), in particular also for DSPG, showing more or less identical heat capacity peaks, but being slightly shifted toward lower temperature as a typical hysteresis effect. Henceforth, PGLa must interact with the DSPG bilayers in the fluid phase. Thus, we conclude that the peptide is not dissolved in the aqueous phase, but is somehow in contact with the lipid bilayer.
An alternative explanation would be that PGLa does not penetrate into the bilayer in the fluid phase, but lies on its top, aligning its positive charges with the negative PG headgroup charges. In fact, Li and Salditt reported an increase of dB in the presence of magainin 2 for DMPC/DMPG mixtures and assumed that the peptide leads to a crowding of DMPG due to electrostatic interactions (37). This is supported by the DPPG order-parameter profile in the presence of PGLa, showing that a change in mobility due to a localization of the peptide in a particular membrane depth is unlikely. Such an effect might thus explain the observed increase in membrane thickness for DMPG and DPPG upon the addition of PGLa (Fig. 7). However, it cannot account for the absence of a thickening effect in DSPG membranes. The area/lipid of DSPG bilayers is ∼3 Å2 larger than that of DPPG (Table 2). Thus, if the PG headgroups were to align with the PGLa charges, a much more pronounced thickening effect would be expected for DSPG. However, the changes in dB and A in DSPG bilayers containing PGLa are insignificant (Table 2). In addition, PGLa aligned with its polar side toward the charged membrane surface would require an aggregation of PGLa to shield its hydrophobic moieties from the aqueous phase. Such peptide oligomerization would then have to be reversed upon the transition into the gel phase to form the quasi-interdigitated phase. This is very unlikely to happen and we can, therefore, also rule out this explanation.
An indication about the present peptide-bilayer interactions can be obtained considering the length of PGLa and hydrophobic matching (41) (see also Fig. 8). Assuming a complete α-helical backbone, the length along its axis turns out to be ∼32 Å (18,21). This value can now be compared to 2dC for DSPG (Table 2), which remarkably turns out to be identical. Thus, PGLa would fit perfectly into the hydrophobic core of DSPG. Due to this perfect hydrophobic match, the DSPG bilayers do not need to deform, which explains our invariant observations from 2H-NMR and x-ray scattering. The hydrophobic core of DPPG is in turn too small to accommodate the peptide with its full length. It therefore responds to the insertion of the peptide by stretching of the hydrocarbon chains, leading to increased order and a larger membrane thickness. Indeed, an increase of chain order and membrane thickness has been observed previously for PC bilayers in the presence of hydrophobic model peptides with N- and C-terminal anchors (42), and this was also attributed to hydrophobic matching. It is interesting to note that the dB of DPPG in the presence of PGLa is about equal to the dB for DSPG (Table 2). In the case of DMPG, the increase of dB upon addition of PGLa is, at ∼1 Å, significantly smaller than that for DPPG. Apparently, the entropic penalty of increasing the membrane thickness to a value that can accommodate PGLa with its helical axis normal to the bilayer surface is too high. We therefore think that the system responds simply by tilting the peptide with respect to the DMPG membrane normal, requiring only a small increase of membrane thickness as an energetically favorable compromise. In fact, in support of this argument, PGLa has been shown by solid-state NMR to insert at an angle of 30° into DMPC/DMPG bilayers (18), where it has been postulated to be most stable as an antiparallel dimer maintaining an overall amphiphilicity.
In conclusion, we note that this explanation also accounts for our DSC results, which showed the presence of a peptide-rich phase that melts/freezes at slightly higher temperature upon heating and cooling, respectively (Fig. 1). The peptides simply change their insertion state from being parallel to the lipid bilayer surface in the gel phase to a transmembrane state in the fluid phase above a given threshold concentration. Peptide insertion is probably facilitated by the strong density fluctuations occurring in the phase transition regime (43,44). The inserted peptides can be either tilted or not in response to the elastic properties of the lipid bilayer. Further, we propose that PGLa, having a net charge of +5, forms pores of the toroidal type, in agreement with theoretical considerations (45) and recent experimental observations (19). It is certain that peptide insertion in the fluid phase occurs only above a particular threshold concentration, L/P* (21), which was not determined in this study. However, our 2H-NMR results suggest that 25 < L/P* < 100 for DPPG, since no effects could be observed in the order-parameter profile for L/P = 100:1 (Fig. 4 A). Recent oriented circular dichroism measurements on PGLa insertion in DMPC bilayers reported a value in the same concentration range (46).
CONCLUSIONS
Our results on the interaction of the antimicrobial frog-skin peptide PGLa with PG model membranes unequivocally demonstrate a complex behavior depending on several parameters, such as acyl chain length, lipid phase state, and peptide concentration. Quasi-interdigitated phases were detected for DPPG and DSPG bilayers below the Tm, being most pronounced for DSPG. This phase is most likely also present in DMPG bilayers, but difficult to detect at the present resolution. In the fluid phase, we observed an increase of the membrane thickness for DMPG and DPPG, whereas DSPG bilayers remained unaffected (Fig. 8). It is therefore not straightforward to extrapolate behavior in the fluid phase from observations in the gel phase. Furthermore, although membrane thinning is at present considered the hallmark of peptide pore formation, it is not a universal response of bilayers to insertion of amphipathic peptides. The system may respond with a tilt of the inserted peptide or an increase of membrane thickness, or may even remain virtually unperturbed, depending on the match of hydrophobic moieties of lipid and peptide. We believe that this is a general result with respect to the currently discussed mechanisms of membrane disruption by AMPs, i.e., the detergentlike carped model and the formation of discrete barrel stave or toroidal pores (for recent reviews, see, e.g., (2,47)). Even a detergentlike disintegration of lipid membranes proceeds through a transient insertion of the peptides into the lipid bilayer. This is where our findings will apply.
Acknowledgments
This work was supported by a Unesco-L'Oréal Women in Science fellowship to A. H., a Fonds zur Förderung der wissenschaftlichen Forschung (Austrian Science Fund) grant (P17112-B03) to G.P., Deutsche Forschungsgemeinschaft Center for Functional Nanostructures and Helmholtz Association grants to S.L.G. and A.S.U., and grants from the Biotechnology and Biological Sciences Research Council and the Molecular Research Center to A.W. We further acknowledge support from Magnex Scientific, UK, and Varian for NMR.
Weiquo Jing's present address is The Rockefeller University, Proteomics Resource Center, 1230 York Ave., New York, NY 10065.
Editor: Thomas J. McIntosh.
References
- 1.Lohner, K., editor. 2001. Development of Novel Antimicrobial Agents: Emerging Strategies. Horizon Scientific, Wymondham, UK.
- 2.Lohner, K., and S. E. Blondelle. 2005. Molecular mechanisms of membrane perturbation by antimicrobial peptides and the use of biophysical studies in the design of novel peptide antibiotics. Comb. Chem. High Throughput Screen. 8:241–256. [DOI] [PubMed] [Google Scholar]
- 3.Lohner, K., E. Sevcsik, and G. Pabst. 2008. Liposome-based biomembrane mimetic systems: implications for lipid-peptide interactions. In Advances in Planar Lipid Bilayers and Liposomes, Vol. 6. A. Leitmannova-Liu, editor. Elsevier, Amsterdam. 103–137.
- 4.Huang, H. W. 2006. Molecular mechanism of antimicrobial peptides: the origin of cooperativity. Biochim. Biophys. Acta. 1758:1292–1302. [DOI] [PubMed] [Google Scholar]
- 5.Lee, M. T., W. C. Hung, F. Y. Chen, and H. W. Huang. 2008. Mechanism and kinetics of pore formation in membranes by water-soluble amphipathic peptides. Proc. Natl. Acad. Sci. USA. 105:5087–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sevcsik, E., G. Pabst, A. Jilek, and K. Lohner. 2007. How lipids influence the mode of action of membrane-active peptides. Biochim. Biophys. Acta. 1768:2568–2595. [DOI] [PubMed] [Google Scholar]
- 7.Sevcsik, E., G. Pabst, W. Richter, S. Danner, H. Amenitsch, and K. Lohner. 2008. Interaction of LL-37 with model membrane systems of different complexity: influence of the lipid matrix. Biophys. J. 94:4688–4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pabst, G., S. Danner, R. Podgornik, and J. Katsaras. 2007. Entropy-driven softening of fluid lipid bilayers by alamethicin. Langmuir. 23:11705–11711. [DOI] [PubMed] [Google Scholar]
- 9.Afonin, S., S. L. Grage, M. Ieronimo, P. Wadhwani, and A. S. Ulrich. 2008. Temperature-dependent transmembrane insertion of the amphiphilic peptide in lipid bilayers observed by solid state 19F-NMR. J. Am. Chem. Soc. In press. [DOI] [PubMed]
- 10.Watts, A., K. Harlos, W. Maschke, and D. Marsh. 1978. Control of the structure and fluidity of phosphatidylglycerol bilayers by pH titration. Biochim. Biophys. Acta. 510:63–74. [DOI] [PubMed] [Google Scholar]
- 11.Hoffmann, W., K. Richter, and G. Kreil. 1983. A novel peptide designated PYLa and its precursor as predicted from cloned mRNA of Xenopus laevis skin. EMBO J. 2:711–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreu, D., H. Aschauer, G. Kreil, and R. B. Merrifield. 1985. Solid-phase synthesis of PYLa and isolation of its natural counterpart, PGLa [PYLa-(4–24)] from skin secretion of Xenopus laevis. Eur. J. Biochem. 149:531–535. [DOI] [PubMed] [Google Scholar]
- 13.Bevins, C. L., and M. Zasloff. 1990. Peptides from frog skin. Annu. Rev. Biochem. 59:395–414. [DOI] [PubMed] [Google Scholar]
- 14.Blondelle, S. E., and K. Lohner. 2001. Interaction of the antimicrobial peptide PGLa and its Ala-substitution analog with membrane-mimetic systems. In Peptides: The Wave of the Future. R. A. Houghten and M. Lebl, editors. American Peptide Society, San Diego. 491–492.
- 15.Jackson, M., H. H. Mantsch, and J. H. Spencer. 1992. Conformation of magainin-2 and related peptides in aqueous solution and membrane environments probed by Fourier transform infrared spectroscopy. Biochemistry. 31:7289–7293. [DOI] [PubMed] [Google Scholar]
- 16.Bechinger, B., M. Zasloff, and S. J. Opella. 1998. Structure and dynamics of the antibiotic peptide PGLa in membranes by solution and solid-state nuclear magnetic resonance spectroscopy. Biophys. J. 74:981–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Latal, A., G. Degovics, R. F. Epand, R. M. Epand, and K. Lohner. 1997. Structural aspects of the interaction of peptidyl-glycylleucine-carboxyamide, a highly potent antimicrobial peptide from frog skin, with lipids. Eur. J. Biochem. 248:938–946. [DOI] [PubMed] [Google Scholar]
- 18.Strandberg, E., P. Wadhwani, P. Tremouilhac, U. H. Durr, and A. S. Ulrich. 2006. Solid-state NMR analysis of the PGLa peptide orientation in DMPC bilayers: structural fidelity of 2H-labels versus high sensitivity of 19F-NMR. Biophys. J. 90:1676–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hickel, A., S. Danner-Pongratz, H. Amenitsch, G. Degovics, M. Rappolt, K. Lohner, and G. Pabst. 2008. Influence of antimicrobial peptides on the formation of nonlamellar lipid mesophases. Biochim. Biophys. Acta. In press. [DOI] [PubMed]
- 20.Konovalov, O., I. Myagkov, B. Struth, and K. Lohner. 2002. Lipid discrimination in phospholipid monolayers by the antimicrobial frog skin peptide PGLa. A synchrotron X-ray grazing incidence and reflectivity study. Eur. Biophys. J. 31:428–437. [DOI] [PubMed] [Google Scholar]
- 21.Tremouilhac, P., E. Strandberg, P. Wadhwani, and A. S. Ulrich. 2006. Conditions affecting the re-alignment of the antimicrobial peptide PGLa in membranes as monitored by solid state 2H-NMR. Biochim. Biophys. Acta. 1758:1330–1342. [DOI] [PubMed] [Google Scholar]
- 22.Tremouilhac, P., E. Strandberg, P. Wadhwani, and A. S. Ulrich. 2006. Synergistic transmembrane alignment of the antimicrobial heterodimer PGLa/magainin. J. Biol. Chem. 281:32089–32094. [DOI] [PubMed] [Google Scholar]
- 23.Hodzic, A., M. Rappolt, H. Amenitsch, P. Laggner, and G. Pabst. 2008. Differential modulation of membrane structure and fluctuations by plant sterols and cholesterol. Biophys. J. In press. [DOI] [PMC free article] [PubMed]
- 24.Pabst, G., S. Danner, S. Karmakar, G. Deutsch, and V. A. Raghunathan. 2007. On the propensity of phosphatidylglycerols to form interdigitated phases. Biophys. J. 93:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pabst, G., A. Hodzic, J. Strancar, S. Danner, M. Rappolt, and P. Laggner. 2007. Rigidification of neutral lipid bilayers in the presence of salts. Biophys. J. 93:2688–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laggner, P., and H. Stabinger. 1976. The partial specific volume changes involved in the thermotropic phase transitions of pure and mixed lecithins. In Colloid and Interface Science. M. Kerker, editor. Academic Press, New York. 91–96.
- 27.McCabe, M. A., and S. R. Wassall. 1997. Rapid deconvolution of NMR powder spectra by weighted fast Fourier transformation. Solid State Nucl. Magn. Reson. 10:53–61. [DOI] [PubMed] [Google Scholar]
- 28.Pabst, G., M. Rappolt, H. Amenitsch, and P. Laggner. 2000. Structural information from multilamellar liposomes at full hydration: full q-range fitting with high quality x-ray data. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Topics. 62:4000–4009. [DOI] [PubMed] [Google Scholar]
- 29.Pabst, G., R. Koschuch, B. Pozo-Navas, M. Rappolt, K. Lohner, and P. Laggner. 2003. Structural analysis of weakly ordered membrane stacks. J. Appl. Cryst. 63:1378–1388. [Google Scholar]
- 30.Pabst, G. 2006. Global properties of biomimetic membranes: perspectives on molecular features. Biophys. Rev. Lett. 1:57–84. [Google Scholar]
- 31.Danner, S., G. Pabst, K. Lohner, and A. Hickel. 2008. Structure and thermotropic behavior of the Staphylococcus aureus lipid lysyl-dipalmitoylphosphatidylglycerol. Biophys. J. 94:2150–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang, Y. P., R. N. Lewis, and R. N. McElhaney. 1997. Calorimetric and spectroscopic studies of the thermotropic phase behavior of the n-saturated 1,2-diacylphosphatidylglycerols. Biophys. J. 72:779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lohner, K., and E. J. Prenner. 1999. Differential scanning calorimetry and X-ray diffraction studies of the specificity of the interaction of antimicrobial peptides with membrane-mimetic systems. Biochim. Biophys. Acta. 1462:141–156. [DOI] [PubMed] [Google Scholar]
- 34.Schindler, H., and J. Seelig. 1975. Deuterium order parameters in relation to thermodynamic properties of a phospholiped bilayer. A statistical mechanical interpretation. Biochemistry. 14:2283–2287. [DOI] [PubMed] [Google Scholar]
- 35.Petrache, H. I., S. W. Dodd, and M. F. Brown. 2000. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys. J. 79:3172–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dimova, R., B. Pouligny, and C. Dietrich. 2000. Pretransitional effects in dimyristoylphosphatidylcholine vesicle membranes: optical dynamometry study. Biophys. J. 79:340–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li, C., and T. Salditt. 2006. Structure of magainin and alamethicin in model membranes studied by x-ray reflectivity. Biophys. J. 91:3285–3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Balgavy, P., M. Dubnickova, N. Kucerka, M. A. Kiselev, S. P. Yaradaikin, and D. Uhrikova. 2001. Bilayer thickness and lipid interface area in unilamellar extruded 1,2-diacylphosphatidylcholine liposomes: a small-angle neutron scattering study. Biochim. Biophys. Acta. 1512:40–52. [DOI] [PubMed] [Google Scholar]
- 40.Petrache, H. I., S. Tristram-Nagle, K. Gawrisch, D. Harries, V. A. Parsegian, and J. F. Nagle. 2004. Structure and fluctuations of charged phosphatidylserine bilayers in the absence of salt. Biophys. J. 86:1574–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Killian, J. A. 2003. Synthetic peptides as models for intrinsic membrane proteins. FEBS Lett. 555:134–138. [DOI] [PubMed] [Google Scholar]
- 42.de Planque, M. R., D. V. Greathouse, R. E. Koeppe, H. Schafer, D. Marsh, and J. A. Killian. 1998. Influence of lipid/peptide hydrophobic mismatch on the thickness of diacylphosphatidylcholine bilayers. A 2H NMR and ESR study using designed transmembrane α-helical peptides and gramicidin A. Biochemistry. 37:9333–9345. [DOI] [PubMed] [Google Scholar]
- 43.Pabst, G., J. Katsaras, V. A. Raghunathan, and M. Rappolt. 2003. Structure and interactions in the anomalous swelling regime of phospholipid bilayers. Langmuir. 19:1716–1722. [Google Scholar]
- 44.Pabst, G., H. Amenitsch, D. P. Kharakoz, P. Laggner, and M. Rappolt. 2004. Structure and fluctuations of phosphtidylcholines in the vicinity of the main phase transition. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 70:021908. [DOI] [PubMed] [Google Scholar]
- 45.Zemel, A., A. Ben Shaul, and S. May. 2005. Perturbation of a lipid membrane by amphipathic peptides and its role in pore formation. Eur. Biophys. J. 34:230–242. [DOI] [PubMed] [Google Scholar]
- 46.Buerck, J., S. Roth, P. Wadhwani, S. Afonin, N. Kanithasen, E. Strandberg, and A. S. Ulrich. 2008. Conformation and membrane orientation of amphiphilic helical peptides by OCD. Biophys. J. 95:3872–3881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sato, H., and J. B. Feix. 2006. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic α-helical antimicrobial peptides. Biochim. Biophys. Acta. 1758:1245–1256. [DOI] [PubMed] [Google Scholar]