

Abstract
Previous studies have demonstrated a calcium-dependent interaction of calmodulin (CaM) and Fas that is regulated during Fas-induced apoptosis in several cell lines, including cholangiocarcinoma, Jurkat cells, and osteoclasts. The binding of CaM and Fas has been identified on residues 231–254 of Fas; the V254N point mutation decreases the CaM/Fas binding, and the C-terminal deletion mutation increases the CaM/Fas binding. Recent studies have shown that CaM is recruited into the Fas-mediated death-inducing signaling complex (DISC) in a calcium-dependent manner. However, the molecular mechanisms whereby Fas mutations and CaM/Fas binding might regulate Fas-mediated DISC formation are unknown. In this study we investigated the binding thermodynamics and conformation of the CaM/Fas complexes with combined explicit solvent molecular-dynamics simulations and implicit solvent binding free-energy calculations. The binding free-energy analysis demonstrated that the Fas V254N point mutation reduced its binding affinity with CaM. In contrast, the Fas mutant with the deletion of the 15 amino acid at the C-terminus increased its binding to CaM. These observations are consistent with previous findings from biochemical studies. Conformational analyses further showed that the Fas V254N mutation resulted in an unstable conformation, whereas the C-terminal deletion mutation stabilized the Fas conformation, and both mutations resulted in changes of the degree of correlation between the motions of the residues in Fas. Analysis of the CaM/Fas complex revealed that CaM/Fas binding stabilized the conformation of both CaM and Fas and changed the degree of correlated motion of the residues of CaM and Fas. The results presented here provide structural evidence for the roles of Fas mutations and CaM/Fas binding in Fas-induced DISC formation. Understanding the molecular mechanisms of CaM/Fas binding in Fas-mediated DISC formation should provide important insights into the function of Fas mutations and CaM in regulating Fas-mediated apoptosis.
INTRODUCTION
Fas death receptor-activated signaling pathways have been shown to regulate apoptosis in a variety of cells, including cholangiocarcinoma, Jurkat cells, and osteoclasts (1,2). Dysfunction of Fas and its signaling pathways has been associated with diseases in both mice and humans (3–6). Mutations of Fas expression and its signaling in humans are associated with autoimmune lymphoproliferative syndrome (ALPS) (3,4,6), a heritable disorder of lymphocyte homeostasis. It has been reported that MRL/lpr−/− mice that are homozygous for Fas mutations develop an ALPS-like disease (5). This is in accord with the observation that numerous types of cancer express mutations of Fas that impair its function (7–12). In human cholangiocarcinoma, down-regulation of Fas is correlated with increased tumor size and a decreased survival time for patients (12–14). Thus, understanding the molecular mechanisms that are responsible for regulating the Fas signaling pathway is potentially important for identifying novel strategies and targets for the treatment of these diseases.
The Fas-activated signaling pathway is well characterized. Fas is a six-α-helix structure with a single hydrophobic site located within helices 5 and 6 (15). The Fas death receptor has the ability to self-associate or bind with other target proteins, including calmodulin (CaM) and the Fas-associated death domain (FADD) (15,16). Upon activation by its ligand, Fas recruits FADD via its death domain (DD), which in turn recruits the downstream signaling molecule caspase-8, or its enzymatic inactive homolog FLICE-like inhibitory protein (FLIP), to form the death-inducing signaling complex (DISC). The formation of the DISC is a crucial step in Fas-mediated signaling. It has been demonstrated that helices 1–2 of Fas associate with CaM (1,2) and that this interaction is regulated during Fas-mediated apoptosis. CaM regulates a variety of cellular Ca2+-dependent processes. The structure of CaM consists of eight helices that form four calcium-binding loops, which exhibit the characteristic EF hand structure (17). The dumbbell-shaped structure of CaM is composed of two globular terminal regions and two helices comprising the interdomain region (18). Upon binding of the calcium ions, the helices at either end of the EF hand rearrange and cause the protein to take on an open conformation, allowing for a greater interaction with target proteins such as Fas (17,19). Recently, CaM was found to be recruited into the Fas-mediated DISC (20); however, the role of CaM/Fas binding in the formation of the Fas-mediated DISC is unknown. It has been postulated that the interaction of Fas with CaM alters the conformation of Fas and therefore affects the binding affinity between Fas and FADD (19). The valine254 to asparagine (V254N) mutation of Fas, which results in a deficiency of Fas function and Fas-mediated signaling (15), has been shown to decrease the binding of Fas to both CaM and FADD (1,15). Furthermore, a C-terminal mutation, a deletion of the last 15 residues of the C-terminal of Fas, enhances the binding of Fas to CaM (19) and FADD (21). These mutant forms of Fas may result in conformational changes of Fas, thus altering its ability to interact with CaM and FADD. Further investigation is warranted to determine the molecular mechanisms underlying CaM/Fas binding and its role in regulating Fas-mediated signaling pathways.
In this study we investigated the conformation and binding thermodynamics of the CaM/Fas complex using explicit solvent molecular dynamics (MD) simulations and implicit solvent binding free-energy calculations. Binding free-energy calculations showed decreased binding between the Fas V254N mutant and CaM, but increased binding between the Fas C-terminal deletion mutant and CaM. These observations are consistent with previous studies that analyzed purified recombinant proteins by means of protein pull-down assays (19). Conformational analyses showed that the Fas V254N mutation resulted in an unstable conformation, whereas the C-terminus deletion mutation stabilized the Fas structure. Both mutations altered the degree of correlation seen between the motions of the residues in Fas. Analysis of the CaM/Fas complex revealed conformation changes and stabilization of both the CaM and Fas proteins, as well as changes in the degree of correlated motion of the residues of the proteins upon the formation of the CaM/Fas complex. Results from this study provide structural evidence in support of the important role of CaM/Fas binding in Fas-induced DISC formation. Understanding the molecular mechanism of CaM/Fas binding in Fas-mediated DISC formation should provide important insights into the function of CaM in regulating Fas-activated signaling pathways.
MATERIALS AND METHODS
Protein complex construction
The initial protein structures of the Fas DD (1ddf) (15) and Ca2+-bound CaM (3cln) (22), which were used for the MD simulations, were obtained from the Protein Data Bank. All mutations in this study were performed with the mutagenesis function of PyMol (23), and the visualizations used in the study were created with Visual Molecular Dynamics (VMD) software (24).
The initial CaM/Fas complex was determined using the docking utilities ZDOCK and RDOCK (25–27). The developers of these programs participate regularly in the Critical Assessment of Prediction of Interactions (CAPRI) Challenge (28–30). This challenge provides developers of protein docking algorithms an opportunity to compare their experimental complexes with known structures. These programs have consistently performed well in such exercises, and during the most recent event, the group predicted five out of six targets within the acceptable threshold; the one unsuccessful target was limited by inaccurate homology models (30). Additionally, published studies have successfully used these docking programs to generate complexes to be subjected to MD simulations and obtain satisfactory research results (31,32). Previous biochemical studies (19) and predictions using the CaM Target Database (17) have determined that the CaM binding region is located within helices 1 and 2 (residues (res) 231–254) of the Fas DD. This region exhibits the common 1-5-10 CaM binding motif (19). With the known binding site used as a docking constraint, the ZDOCK docking software was used to generate 2000 possible complexes that were scored based on their geometry, desolvation, and electrostatics (25). The RDOCK program used CHARMM to minimize the structures and determine the electrostatic, van der Waals, and contact energies associated with each of the proposed complexes (26). On the basis of these results, the 2000 predicted structures were ranked, and the highest-ranking structure was chosen to be the most probable protein complex. The structures generated for the CaM and Fas complexes, from CaM, Fas, and Fas mutants are shown in Fig. 1.
FIGURE 1.

Constructed complexes of CaM and Fas. (A) Complex of WT CaM and Fas. (B) Complex of CaM and Fas V254N mutant. (C) Complex of CaM and Fas C-terminal mutant.
MD simulation
To elucidate the molecular mechanisms underlying CaM and Fas interactions, a total of seven MD simulations were performed using the AMBER 9 package (33) (Table 1). The “parm99” parameters of the AMBER force field were assigned to the atoms in the simulated system, and a standard MD simulation protocol was conducted for the seven systems. The protein, or protein complex, was first minimized and then solvated in a TIP3P (34) water box with a 150 mM NaCl physiological salt concentration. A buffer of 10 Å was set between the protein and the box boundaries to reduce potential artifacts arising from periodicity. Additional ions were added to each system to neutralize the charge of the protein or protein complex. The AMBER 9 program Leap was used to solvate and neutralize the system. The solvent was then minimized and equilibrated for 10 ps while the proteins and ions were constrained. The entire system was then heated up to 300K by steps of 50K, lasting 10 ps each, at a constant temperature and volume, and 30 ns simulations at 300K constant temperature and 1 atm constant pressure were conducted for each system. The first 15 ns of each simulation served as the equilibration of the system, and the last 15 ns simulations served as the production run for data analysis. Periodic boundary conditions were applied to the simulated systems. The SHAKE constraints were applied to all hydrogen-heavy bonds to permit a dynamics time step of 2 fs. Electrostatic interactions were calculated with the particle-mesh Ewald method (PME) (35). Both the direct space PME and Lennard-Jones cutoffs were set at 10 Å. The data for the MD simulations were collected every 2 ps. The simulations were performed on a local BlueGene cluster using 32 processors for each simulation.
TABLE 1.
Seven systems simulated in this study
| System | Calmodulin (CaM) | Fas death domain (Fas DD) | CaM/Fas complex |
|---|---|---|---|
| 1 | Wild-type (WT) | ||
| 2 | WT | ||
| 3 | V254N mutant | ||
| 4 | C-terminal deletion mutant | ||
| 5 | CaM WT – Fas WT | ||
| 6 | CaM WT – Fas V254N mutant | ||
| 7 | CaM WT – Fas C-terminal deletion mutant |
Binding free-energy calculations
To verify the equilibration of the protein complex obtained using the docking programs, and to better understand the effects of protein mutations and conformational changes on the binding affinity, binding free energies were calculated for the three CaM/Fas complexes in this study. The binding free energy was calculated with the molecular mechanics-Poisson Bolzmann surface area (MM-PBSA) method as described in previous studies (18,36,37), which was implemented using the AMBER 9 molecular dynamics software package. The MM-PBSA method combines molecular mechanics, continuum electrostatics, solvent-accessible surface area calculations, and normal mode analyses for entropy to calculate the binding free energy from a series of snapshots obtained from the trajectories of the MD simulations.
As stated in Eq. 1, the change of any energy component of a system can be determined from the various components of the complex:
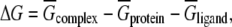 |
(1) |
where 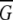 is the average value over the production trajectory.
is the average value over the production trajectory.
The binding energy is the sum of the molecular mechanics contribution to the energy (also known as the gas phase energy) and the solvation free energy of the system (Eq. 2):
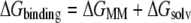 |
(2) |
 |
(3) |
The solvation free energy is made up of both polar and nonpolar contributions (Eq. 3). The polar contribution to the solvation free energy is based on the electrostatics of the system and calculated using the Poisson-Boltzman equation. In AMBER, this term is computed with the pbsa solver. The nonpolar component is dependent on the surface area of the molecule that is accessible to the solvent.
The gas phase contribution to the binding free energy is equal to the difference between the molecular mechanics energy and the entropy of the system (Eq. 4). The molecular mechanics portion of the binding energy calculation (Eq. 5) is equal to the sum of the van der Waals energy, electrostatic energy, and internal energy resulting from bond, angle, and torsion energies. T represents the temperature and S is the entropy of the system. The entropy of the system was computed using normal mode analysis. All structures were completely energy-minimized before analysis using the nmode program of AMBER.
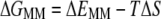 |
(4) |
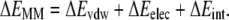 |
(5) |
The AMBER program mm_pbsa was used to determine the binding free energy for the complexes. Snapshots with a 10 ps time step from the MD production trajectories were used for the binding free-energy calculations. The binding free energy was calculated by finding the energy difference between the complex and the receptor and ligand (Eq. 1). The complex for each of the frames in the production trajectory was decomposed into receptor and ligand components. The resulting three components (complex, receptor, and ligand) were then used for the binding free-energy analysis. For the determination of polar solvation energy with the Poisson-Boltzman equation, values of 1 and 78.4 (18) were used for the dielectrics of the solute and solvent, respectively. Also, an ionic strength of 150 mM and a temperature of 300K were defined based on the conditions for the MD simulations along with a scale of two grids per Å. Due to the computational cost of calculating the entropy term, a subset of 1154 frames from the 30 ns production trajectories was used for normal mode analysis to obtain the entropy of the system. On the basis of the mean and standard deviation (SD) from the binding free-energy calculations, a Student's t-test with 95% confidence was conducted to determine whether the effect of Fas mutations on the binding affinity between CaM and Fas was statistically significant.
Conformation analyses
For this study a series of analyses were carried out to better understand the conformational character of CaM and the various Fas mutants, and the conformational changes induced by their interaction. The root mean-square deviation (RMSD) of protein backbone atoms was analyzed in conjunction with the binding free energy results to determine the systems' equilibration tendencies and its convergence. Root mean-square fluctuations (RMSFs) of the proteins were calculated on a residue-by-residue basis and averaged over the production simulation trajectories to observe the conformation fluctuation of protein domains. Dynamical cross-correlation maps (normalized covariance matrices) between residues provided insight into which residues have general movements correlating to the motion of other residues, and the degree of this correlation. These analyses were performed using the ptraj program of AMBER, and MATLAB (The MathWorks, Natick, MA) was used to generate the cross-correlation plots.
Statistical methods
To determine the effect of Fas mutations on binding affinity, the average values and the SDs of the binding free energy were calculated for the three CaM/Fas complexes. Because the adjacent snapshots in MD trajectories have a tendency to be correlated with each other, the autocorrelation times (38–40) for the binding free energy and the components of the binding free energy were obtained and the trajectories were resampled into statistically independent periods to calculate the statistical SD. The autocorrelation time τ was calculated as in previous studies (39,40). To avoid the potential issue that the individual quantity could have a different autocorrelation time and could couple to the longest-time-scale system property, we chose to calculate the autocorrelation time τ for the system RMSD as a whole (40). The longest decorrelation times of 2τ based on the RMSD of the three CaM/Fas complexes were chosen as the decorrelation times to calculate the SDs of the binding free energy and its components (40). With the obtained decorrelation times, bootstrap analyses (41) were performed with the use of a protocol similar to that of Chen and Pappu (42): 1), the first set of frames of the trajectory within 2τ time was not used for the analyses; 2), a random frame after 2τ time was chosen as the starting point and new resampled data sets within the 2τ time period were generated; and 3), steps 1 and 2 were repeated 1000 times. Each subsample data set that was obtained was used to calculate the averages of the subsamples, and the averages over all the subsamples were used to calculate the SD of the studied variables. With the mean and SDs of the binding free energy for the three CaM/Fas complexes, Student's t-test (44), with 95% confidence, was used to determine whether the effect of Fas mutations on the binding affinity of the CaM/Fas complex was significant.
RESULTS AND DISCUSSION
The binding free energy of the complexes over the 30 ns MD simulations, which was calculated as the ensemble-averaged value over the production trajectory, showed that the energy of the systems converged after the first 15 ns of the production simulation (Fig. 2). The RMSD of CaM (for both the N- and C-termini) and the Fas DD protein core (res 225–318) from the CaM/Fas complex (Fig. 3) over the 30 ns MD simulations also showed that the systems reached initial equilibration after the first 15 ns. It is important to note that although large RMSD values (+4 Å) are observed for the Fas protein core from the Fas/CaM complex, the same degree of RMSD was also observed in the individual Fas protein (Fig. 3 A). This is indicative not of protein complex rearrangement, but rather the natural flexibility of the core of the Fas protein. The remaining 15 ns were then used for the analyses of Fas and CaM conformational changes and binding thermodynamics.
FIGURE 2.

Binding free energy of the complexes of CaM and Fas over the 30 ns MD simulations. (A) Binding free energy without the entropy component over time. (B) The entropy component of binding free energy over time. Because of the computational cost to calculate the entropy term, the number of sampling points for the entropy calculation was different from that for the calculations of other components of binding free energy; thus, the entropy term and other components of the binding free energy are shown in two figures.
FIGURE 3.

RMSD for the Fas DD protein core (res 225–318) and CaM in its WT and in the CaM/Fas complex over the 30 ns MD simulation for different simulation cases: (A) the Fas DD protein core, (B) the N-terminal of CaM (res 1–77), and (C) the C-terminal of CaM (res 78–148).
Previous biochemical studies showed that the V254N mutation of Fas decreases the ability of Fas to bind with CaM (19), and a C-terminal mutation, a deletion of the last 15 residues of the C-terminus of Fas, enhances the binding of CaM and Fas (19). Using the MD simulation trajectories, we performed binding free-energy calculations and compared the results with those from experimental observations. After validating our binding affinity results with the experimental observations, we investigated the conformation changes of Fas induced by its mutations, as well as the structural changes of Fas and CaM that resulted from the formation of the CaM/Fas complex.
Binding thermodynamics
To determine the effect of the Fas V254N and C-terminal deletion mutations on the ability of Fas to bind with CaM, binding free-energy calculations were performed on the Fas/CaM complexes that consisted of CaM bound to three different Fas variations. All of the resulting energy calculations, including the values averaged over the 15–30 ns trajectories and the SDs obtained by statistical analyses, are shown in Table 2.
TABLE 2.
Free energy calculations for the binding of CaM to Fas
| Energy | Fas WT | Fas V254N mutant | Fas C-terminal deletion mutant |
|---|---|---|---|
| ΔEvdw | −43.38 ± 4.23 | −46.26 ± 2.93 | −95.97 ± 2.19 |
| ΔEelec | −484.78 ± 53.51 | −604.05 ± 22.15 | −810.77 ± 27.22 |
| ΔGpolar | 476.94 ± 49.87 | 609.65 ± 23.54 | 835.65 ± 30.48 |
| ΔGnonpolar | −8.39 ± 0.64 | −9.32 ± 0.27 | −15.59 ± 0.34 |
| TΔS | −52.50 ± 2.94 | −49.65 ± 1.92 | −56.81 ± 4.04 |
| ΔGbinding | −7.11 ± 4.67 | −0.33 ± 4.73 | −29.87 ± 3.41 |
| ΔΔGbinding | 6.78* | −22.74* | |
| Results from biochemical binding studies (19) | Strong | Weaker compared to the WT | Stronger compared to the WT |
All values in this table are expressed in terms of kcal/mol. ΔEvdw is the van der Waals energy, ΔEelec is the electrostatic energy, ΔGpolar is the polar contribution based on the solution to the Poisson-Boltzmann equation, ΔGnonpolar is the nonpolar contribution to the binding energy based on molecular surface area, TΔS is the solute entropic contribution, ΔGbinding is the overall binding free energy for the complex, and ΔΔGbinding is relative binding free energies with respect to the WT CaM/Fas complex.
Differences that are statistically significant (Student's t-test, p < 0.05).
The calculated relative binding free energies of the CaM/Fas mutants with respect to the wild-type (WT) CaM/Fas complex revealed a binding free-energy increase of 10.07 kcal/mol by the Fas V254N mutation and a binding free-energy decrease of 20.59 kcal/mol by the deletion of the 15 C-terminal amino acids of the Fas DD (Table 2). The relative binding free-energy analyses showed that the deletion of the 15 C-terminal amino acids of the Fas DD had a significant positive effect on the binding affinity of the CaM/Fas complex, whereas the V254N point mutation significantly reduced the binding affinity for the complex.
The calculated results were consistent with the binding affinity trends observed in previous biochemical binding studies (19). By analyzing the energy components, one can see where the majority of the energy contributions originate. The van der Waals energy for the first two cases is relatively constant, whereas the value for the C-terminal mutation is two times less than this value. This is to be expected because the first two complexes differ by a single point mutation and the latter is lacking a total of 15 residues. Fas mutations also resulted in significantly different electrostatic potential energy and polar solvation energy components for CaM/Fas mutant complexes compared to the WT CaM/Fas complex. Conformational changes of Fas by its mutations could contribute to the changes seen in each energy component of the binding free energy, and thus could have an effect on the binding affinity of the complex.
After validating our binding free-energy analysis results by comparison with those obtained from experimental studies, we further investigated the conformation changes of Fas induced by its mutations, as well as the structural changes of Fas and CaM resulting from the formation of the CaM/Fas complex.
Conformational changes in Fas
Previous experiments have demonstrated that the V254N mutation of Fas decreases Fas binding to CaM (19), and a deletion of the last 15 residues of the C-terminus of Fas enhances the binding of Fas to both CaM (19) and FADD (21). However, the molecular mechanism underlying CaM and Fas binding, and how the mutations contribute to the binding of Fas to CaM are unknown. RMSF comparisons of Fas (Fig. 4) and dynamical cross-correlation maps for the comparison of correlated motion of residues in Fas (Fig. 5) for different conditions showed that Fas conformations were changed by its mutations and by its binding with CaM.
FIGURE 4.

(A) RMSF comparison of the Fas DD in its WT, V254N mutation, and C-terminal deletion mutation. (B) RMSF comparison of the Fas DD after Fas binding to CaM with Fas in its WT, V254N mutation, and C-terminal deletion mutation. (C) RMSF comparison of Fas DD and the Fas DD after its binding to CaM.
FIGURE 5.

Dynamical cross-correlation maps to compare the degree of correlated motion of the residues in the Fas DD (white: correlation between residues; black: anticorrelation between residues). (A) WT Fas DD (top left) compared to the Fas DD after its binding with CaM (bottom right) (circled regions show the correlated motion between Fas N-terminal and C-terminal). (B) Fas DD V254N mutant (top left) compared to the Fas DD V254N mutant after its binding with CaM (bottom right) (circled regions showed the correlated motion between res 271–332 and res 221–241, and between res 235–265 and res 221–241). (C) Fas DD C-terminal mutant (top left) compared to the Fas DD C-terminal mutant after its binding with CaM (bottom right).
For Fas in its WT state, V254N mutation, and C-terminal deletion mutation, the RMSF (Fig. 4, A and B) showed that the V254N mutation caused large conformation fluctuations on or near its binding site with CaM (res 231–270), and the C-terminal deletion mutation resulted in an overall lesser degree of conformational fluctuation. These were observed in both single Fas (Fig. 4 A) and the Fas/CaM complex (Fig. 4 B). Upon binding with CaM, Fas experienced an overall lesser degree of conformational fluctuation compared to that of the single Fas. Dynamical cross-correlation maps that were used to investigate the correlation between the motion of the residues in the protein provided further insight into the effect of Fas mutations and Fas binding with CaM (Fig. 5). The top left halves of Fig. 5, A–C, are the cross-correlation maps for the Fas WT, V254N mutation, and C-terminal mutation. The results show that the Fas V254N mutation increased the degree of correlated motion (white) between res 271–332 and res 221–241 compared to that of the Fas WT. The C-terminal deletion mutation resulted in an overall lesser degree of both correlated (white) and anticorrelated (black) movement for the residues of Fas compared to the WT case. These results indicate that the Fas mutations directly affected the degree of correlated motion within the residues of Fas. Taken together, the conformational changes of Fas and the changes in the degree of correlated motion of the protein as a result of the V254N and C-terminal mutations show that these Fas mutations may contribute to the binding of Fas with CaM observed in previous experiments (19).
Fig. 5 A is the cross-correlation map for Fas in its WT state (top left) and after binding with CaM (bottom right). It shows that the degree of both correlated (white) and anticorrelated movement (black) in Fas is reduced by binding with CaM, especially the Fas N-terminal and C-terminal regions (circled). Fig. 5 B is the cross-correlation map for the Fas V254N mutant (top left) and after formation of the CaM/Fas complex (bottom right). It shows an increased degree of correlation (white) between the movements of res 271–332 and res 221–241, and an increased degree of anticorrelation (black) between the movements of res 235–265 and res 221–241. Fig. 5 C is the cross-correlation map for the Fas C-terminal mutant (top left) and after Fas binding with CaM (bottom right). It shows that an overall decreased degree of both correlated and anticorrelated movement for the residues in Fas resulted from binding with CaM.
In summary, Fas conformational fluctuations and the degree of correlation seen between the motions of the residues in Fas induced by the Fas mutations provide structural evidence to support previous experimental observations that the Fas V254N mutation decreases the ability of Fas to bind with CaM, and the Fas C-terminal deletion mutation enhances the binding of Fas to CaM (19). The changes in conformation and degrees of correlation provide structural insight into the effect of the formation of the CaM/Fas complex on the structure of Fas, and the ability of Fas to bind with FADD to form the DISC (21).
Conformational changes in CaM
The association that occurred between the WT Fas DD and CaM affected not only the conformation of Fas and correlation of the residues of Fas, as described above, but also the conformation of CaM and the degree of correlated motion of the residues in CaM (Fig. 6, A and B). With the calculation of RMSF values for each of the residues of CaM in both the WT and complex states (Fig. 6 A), the formation of the CaM/Fas complex was shown to have a stabilizing effect on the residues of CaM. During the control run of CaM, in the absence of complex formation with Fas, the residue fluctuations varied greatly and exhibited peaks at residues in the following regions: the four calcium-binding loops circled by the dashed line in Fig. 6 A (res 20–28, 56–64, 93–101, and 129–137), and the two linker regions circled by the solid line in Fig. 6 A (res 39–44 and 112–117) connecting the two EF hands in each of the globular regions (44). These six domains that exhibited the greatest movement were the nonhelical components of CaM. Upon formation of the complex with the Fas WT, the overall structure of CaM was stabilized and many of these nonhelical components experienced a lesser degree of fluctuation. The cross-correlation maps (Fig. 6 B) show that the control simulations of CaM exhibit a high degree of correlation (white) between the first and second loops (res 20–28 and 56–64) and between the third and fourth loops (res 93–101 and 129–137; top left half of Fig. 6 B), and showed a high degree of anticorrelation (black) between the two globular domains at the N- and C-terminals (res 20–64 and 93–137). The degree of these correlations and anticorrelations was seen to lessen upon formation of the Fas/CaM complex (bottom right half of Fig. 6 B). The conformational change of CaM and the changes of the degree of correlated motion of residues in CaM, caused by the interaction with Fas, may explain the increased interaction of CaM with other proteins (such as the FLICE-like inhibitory protein (FLIP)) after association with Fas (45).
FIGURE 6.

(A) RMSF comparison of single CaM with CaM after binding to Fas. The dashed circles indicate the calcium-binding loops in CaM; solid circles indicate the linker regions connecting the two EF hands in each of the globular regions of CaM. (B) Dynamical cross-correlation maps to compare the degrees of correlated motion of the residues of the WT CaM (top left) and CaM after its binding to Fas (bottom right) (white: correlation between residues; black: anticorrelation between residues); circled regions showed the correlated motions between res 20–28 and 56–64, between res 93–101 and 129–137, and between the two globular domains at the N- and C-terminals (res 20–64 and 93–137).
The formation of the CaM/Fas complex and Fas mutations not only caused Fas to experience different conformations and different degrees of correlated motion, as described above, it also caused CaM to undergo a stabilizing conformation change and changes in the degree of correlated motion of the residues of CaM (Fig. 6). The effect of Fas mutations on the conformation of CaM and Fas was also visible in the entropy values obtained for the CaM/Fas complexes (Fig. 2 B).
CONCLUSIONS
Taken together, these data demonstrate that the binding of CaM to Fas results in conformational changes of both CaM and Fas that stabilize their structure, and affects the degree of correlated motion of the residues in both CaM and Fas. The Fas C-terminal deletion mutation stabilized the structure of Fas, resulted in an overall lesser degree of both correlated and anticorrelated movement, and increased the CaM/Fas binding affinity. In contrast, the Fas V254N mutation destabilized the structure of Fas, resulted in changes in the degree of correlated motion of residues, and decreased CaM/Fas binding affinity. These results provide important molecular evidence for the structural consequences of the binding of Fas and CaM, and the effects on Fas resulting from its mutations. Also, these results will facilitate further understanding of the function of Fas mutations and CaM/Fas binding in the regulation of Fas-induced DISC formation and Fas-mediated signaling pathways. These studies should provide important insights into the identification of strategies and targets to treat diseases characterized by defective Fas signaling, such as autoimmune diseases, osteoporosis, and cancer.
Acknowledgments
The authors thank Di Pan for helping with the statistical analyses performed in this study, and Nathan A. Baker and the anonymous reviewers for their helpful comments.
This work was supported in part by the National Science Foundation-sponsored UAB ADVANCE program (Y. H. Song), the Mr. & Mrs. Kwok-Chong Woo Grant (J. D. Suever), and a VA Merit Award (J. M. McDonald).
Editor: Bertrand Garcia-Moreno.
References
- 1.Wu, X., M. A. McKenna, X. Feng, T. R. Nagy, and J. M. McDonald. 2003. Osteoclast apoptosis: the role of Fas in vivo and in vitro. Endocrinology. 144:5545–5555. [DOI] [PubMed] [Google Scholar]
- 2.Chen, Y., J. Xu, N. Jhala, P. Pawar, Z. B. Zhu, L. Ma, C. H. Byon, and J. M. McDonald. 2006. Fas-mediated apoptosis in cholangiocarcinoma cells is enhanced by 3,3′-diindolylmethane through inhibition of AKT signaling and FLICE-like inhibitory protein. Am. J. Pathol. 169:1833–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher, G. H., F. J. Rosenberg, S. E. Straus, J. K. Dale, L. A. Middleton, A. Y. Lin, W. Strober, M. J. Lenardo, and J. M. Puck. 1995. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 81:935–946. [DOI] [PubMed] [Google Scholar]
- 4.Rieux-Laucat, F., F. Le Deist, C. Hivroz, I. Roberts, K. Debatin, A. Fischer, and J. de Villartay. 1995. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 268:1347–1349. [DOI] [PubMed] [Google Scholar]
- 5.Puck, J., S. Straus, F. Le Deist, F. Rieux-Laucat, and A. Fischer. 1999. Inherited disorders with autoimmune and defective lymphocyte regulation. In Primary Immunodeficiency Diseases: A Molecular and Genetic Approach. H. Ochs, C. I. E. Smith, and J. M. Puck, editors. Oxford University Press, New York. 339–352.
- 6.Bi, L. L., G. Pan, T. P. Atkinson, L. Zheng, J. K. Dale, C. Makris, V. Reddy, J. M. McDonald, R. M. Siegel, J. M. Puck, M. J. Lenardo, and S. E. Straus. 2007. Dominant inhibition of Fas ligand-mediated apoptosis due to a heterozygous mutation associated with autoimmune lymphoproliferative syndrome (ALPS) Type Ib. BMC Med. Genet. 8:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weller, M., U. Malipiero, A. Rensing-Ehl, P. J. Barr, and A. Fontana. 1995. Fas/APO-1 gene transfer for human malignant glioma. Cancer Res. 55:2936–2944. [PubMed] [Google Scholar]
- 8.Keane, M. M., S. A. Ettenberg, G. A. Lowrey, E. K. Russell, and S. Lipkowitz. 1996. Fas expression and function in normal and malignant breast cell lines. Cancer Res. 56:4791–4798. [PubMed] [Google Scholar]
- 9.Nambu, Y., S. J. Hughes, A. Rehemtulla, D. Hamstra, M. B. Orringer, and D. G. Beer. 1998. Lack of cell surface Fas/APO-1 expression in pulmonary adenocarcinomas. J. Clin. Invest. 101:1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maeda, T., Y. Yamada, R. Moriuchi, K. Sugahara, K. Tsuruda, T. Joh, S. Atogami, K. Tsukasaki, M. Tomonaga, and S. Kamihira. 1999. Fas gene mutation in the progression of adult T cell leukemia. J. Exp. Med. 189:1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan, G., S. M. Vickers, A. Pickens, J. O. Phillips, W. Ying, J. A. Thompson, G. P. Siegal, and J. M. McDonald. 1999. Apoptosis and tumorigenesis in human cholangiocarcinoma cells. Involvement of Fas/APO-1 (CD95) and calmodulin. Am. J. Pathol. 155:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Que, F. G., V. A. Phan, V. H. Phan, A. Celli, K. Batts, N. F. LaRusso, and G. J. Gores. 1999. Cholangiocarcinomas express Fas ligand and disable the Fas receptor. Hepatology. 30:1398–1404. [DOI] [PubMed] [Google Scholar]
- 13.Shimonishi, T., K. Isse, F. Shibata, I. Aburatani, K. Tsuneyama, H. Sabit, K. Harada, K. Miyazaki, and Y. Nakanuma. 2000. Up-regulation of fas ligand at early stages and down-regulation of Fas at progressed stages of intrahepatic cholangiocarcinoma reflect evasion from immune surveillance. Hepatology. 32:761–769. [DOI] [PubMed] [Google Scholar]
- 14.Jhala, N. C., S. M. Vickers, P. Argani, and J. M. McDonald. 2005. Regulators of apoptosis in cholangiocarcinoma. Arch. Pathol. Lab. Med. 129:481–486. [DOI] [PubMed] [Google Scholar]
- 15.Huang, B., M. Eberstadt, E. T. Olejniczak, R. P. Meadows, and S. W. Fesik. 1996. NMR structure and mutagenesis of the Fas (APO-1/CD95) death domain. Nature. 384:638–641. [DOI] [PubMed] [Google Scholar]
- 16.Imtiyaz, H. Z., Y. Zhang, and J. Zhang. 2005. Structural requirements for signal-induced target binding of FADD determined by functional reconstitution of FADD deficiency. J. Biol. Chem. 280:31360–31367. [DOI] [PubMed] [Google Scholar]
- 17.Yap, K. L., J. Kim, K. Truong, M. Sherman, T. Yuan, and M. Ikura. 2000. Calmodulin target database. J Struct Funct Genomics. 1:8–14. [DOI] [PubMed] [Google Scholar]
- 18.Ganoth, A., R. Friedman, E. Nachliel, and M. Gutman. 2006. A molecular dynamics study and free energy analysis of complexes between the Mlc1p protein and two IQ motif peptides. Biophys. J. 91:2436–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahn, E. Y., S. T. Lim, W. J. Cook, and J. M. McDonald. 2004. Calmodulin binding to the Fas death domain. Regulation by Fas activation. J. Biol. Chem. 279:5661–5666. [DOI] [PubMed] [Google Scholar]
- 20.Chen, Y., P. Pawar, G. Pan, L. Ma, H. Liu, and J. M. McDonald. 2008. Calmodulin binding to the Fas-mediated death-inducing signaling complex in cholangiocarcinoma cells. J. Cell. Biochem. 103:788–799. [DOI] [PubMed] [Google Scholar]
- 21.Chinnaiyan, A. M., K. O'Rourke, M. Tewari, and V. M. Dixit. 1995. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 81:505–512. [DOI] [PubMed] [Google Scholar]
- 22.Babu, Y. S., J. S. Sack, T. J. Greenhough, C. E. Bugg, A. R. Means, and W. J. Cook. 1985. Three-dimensional structure of calmodulin. Nature. 315:37–40. [DOI] [PubMed] [Google Scholar]
- 23.DeLano, W. L. 2002. The PyMOL Molecular Graphics System. DeLano Scientific, Palo Alto, CA.
- 24.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- 25.Chen, R., and Z. Weng. 2003. A novel shape complementarity scoring function for protein-protein docking. Proteins. 51:397–408. [DOI] [PubMed] [Google Scholar]
- 26.Li, L., R. Chen, and Z. Weng. 2003. RDOCK: refinement of rigid-body protein docking predictions. Proteins. 53:693–707. [DOI] [PubMed] [Google Scholar]
- 27.Chen, R., and Z. Weng. 2002. Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins. 47:281–294. [DOI] [PubMed] [Google Scholar]
- 28.Chen, R., W. Tong, J. Mintseris, L. Li, and Z. Weng. 2003. ZDOCK predictions for the CAPRI challenge. Proteins. 52:68–73. [DOI] [PubMed] [Google Scholar]
- 29.Wiehe, K., B. Pierce, J. Mintseris, W. W. Tong, R. Anderson, R. Chen, and Z. Weng. 2005. ZDOCK and RDOCK performance in CAPRI rounds 3, 4, and 5. Proteins. 60:207–213. [DOI] [PubMed] [Google Scholar]
- 30.Wiehe, K., B. Pierce, W. W. Tong, H. Hwang, J. Mintseris, and Z. Weng. 2007. The performance of ZDOCK and ZRANK in rounds 6–11 of CAPRI. Proteins. 69:719–725. [DOI] [PubMed] [Google Scholar]
- 31.Wu, Y., Z. Cao, H. Yi, D. Jiang, X. Mao, H. Liu, and W. Li. 2004. Simulation of the interaction between ScyTx and small conductance calcium-activated potassium channel by docking and MM-PBSA. Biophys. J. 87:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi, H., S. Qiu, Z. Cao, Y. Wu, and W. Li. 2008. Molecular basis of inhibitory peptide maurotoxin recognizing Kv1.2 channel explored by ZDOCK and molecular dynamic simulations. Proteins. 70:844–854. [DOI] [PubMed] [Google Scholar]
- 33.Case, D. A., T. E. Cheatham 3rd, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr., A. Onufriev, C. Simmerling, B. Wang, and R. J. Woods. 2005. The Amber biomolecular simulation programs. J. Comput. Chem. 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 35.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 36.Kollman, P. A., I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan, W. Wang, O. Donini, P. Cieplak, J. Srinivasan, D. A. Case, and T. E. Cheatham 3rd. 2000. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33:889–897. [DOI] [PubMed] [Google Scholar]
- 37.Wang, W., W. A. Lim, A. Jakalian, J. Wang, J. Wang, R. Luo, C. I. Bayly, and P. A. Kollman. 2001. An analysis of the interactions between the Sem-5 SH3 domain and its ligands using molecular dynamics, free energy calculations, and sequence analysis. J. Am. Chem. Soc. 123:3986–3994. [DOI] [PubMed] [Google Scholar]
- 38.Allen, M. P., and D. J. Tildesley. 1987. Computer Simulation of Liquids. Oxford University Press, New York.
- 39.Lee, S. J., Y. Song, and N. A. Baker. 2008. Molecular dynamics simulations of asymmetric NaCl and KCl solutions separated by phosphatidylcholine bilayers: potential drops and structural changes induced by strong Na+-lipid interactions and finite size effects. Biophys. J. 94:3565–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu, Y., D. Pan, S. L. Bellis, and Y. Song. 2008. Effect of altered glycosylation on the structure of the I-like domain of beta1 integrin: a molecular dynamics study. Proteins. 73:989–1000. [DOI] [PubMed] [Google Scholar]
- 41.Efron, B., and R. J. Tibshirani. 1998. An Introduction to the Bootstrap. Chapman & Hall, New York.
- 42.Chen, A. A., and R. V. Pappu. 2007. Quantitative characterization of ion pairing and cluster formation in strong 1:1 electrolytes. J. Phys. Chem. 111:6469–6478. [DOI] [PubMed] [Google Scholar]
- 43.Bailey, N. T. J. 1995. Statistical Methods in Biology. Cambridge University Press, New York.
- 44.Wriggers, W., E. Mehler, F. Pitici, H. Weinstein, and K. Schulten. 1998. Structure and dynamics of calmodulin in solution. Biophys. J. 74:1622–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pawar, P. S., K. J. Micoli, J. M. McDonald, and Y. Chen. 2007. Interaction between calmodulin (CaM) and cellular FLICE-like inhibitory protein (c-FLIP) modulates Fas-induced signaling in cholangiocarcinoma cells. Proc. Am. Assoc. Cancer Res., 98th. 295.