

Abstract
Spreading depression (SD), a propagating wave of electrical silence in the cortex and archicortex, involves depolarization of neurons and astrocytes for ∼1 min, due principally to a large increase in extracellular K+. SD is accompanied by large increases in extracellular ATP and is blocked by glutamate N-methyl-D-aspartate receptor antagonists. As a principal means of transmission between astrocytes is through their release of ATP, we have investigated if a model in which SD is driven by the effects of astrocyte waves of ATP interacting with waves of glutamate release from neurons and astrocytes can give a quantitative account of experimental observations on SD. We show that the characteristics of SD and the accompanying extracellular ionic changes can be accommodated by such a model—whether astrocyte transmission is principally through the release of ATP, as in archicortex (hippocampus) and spinal cord, or via gap junctions, as in the neocortex. Furthermore, these models give quantitative accounts of the effects on the characteristics of SD of agents toxic for astrocytes and of gap-junction blockers. Finally, an additional series of critical tests of the model is suggested.
INTRODUCTION
Spreading depression (SD) was first measured as a propagating wave of electrical silence (1,2). It involves a depolarization of neurons and glial cells that lasts for ∼1 min at any particular site in the cortex or archicortex and propagates at ∼40 μm s−1 (3,4). The following facts have been established about SD: First, it is blocked by antagonists to N-methyl-D-aspartate (NMDA) receptors but not to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors (5–8). Second, it is associated with the release of copious amounts of ATP (9,10), due to the vesicular release of ATP from astrocytes (9). Third, Ca2+ waves in astrocytes precede SD (in hippocampus), but blocking these waves does not prevent SD (8,11,12). Fourth, SD is accompanied by large increases in extracellular K+ and concomitant decreases in Na+, Cl−, and Ca2+ (6).
These observations implicate neurons and astrocytes in a synergistic activity involving the release of ATP and glutamate. It is known that glutamate release from neurons activates Ca2+ waves in astrocytes by acting on (+)-α-methyl-4-carboxyphenylglycine) antagonized receptors (13), and that glutamate is released from astrocytes after activation of their purinergic receptors with ATP (7,14–16). On the other hand, glutamate acting on AMPA receptors (17), and on both AMPA and NMDA receptors under certain conditions (18), can stimulate the release of ATP from astrocytes. Glutamate can also act on neuronal NMDA receptors to regulate glutamate release (19).
Pharmacological antagonism of different receptors to ascertain their role in SD indicate the following: SD is independent of AMPA receptors (5), so all the glutamatergic synapses are most likely of the NMDA type between astrocytes and neurons. Furthermore, as there is no depolarization in either neurons or astrocytes that precedes the activation of NMDA receptors, these must be relatively Mg2+-insensitive, which is the case if these cells possess NR2C/NR2D subunits (20,21). Also, since blocking Ca2+ in astrocytes does not affect SD, then both ATP and glutamate release from these must be calcium-independent. There is good evidence that glutamate and ATP can be released by Ca2+ independent pathways ((22); for reviews, see Vesce et al. (23) and Halassa et al. (24)). The release of the transmitters could be via a phospholipase C (PLC) cytochrome P450 arachidonate epoxygenase activated by group 1 (mGluR1 and mGluR5) NMDA receptors (25). This work explores the properties of models that incorporate these observations.
METHODS
The above observations lead to the following aspects to be incorporated into a model of SD: Ca2+ waves are transmitted between astrocytes by the release of ATP and by gap junctions (GJs), with the former dominating in the archicortex and spinal cord ((9,26); see also Theis et al. (27)) and the latter in the neocortex (8,28). Quantitative models of this propagation of the Ca2+ wave that account for experimental observations on astrocyte transmission are available (29,30). We propose that the wave of ATP that propagates with the astrocyte Ca2+ wave, but which is not dependent on it (28), (see also Montana et al. (31)), is responsible for an accompanying wave of glutamate release from the astrocytes (14,32). This glutamate acts on NMDA receptors of neurons to trigger a large depolarization (responsible for SD) and the release of glutamate from the neurons, which acts back on the other astrocytes to trigger their further release of ATP. In this way, a propagating wave of glutamate and ATP release from astrocytes and neurons is effected, which is responsible for SD and which is, of course, accompanied by normal glutamatergic transmission between the neurons utilizing AMPA receptors.
The model consists of a single lane of astrocytes, represented by cubes of side 25 μm placed with their centers 50 μm apart, thus leaving a 25 μm gap between neighboring astrocytes (Fig. 1 A). The diffusion of ATP in the extracellular space is treated numerically as in one of our earlier works (29). There is a one-to-one correspondence between astrocytes and neurons (Fig. 1 B); the precise spatial placement of the neurons is not relevant, as in the model the communication between neurons and astrocytes does not involve transmitter diffusion. For neuron-to-astrocyte communication, the astrocyte processes ensheath synaptic contacts between neurons and rapidly uptake any glutamate spillover (33). For astrocyte-to-neuron communication, the NMDA receptors on the neurons are in close proximity to the glutamate release sites on the astrocytes, so again diffusion is not the dominant mechanism (32).
FIGURE 1.

(A) Lane of model astrocytes, represented by cubes of side 25 μm, separated by spaces of width 25 μm. The cubes have their centers in the x, y plane (z = 0) and are aligned parallel to the x axis. ATP diffuses in the extracellular space and binds to receptors that are uniformly distributed on the surfaces of the cubes. The neurons are not shown; although there is a one-to-one correspondence with the astrocytes, their interaction is via glutamate and, as explained in the text, it is not necessary to explicitly include glutamate diffusion in the model. Thus their spatial position is not needed for the calculations. (B) Neuron-astrocyte model network in which astrocytic transmission is effected by ATP and neuronal transmission by glutamate, as follows: from neuron to neuron, glutamate (GluC) acting on AMPA receptors; from astrocyte to neuron, glutamate (GluA) acting on NMDA receptors; from neuron to astrocyte, glutamate (GluB) acting on metabotropic receptors; from astrocyte to astrocyte, ATP acting on P2Y receptors. (C) Neuron-astrocyte model network in which astrocytic transmission is principally effected by GJs, together with a small component due to released ATP. (The ATP production rate is reduced to one-tenth of that in the model of part A.) The remainder of the network is as in part A.
Fig. 1 B shows the various communication paths between the astrocytes and neurons where ATP is the transmission agent between astrocytes; glutamate involved in the different pathways is labeled GluA, GluB, and GluC for convenience. The processes involved are:
- Glutamate (GluA) is released from astrocytes, as a result of the action of local ATP, according to
The first term in this equation comes from a least-squares fit to the experimental data in Fig. 1 b of (35) (see the Supplementary Material, Data S1, for details) and the second term accounts for the removal of excess glutamate.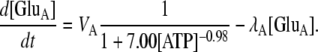
(1) Glutamate (GluA) acts on NMDA receptors on neurons. The membrane potential Vm is given by the standard equation
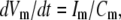 where Im (A cm−2) is the density of current into the neuron, Cm = 10−6 F cm−2 is the membrane capacitance, and the resting membrane potential is −70 mV. The input current Im is the sum of all currents into the neuron and is given explicitly by the formulas in Data S1. The same equation is also used to calculate the membrane potential of the astrocytes, using the same parameter values except that the resting membrane potential is now −85 mV.
where Im (A cm−2) is the density of current into the neuron, Cm = 10−6 F cm−2 is the membrane capacitance, and the resting membrane potential is −70 mV. The input current Im is the sum of all currents into the neuron and is given explicitly by the formulas in Data S1. The same equation is also used to calculate the membrane potential of the astrocytes, using the same parameter values except that the resting membrane potential is now −85 mV.- Glutamate (GluB) released from neurons acts on metabotropic receptors on astrocytes. We follow the formalism of an earlier work (29), which was for the action of ATP on metabotropic purinergic receptors. The fraction of activated G-protein is
where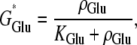
(3) 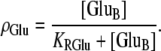
(4) - ATP is released from astrocytes as a result of metabotropic receptor activation. An intermediate step is the release of IP3 from internal stores according to
The remaining steps, including the diffusion of IP3 inside the cells and its degradation, are as in our earlier work (29).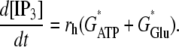
(7) Glutamate (GluC) released from neurons acts on AMPA receptors on neurons. This is described by equations in Destexhe et al. (36); however, because of the relatively fast kinetics of this receptor, its inclusion does not affect the theoretical SD results. We also note that experimentally blocking the AMPA receptor does not prevent SD (7,11). Thus this receptor was not included in the final model.
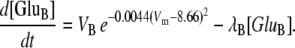
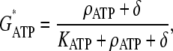
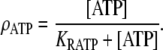
FIGURE 5.

Ionic changes accompanying propagation of the wave of spreading depression in the neuron-astrocyte network of Fig. 1 B. Column 1 gives intracellular concentrations in each of seven successive neurons in the network of K+ (A), Na+ (D), Ca2+ (G), and Cl− (J). Column 2 (middle) gives the corresponding extracellular concentrations, and column 3 gives the corresponding intracellular concentrations in each of seven successive astrocytes in the network.
Fig. 1 C shows the changes made when communication between the astrocytes is principally by GJs, with only a small contribution from ATP diffusion, indicated by the broken lines. Following Hofer et al. (37), the concentration change in IP3 in astrocyte i due to the GJ with astrocyte i − 1 is
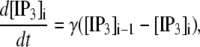 |
(8) |
where the concentration are measured at the neighboring boundaries of astrocytes i − 1 and i. Also, IP3 is now produced by an additional process involving PLCδ activity, assumed to take place at the surface of the astrocytes. This is modeled by including in Eq. 7 the additional term 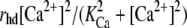 (37). Parameter values for the above processes are given in Table 1.
(37). Parameter values for the above processes are given in Table 1.
TABLE 1.
Model parameter values
| Value | Value | |||
|---|---|---|---|---|
| Symbol | Definition | ATP model | GJ model | |
| VA (mM s−1) | Glu release rate (astrocyte) | 600 | 500 | Fitted |
| λA (s−1) | Decay rate | 10 | 10 | Fitted |
| VB (mM s−1) | Glu release rate (neuron) | 600 | 500 | Fitted |
| λB (s−1) | Decay rate | 10 | 10 | Fitted |
| KGlu | G-protein activation constant | 8.82 | 8.82 | cf. (29). |
| KRGlu (μM) | G-protein binding constant | 5 | 5 | Fitted |
| KATP | G-protein activation constant | 8.82 | 8.82 | (29) |
| KRATP (μM) | G-protein binding constant | 2.5 | 2.5 | (29) |
| rh (μmol μm−2 s−1) | IP3 production rate (ATP) | 2 × 10−14 | 7 × 10−14 | (29) |
| γ (s−1) | G-J strength | 3 | Fitted | |
| KCa (μM) | PLCδ activation constant | 0.3 | (37) | |
| rhd (μmol μm−2 s−1) | IP3 production rate (PLCδ) | 5 × 10−16 | Fitted | |
| DNa (μm2 s−1) | Diffusion coefficient for Na+ | 1.33 × 103 | 1.33 × 103 | (48) |
| DK (μm2 s−1) | Diffusion coefficient for K+ | 1.96 × 103 | 1.96 × 103 | (48) |
| DCl (μm2 s−1) | Diffusion coefficient for Cl− | 2.03 × 103 | 2.03 × 103 | (48) |
| DCa (μm2 s−1) | Diffusion coefficient for Ca2+ | 0.79 × 103 | 0.79 × 103 | (48) |
Some values are available in the literature, as indicated; others have been chosen to fit experimental data.
For comparison with experimental results, it is necessary to compute the changes in ionic concentrations and potential, especially in the extracellular fluid, that accompany the SD wave. Fig. 2 shows the ionic currents that occur; the detailed equations governing these currents are given in Data S1. For the neurons, the most important currents are due to NMDA, KDR, NaF, and CaHVA, as well as associated pumps and leaks. For the astrocytes, the dominant currents are due to KDR, BK, NaF, and CaHVA, associated pumps and leaks, and the Na+/K+/Cl− transporter. We have not incorporated the putative large, unidentified channel in neuronal membrane recently suggested to be relevant in SD (38).
FIGURE 2.

Ionic currents between neurons, astrocytes, and the extracellular space. There are five K+ currents (indicated here by a single arrow): KDR (delayed rectifier), BK (noninactivating calcium-dependent), KM (noninactivating muscarinic), SK (voltage-independent Ca2+-dependent), and IK (K2 Ca2+-dependent); two Na+ currents (indicated by a single arrow): NaF (fast Na+) and NaP (persistent Na+); one Ca2+ current (high-voltage activated, P-type) and the NMDA receptor currents, present only in the neurons. The pumps and exchangers are the Cl−, Ca2+, and Na+/K+ pumps, the Na+/Ca2+ exchanger, and the K+/Cl− and Na+/K+/Cl− transporters, the last one present only in the astrocytes. The remaining currents are the K+, Na+, Ca2+, and Cl− leak currents. The detailed equations and parameter values for all these currents are given in Data S1.
The extracellular potential VSD is calculated using the following expression for the potential gradient (cf. (39), Eq. 5):
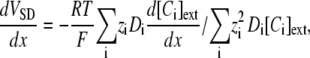 |
(9) |
where zi is the valence and Di is the diffusion coefficient for ionic species Ci and the sum is over the four ions Na+, K+, Cl−, and Ca2+. Details of the application of this formula are given in Data S1, where there are also more details on the model and computation methods.
RESULTS
Propagation of spreading depression in hippocampus and spinal cord
The appropriate model for this, shown in Fig. 1 B, incorporates transmission between astrocytes mediated by their release of ATP. Networks consisting solely of astrocytes using ATP as their transmitter have been previously analyzed by us (29,30) and are used here. Thus, removing neurons from the network of Fig. 1 B and initiating an ATP transient in an astrocyte gives rise to the propagation of a wave of increased extracellular ATP at successive astrocytes that stabilizes in amplitude after about the first two astrocytes (Fig. 3 A). Concomitantly, the increases in IP3 (Fig. 3 B) and in intracellular Ca2+ (Fig. 3 C) also settle to about the same peak values by the third astrocyte in the network chain. These results are consistent with the fact that blocking NMDA receptors, and therefore eliminating neurons from the network in Fig. 1 B, blocks SD (see the Introduction) but does not block astrocytic Ca2+ waves.
FIGURE 3.

Effect of removing all the neurons (simulating neurotoxic cell death) on the propagation of the purinergic (ATP) and Ca2+ waves by astrocytes in the model (see Fig. 1 B). Shown are changes in ATP concentration at 7 successive astrocytes (A), after initiation by a rectangular pulse of ATP of amplitude 15 μM and duration 5 s, applied to the first astrocyte, together with changes in IP3 (B) and intracellular Ca2+ (C). The propagation speed of the ATP wave is ∼20 μm s−1.
Incorporation of the neuronal chain into the network now allows for the feedback pathway from astrocyte to neuron to astrocyte, mediated by glutamate, to be established (Fig. 1 B). This results in an approximate doubling in amplitude of the astrocytic waves of ATP, IP3, and intracellular Ca2+ and an increase in their velocity from ∼20 μm s−1 to ∼41 μm s−1 (compare Fig. 4, A–C, with Fig. 3, A–C). These effects are due to the action of glutamate released from the neurons (GluB in Fig. 1 B; see Fig. 4 E), which has in turn been released by the action of glutamate from astrocytes (GluA in Fig. 1 B; see Fig. 4 D), to release ATP from the astrocytes in addition to that already released through purely purinergic mechanisms (Fig. 4 A). The action of astrocytic release of glutamate onto the neuronal NMDA receptors (GluA in Fig. 1 B) is to trigger a plateau-type depolarization of ∼50 mV in all the neurons in the chain lasting for ∼45 s (Fig. 4 F) and of ∼40 mV in the astrocytes lasting for the same time (Fig. 4 G). These values are within the experimental ranges of 20–55 mV for neurons and 20–50 mV for glial cells, with durations of 30–120 s (40,41).
FIGURE 4.

Cellular events driving propagation of the wave of spreading depression in the neuron-astrocyte network of Fig. 1 B. Shown are the changes in ATP concentration at seven successive astrocytes (A) after initiation of the purinergic wave at the first astrocyte in the chain (see Fig. 1 B), together with the accompanying changes in intracellular IP3 (B) and intracellular Ca2+ (C) within the astrocytes. The concentration of glutamate released from these astrocytes onto successive neurons (GluA in Fig. 1 B) and of glutamate released from the neurons onto successive astrocytes (GluB in Fig. 1 B) is given in D and E, respectively. The effect of these transmitters on the transmembrane potential of successive neurons (F) and astrocytes (G) is given together with that of the extracellular potential change at successive sites along the network chain of Fig. 1 B, indicating the wave of propagating SD (H); increasing DK by a factor of 4 gives the larger potential shown in (I). The propagation speed of the ATP wave is now ∼41 μm s−1, double that of Fig. 3 A where no neurons are present.
The extracellular potential in the model, VSD, measured as the SD potential change, consists of a (negative) peak of ∼2.7 mV, which is maintained as a plateau for ∼20 s (Fig. 4 H). The wave has a total duration of ∼60 s and propagates with a velocity of ∼41 μm s−1. The amplitude is rather less than that observed experimentally (see Table 2); increasing the diffusion coefficient of K+, DK, to four times the value given in Table 1 now gives a peak of ∼10 mV, with other characteristics remaining approximately the same (Fig. 4 I). This increase in DK is to take into account the accelerated uptake of K+ from the extracellular space by astrocytes through the mechanisms of spatial buffering and siphoning (42); further comments on this are made in the Discussion section below. The extracellular potential wave is led by the astrocyte Ca2+ wave, as is observed experimentally (8). Table 2 shows that the calculated properties of the SD wave are similar to those measured in retina, hippocampus, and cerebellum, where propagation of the astrocyte Ca2+ waves is known to be dependent on purinergic transmission.
TABLE 2.
Comparison between experimental and model properties of SD
Propagation of SD is accompanied by changes in ionic concentrations in the extracellular space (Fig. 5 column 2), initiated principally by the opening of neuronal NMDA receptors. These changes are an increase in extracellular K+ (Fig. 5 B), accompanied by decreases in extracellular Na+ (Fig. 5 E), Ca2+ (Fig. 5 H), and Cl− (Fig. 5 K). The changes in each of these follows the neuronal membrane potential change (Fig. 4 F), consequent on the effects of glutamate on the NMDA receptors (GluA; Fig. 4 D). Changes in the corresponding intracellular neuronal ionic concentrations are shown in column 1 of Fig. 5; they have temporal shapes that are almost the inverse of those for the extracellular concentrations (column 2 of Fig. 5). The corresponding intracellular changes in the astrocytic concentrations of these ions are shown in column 3 of Fig. 5. Table 3 presents a comparison between the observed changes in K+, Na+, Cl−, and Ca2+ during SD in the retina and cerebellum with the results of the present model. The model values are compatible with the experimental values; in particular, the predicted change in K+ concentration is less than that of Na+, in agreement with observation (see Table 3).
TABLE 3.
Comparison between experimental and model results for ion movements in SD
The effect of toxic agents on SD, thought to be reasonably specific for astrocytes, has been studied in detail in the hippocampus (43). This study shows that SD still occurs and has a similar amplitude (15–20 mV peak) but increased duration and about half the velocity of normal SD (see Fig. 1 in Largo et al. (43)). If in the present model every second astrocyte is removed, to simulate the effect of toxic destruction of a large number of astrocytes, then the results illuminate how the change in velocity of SD might originate. Fig. 6 A shows that propagation of the ATP wave along the chain of astrocytes is slowed, as are also the changes in IP3 (Fig. 6 B) and intracellular Ca2+ (Fig. 6 C) due to the increased distance that released ATP has now to diffuse between astrocytes. This has the consequences that the glutamate feedback pathways, initiated by ATP triggering the release of glutamate from astrocytes (GluA in Fig. 1 B), are delayed (Fig. 6, D and E). However, the duration of the action of glutamate on the neuronal NMDA receptors, giving rise to changes in the membrane potential of the neurons (Fig. 6 F) and astrocytes (Fig. 6 G), is unchanged. The result is that the peak of the SD potential is similar to that of the control, as is the duration (compare Fig. 6, H and I, with Fig. 4, H and I, but the velocity is decreased from ∼41 μm s−1 to ∼26 μm s−1. Intracellular and extracellular ion changes are the same as those in the full network but delayed in onset to the same extent as the ATP wave. Thus the model accounts for the constant peak amplitude and slowing velocity of SD as astrocytes are removed from the network, but does not give the increased duration of the SD wave.
FIGURE 6.

As for Fig. 4, only now every second astrocyte has been removed to simulate the effects of toxic cell-death of some astrocytes. The shape of the SD wave, (H) and (I), is little affected by removing every second astrocyte from the network (compare with Fig. 4, H and I).
Propagation of spreading depression in the neocortex
The appropriate model for this is shown in Fig. 1 C, which incorporates transmission between astrocytes mediated by GJs with a small component due to their release of ATP. This relatively small triggered release gives rise to a very low concentration of ATP in the extracellular space surrounding sequential astrocytes in the network chain (compare Fig. 7 A with Fig. 4 A). However, the steady-state IP3 concentration reached by about the third astrocyte in successive astrocytes in this model is about the same as that in the previous model, because of diffusion of IP3 between astrocytes through their GJs and also additional IP3 release due to PLCδ activation (compare Fig. 7 B with Fig. 4 B). The result is that the peak intracellular Ca2+ concentrations are much the same in the two models (compare Fig. 7 C with Fig. 4 C). However, as the glutamate release from astrocytes (GluA in Fig. 1 C) is dependent on the action of ATP on these cells in the network, the decreased ATP release (Fig. 7 A) leads to a concomitant decrease in glutamate (GluA) release (Fig. 7 D). This results in opening of the NMDA receptor channels in the neurons for a shorter period, thus decreasing the duration of the neuronal membrane potential change (compare Fig. 7 F with Fig. 4 F). The neurons then release glutamate onto astrocytes (GluB in Fig. 1 C) for a shorter period of time compared with the first model (Fig. 1 B), resulting in a shortening of the duration of the astrocyte membrane potential change (compare Fig. 7 G with Fig. 4 G).
FIGURE 7.

As for Fig. 4, only now using the neuron-astrocyte network of Fig. 1 C. The SD wave, (H) and (I), is similar to that which occurs when there is only ATP transmission between astrocytes, although it is of shorter duration (compare with Fig. 4, H and I).
This decrease in the amount of triggered ATP release also leads to decreases in the duration of the intracellular ion changes in the neurons (compare column 1 of Fig. 8 with column 1 of Fig. 5), and astrocytes (compare column 3 of Fig. 8 with column 3 of Fig. 5) and in the extracellular ion changes (compare column 2 of Fig. 8 with column 2 of Fig. 5). These changes in the extracellular ionic concentrations give rise to a SD of substantially shorter duration in this model compared to the previous one (compare Fig. 7 H with Fig. 4 H),but only a small decrease in propagation velocity from ∼41 μm s−1 to ∼37 μm s−1.
FIGURE 8.

As for Fig. 5, only now using the neuron-astrocyte network of Fig. 1 C.
Blocking GJ connexins in the neocortex does not block SD, but slows it (8). In the present model, blocking GJs did not block SD, as the small ATP transmission between astrocytes is sufficient to maintain propagation of the Ca2+ wave, but at a much lower velocity of 22 μm s−1, which determines the much slower rate of SD propagation (Fig. 9).
FIGURE 9.

As for Fig. 7, only now the GJs have been blocked and transmission is via the reduced ATP present in the GJ model.
Blocking NMDA receptors in the neocortex blocks SD, but leaves propagation of Ca2+ and ATP waves, albeit at a reduced rate (8). This also occurs in the model, with the velocity reduced from ∼37 μm s−1 to ∼20 μm s−1 (compare Fig. 10, A–C, with Fig. 7, A–C). Blocking NMDA receptors and GJs of course blocks SD but still leaves some Ca2+ wave propagation in the astrocyte chain (Fig. 10 F), which, however, dies out after two more cells in the chain. This occurs because of the small ATP release maintaining transmission between the astrocytes (Fig. 10 D) and generating sufficient IP3 (Fig. 10 E) to continue release of intracellular Ca2+ along the astrocyte chain (Fig. 10 F).
FIGURE 10.

Top row shows the effect of blocking the NMDA receptors on neurons (cf. Fig. 3); transmission is via GJs and reduced ATP. The bottom row shows the effect of additionally blocking the GJs.
Removal of the Ca2+ wave in the model does not affect SD, as the latter is dependent on the ATP wave and that in turn depends on IP3, but is independent of the changes in intracellular Ca2+ release (29), a phenomenon that is observed experimentally (8).
DISCUSSION
Criticisms of the model
The main objection to the present model is the experimental observation that purports to show that SD still occurs in the absence of astrocytes. The gliotoxin fluorocitrate, when perfused into the hippocampus, leads to the destruction of many astrocytes at 4 h and the loss of action potential firing capacity of neurons, but the continuance of a form of SD that is ∼50% normal before the neurons are also toxically destroyed (43). Heptanol, which is known to block the release of ATP from astrocytes (44), completely blocks SD in the hippocampus whereas fluorocitrate only produces a partial (∼50%) change (43). The fact that blocking ATP release with the heptanol blocks SD is consistent with our model. The possibility exists that the failure to block SD after widespread toxic effects of fluorocitrate may be put down to the remaining viable astrocytes and neurons being able to still sustain the ionic mechanisms that drive SD, as the results of Fig. 6 suggest.
The extracellular potential
Our method of calculating the extracellular potential, VSD, uses the formula of Almeida et al. (39) (see Eq. 9 above; also Data S1). Using the standard diffusion coefficients for K+, Na+, and Cl−, as given in Table 1, this produces a potential with a smaller amplitude than that observed experimentally. This will inevitably happen if the change in [K+]ext is less than the change in [Na+]ext, as is the case in our model and also experimentally (Table 3). Almeida et al. (39) obtain a larger potential change (13 mV) but this is because their [K+]ext change is twice their [Na+]ext change (see Table 3), a result achieved by using a phenomenological equation for K+ removal, rather than the known ion channels and exchangers. When these are incorporated correctly, as in our model, it is clear that the change in [Na+]ext will always be greater than the change in [K+]ext. (K+ is removed from the extracellular space by the NaK exchanger on neurons and astrocytes and the NaKCl transporter on astrocytes; the former releases 3 Na+ for 2 K+ removed, and the latter removes Na+ and K+ at the same rate.) The model of Shapiro (45) does predict a large amplitude for VSD (see Table 2), but no details are provided as to how this is calculated.
Other factors, such as cell swelling or the presence of additional ions, will have only a minor effect on VSD, so we are forced to the conclusion that the effective diffusion coefficient for K+, DK, is larger than that for free diffusion. This will be true if there are additional mechanisms acting to remove K+ more rapidly from the local extracellular space, and these could be the astrocyte-mediated spatial buffering and siphoning to capillaries, as described by Somjen (42).
Comparison between model predictions and experimental results
The present model incorporates the following experimental observations: First, that the Ca2+ astrocytic wave leads SD (8,11). Second, that blocking NMDA receptors reduces the velocity but does not block the Ca2+ wave in astrocytes (5,8,13), which follows from the critical role the astrocyte release of glutamate onto neuronal NMDA receptors has in the feedback pathway assisting astrocytic Ca2+ propagation in the model. Third, that blocking the Ca2+ wave in astrocytes does not block SD (8), a result that is expected from the model because the release of ATP is not dependent on global changes in astrocyte intracellular Ca2+ (28). Fourth, that blocking NMDA receptors blocks SD (5,6) as expected from the essential role of astrocyte-released glutamate acting on neuronal NMDA receptors in the model. Fifth, blocking GJs or using connexin-43 knock-out mice reduces the Ca2+ wave velocity of astrocytes without blocking SD (8), a result to be expected, given that ATP can affect transmission between cortical astrocytes if their CX43 are blocked (28). There is, however, no decrease in SD velocity in the hippocampus after the block of GJs (27), as transmission between astrocytes is not due to connexins but to ATP in this area (28).
Modeling spreading depression
There are several different kinds of models of SD. One class of these consists of neurons and astrocytes, each with an array of Na+, K+, Cl−, and Ca2+ voltage-dependent channels and ion pumps, together with transmitter-receptors allowing further ion movement. In one model of this class, neurons are coupled via GJs to allow for the diffusion of K+ between neurons (45). This gives rise to appropriate changes in concentration of K+, Na+, and Ca2+ with realistic SD velocity, but unfortunately there is no evidence for widespread GJ coupling between neurons. In another model of this class, the emphasis is on K+ diffusion within single astrocytes to the ends of their processes, where a remote release of K+ occurs into the extracellular space and by this means propagates the rise in extracellular K+ (46). However, this model has not been used to produce quantitative predictions of the characteristics of SD. In a different class of models, electrodiffusion equations are used to describe the movements of ions, coupled by the electrical field in the extracellular space, to give accurate accounts of amplitude and temporal changes in Na+, K+, Cl− and Ca2+ that accompany SD in different layers of the retina (39,47). This work does not incorporate the activity of astrocytes or of NMDA receptors.
The work presented here is based on our previous model of Ca2+ and ATP wave propagation among astrocytes, in which ATP is the transmitter (29,30), as seems to be the case for both hippocampal and spinal cord astrocytes (28). However, in the present astrocyte network model, IP3 diffusion between astrocytes, facilitated by GJs, has also been considered as a mode of astrocyte transmission. This has been introduced as it is the dominant form of astrocyte transmission in the neocortex, although ATP transmission is also present as a default mechanism should GJs fail. In principal, we could find little difference in the characteristics of SD using either mode of astrocytic communication, a result that can be partially attributed to the similar diffusion rates of ATP and IP3 (29).
Further tests of the present model
Many aspects of the present neuronal-astrocyte hypothesis for SD are open to direct experimental test, of which the most important is to examine the effects of purinergic antagonists on SD. Blocking ATP receptors on astrocytes that mediate propagation in the astrocyte system in the archicortex, neocortex, and spinal cord, and the release of glutamate onto adjacent neurons, should completely block SD according to the present model (see Fig. 1, B and C). Another test would be to use apyrase to remove any released ATP. A further important aspect of the model that requires experimental investigation concerns whether the NMDA receptors on neurons affected by astrocyte-derived glutamate are of the relatively Mg2+-independent type; that is, incorporate NR2D and NR2C subunits (20,21).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
This work was supported by Australian Research Council grant DP0559268.
Editor: Arthur Sherman.
References
- 1.Leao, A. A. P. 1944. Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 7:359–390. [DOI] [PubMed] [Google Scholar]
- 2.Leao, A. A. 1951. The slow voltage variation of cortical spreading depression of activity. Electroencephalogr. Clin. Neurophysiol. 3:315–321. [DOI] [PubMed] [Google Scholar]
- 3.Somjen, G. G. 2001. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol. Rev. 81:1065–1096. [DOI] [PubMed] [Google Scholar]
- 4.Kunkler, P. E., R. E. Hulse, M. W. Schmitt, C. Nicholson, and R. P. Kraig. 2005. Optical current source density analysis in hippocampal organotypic culture shows that spreading depression occurs with uniquely reversing currents. J. Neurosci. 25:3952–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruger, H., U. Heinemann, and H. J. Luhmann. 1999. Effects of ionotropic glutamate receptor blockade and 5–HT1A receptor activation on spreading depression in rat neocortical slices. Neuroreport. 10:2651–2656. [DOI] [PubMed] [Google Scholar]
- 6.Martins-Ferreira, H., M. Nedergaard, and C. Nicholson. 2000. Perspectives on spreading depression. Brain Res. Brain Res. Rev. 32:215–234. [DOI] [PubMed] [Google Scholar]
- 7.Anderson, T., and R. Andrew. 2002. Spreading depression: imaging and blockade in the rat neocortical brain slice. J. Neurophysiol. 88:2713–2725. [DOI] [PubMed] [Google Scholar]
- 8.Peters, O., C. Schipke, Y. Hashimoto, and H. Kettenmann. 2003. Different mechanisms promote astrocyte Ca2+ waves and spreading depression in the mouse neocortex. J. Neurosci. 23:9888–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowser, D. N., and B. S. Khakh. 2007. Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J. Gen. Physiol. 129:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schock, S. C., N. Munyao, Y. Yakubchyk, L. A. Sabourin, A. M. Hakim, E. C. G. Ventureyra, and C. S. Thompson. 2007. Cortical spreading depression releases ATP into the extracellular space and purinergic receptor activation contributes to the induction of ischemic tolerance. Brain Res. 1168:129–138. [DOI] [PubMed] [Google Scholar]
- 11.Basarsky, T. A., S. Duffy, R. D. Andrew, and B. A. MacVicar. 1998. Imaging spreading depression and associated calcium dynamics in brain slices. J. Neurosci. 18:7189–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chuquet, J., L. Hollander, and E. A. Nimchinsky. 2007. High-resolution in vivo imaging of the neurovascular unit during spreading depression. J. Neurosci. 27:4036–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rao, S. P., and S. K. Sidar. 2006. Astrocytes in 17b-estradiol treated mixed hippocampal cultures show attenuated calcium response to neuronal activity. Glia. 53:817–826. [DOI] [PubMed] [Google Scholar]
- 14.Takano, T., J. Kang, J. K. Jaiswal, S. M. Simon, J. H. C. Lin, Y. Yu, Y. Li, J. Yang, G. Dienel, H. R. Zielke, et al. 2005. Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc. Natl. Acad. Sci. USA. 102:16466–16471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiga, H., J. Murakami, T. Nagao, M. Tanaka, K. Kawahara, I. Matsuoka, and E. Ito. 2006. Glutamate release from astrocytes is stimulated via the appearance of exocytosis during cyclic AMP-induced morphologic changes. J. Neurosci. Res. 84:338–347. [DOI] [PubMed] [Google Scholar]
- 16.Fellin, T., T. Pozzan, and G. Carmignoto. 2006. Purinergic receptors mediate two distinct glutamate release pathways in hippocampal astrocytes. J. Biol. Chem. 281:4274–4284. [DOI] [PubMed] [Google Scholar]
- 17.Coco, S., F. Calegari, E. Pravettoni, D. Pozzi, E. Taverna, P. Rosa, M. Matteoli, and C. Verderio. 2003. Storage and release of ATP from astrocytes in culture. J. Biol. Chem. 278:1354–1362. [DOI] [PubMed] [Google Scholar]
- 18.Werry, E. L., G. J. Liu, and M. R. Bennett. 2006. Glutamate-stimulated ATP release from spinal cord astrocytes is potentiated by substance P. J. Neurochem. 99:924–936. [DOI] [PubMed] [Google Scholar]
- 19.Robert, A., J. A. Black, and S. G. Waxman. 1998. Endogenous NMDA-receptor activation regulates glutamate release in cultured spinal neurons. J. Neurophysiol. 80:196–208. [DOI] [PubMed] [Google Scholar]
- 20.Monyer, H., R. Sprengel, R. Schoepfer, A. Herb, M. Higuchi, H. Lomeli, N. Burnashev, B. Sakmann, and P. Seeburg. 1992. Heteromeric NMDA receptors: Molecular and functional distinction of subtypes. Science. 256:1217–1221. [DOI] [PubMed] [Google Scholar]
- 21.Binshtok, A. M., I. A. Fleidervish, R. Sprengel, and M. J. Gutnick. 2006. NMDA receptors in layer 4 spiny stellate cells of the mouse barrel cortex contain the NR2C subunit. J. Neurosci. 26:708–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Queiroz, G., D. K. Meyer, A. Meyer, K. Starke, and I. von Kugelgen. 1999. A study of the mechanism of the release of ATP from rat cortical astroglial cells evoked by activation of glutamate receptors. Neuroscience. 91:1171–1181. [DOI] [PubMed] [Google Scholar]
- 23.Vesce, S., D. Rossi, L. Brambilla, and A. Volterra. 2007. Glutamate release from astrocytes in physiological conditions and in neurodegenerative disorders characterized by neuroinflammation. Int. Rev. Neurobiol. 82:57–71. [DOI] [PubMed] [Google Scholar]
- 24.Halassa, M. M., T. Fellin, and P. G. Haydon. 2007. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol. Med. 13:54–63. [DOI] [PubMed] [Google Scholar]
- 25.Gebremedhin, D., K. Yamaura, C. Zhang, J. Bylund, R. C. Koehler, and D. R. Harder. 2003. Metabotropic glutamate receptor activation enhances the activities of two types of Ca2+-activated K+ channels in rat hippocampal astrocytes. J. Neurosci. 23:1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gallagher, C. J., and M. W. Salter. 2003. Differential properties of astrocyte calcium waves mediated by P2Y1 and P2Y2 receptors. J. Neurosci. 23:6728–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Theis, M., R. Jauch, L. Zhuo, D. Speidel, A. Wallraff, B. Döring, C. Frisch, G. Söhl, B. Teubner, C. Euwens, et al. 2003. Accelerated hippocampal spreading depression and enhanced locomotory activity in mice with astrocyte-directed inactivation of connexin43. J. Neurosci. 23:766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haas, B., C. G. Schipke, O. Peters, G. Söhl, K. Willecke, and H. Kettenmann. 2006. Activity-dependent ATP-waves in the mouse neocortex are independent from astrocytic calcium waves. Cereb. Cortex. 16:237–246. [DOI] [PubMed] [Google Scholar]
- 29.Bennett, M. R., L. Farnell, and W. G. Gibson. 2005. A quantitative model of purinergic junctional transmission of calcium waves in astrocyte networks. Biophys. J. 89:2235–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bennett, M. R., V. Buljan, L. Farnell, and W. G. Gibson. 2006. Purinergic junctional transmission and propagation of calcium waves in spinal cord astrocyte networks. Biophys. J. 91:3560–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montana, V., E. Malarkey, C. Verderio, M. Matteoli, and V. Parpura. 2006. Vesicular transmitter release from astrocytes. Glia. 54:700–715. [DOI] [PubMed] [Google Scholar]
- 32.Jourdain, P., L. H. Bergersen, K. Bhaukaurally, P. Bezzi, M. Santello, M. Domercq, C. Matute, F. Tonello, V. Gundersen, and A. Volterra. 2007. Glutamate exocytosis from astrocytes controls synaptic strength. Nat. Neurosci. 10:331–339. [DOI] [PubMed] [Google Scholar]
- 33.Magistretti, P., and L. Pellerin. 1999. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354:1155–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reference deleted in proof.
- 35.Domercq, M., L. Brambilla, E. Pilati, J. Marchaland, A. Volterra, and P. Bezzi. 2006. P2Y1 receptor-evoked glutamate exocytosis from astrocytes. J. Biol. Chem. 281:30684–30696. [DOI] [PubMed] [Google Scholar]
- 36.Destexhe, A., Z. F. Mainen, and T. J. Sejnowski. 1994. Synthesis of models for excitable membranes, synaptic transmission and neuromodulation using a common kinetic formalism. J. Comp. Neurol. 1:195–230. [DOI] [PubMed] [Google Scholar]
- 37.Hofer, T., L. Venance, and C. Giaume. 2002. Control and plasticity of intercellular calcium waves in astrocytes: a modeling approach. J. Neurosci. 22:4850–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makarova, J., J. M. Ibarz, S. Canals, and O. Herreras. 2007. A steady-state model of spreading depression predicts the importance of an unknown conductance in specific dendritic domains. Biophys. J. 92:4216–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Almeida, A. C., H. Z. Texeira, M. A. Duarte, and A. F. Infantosi. 2004. Modeling extracellular space electrodiffusion during Leao's spreading depression. IEEE Trans. Biomed. Eng. 51:450–458. [DOI] [PubMed] [Google Scholar]
- 40.Higashida, H., G. Mitarai, and S. Watanabe. 1974. A comparative study of membrane potential changes in neurons and neuroglial cells during spreading depression in the rabbit. Brain Res. 65:411–425. [DOI] [PubMed] [Google Scholar]
- 41.Sugaya, E., M. Takato, and Y. Noda. 1975. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J. Neurophysiol. 38:822–841. [DOI] [PubMed] [Google Scholar]
- 42.Somjen, G. G. 2004. Ions in the Brain. Oxford, New York.
- 43.Largo, C., J. M. Ibarz, and O. Herreras. 1997. Effects of the gliotoxin fluorocitrate on spreading depression and glial membrane potential in rat brain in situ. J. Neurophysiol. 78:295–307. [DOI] [PubMed] [Google Scholar]
- 44.Suadicani, S. O., C. F. Brosnan, and E. Scemes. 2006. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J. Neurosci. 26:1378–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shapiro, B. E. 2001. Osmotic forces and gap junctions in spreading depression: a computational model. J. Comput. Neurosci. 10:99–120. [DOI] [PubMed] [Google Scholar]
- 46.Smith, J. M., D. P. Bradley, M. F. James, and C. L. H. Huang. 2006. Physiological studies of cortical spreading depression. Biol. Rev. Camb. Philos. Soc. 81:457–481. [DOI] [PubMed] [Google Scholar]
- 47.Teixeira, H. Z., A. C. G. Almeida, A. F. C. Infantosi, M. A. Vasconcelos, and M. A. Duarte. 2004. Simulation of the effect of Na and Cl on the velocity of a spreading depression wave using a simplified electrochemical model of synaptic terminals. J. Neural Eng. 1:117–126. [DOI] [PubMed] [Google Scholar]
- 48.Hille, B. 1992. Ionic Channels of Excitable Membranes. Sinauer, Sunderland, MA.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.