

Abstract
Calmodulin (CaM) is the major pathway that transduces intracellular Ca2+ increases to the activation of a wide variety of downstream signaling enzymes. CaM and its target proteins form an integrated signaling network believed to be tuned spatially and temporally to control CaM's ability to appropriately pass signaling events downstream. Here, we report the spatial diffusivity and availability of CaM labeled with enhanced green fluorescent protein (eGFP)-CaM, at basal and elevated Ca2+, quantified by the novel fluorescent techniques of raster image scanning spectroscopy and number and brightness analysis. Our results show that in basal Ca2+ conditions cytoplasmic eGFP-CaM diffuses at a rate of 10 μm2/s, twofold slower than the noninteracting tracer, eGFP, indicating that a significant fraction of CaM is diffusing bound to other partners. The diffusion rate of eGFP-CaM is reduced to 7 μm2/s when a large (646 kDa) target protein Ca2+/CaM-dependent protein kinase II is coexpressed in the cells. In addition, the presence of Ca2+/calmodulin-dependent protein kinase II, which can bind up to 12 CaM molecules per holoenzyme, increases the stoichiometry of binding to an average of 3 CaMs per diffusive molecule. Elevating intracellular Ca2+ did not have a major impact on the diffusion of CaM complexes. These results present us with a model whereby CaM is spatially modulated by target proteins and support the hypothesis that CaM availability is a limiting factor in the network of CaM-signaling enzymes.
INTRODUCTION
Ca2+ signaling is one of the best studied second messenger pathways in cells because of its ubiquitous role in modulating a wide variety of systems, from cell division to muscle contraction to neuronal communication, among many others (1,2). The major pathway for mediating responses to Ca2+ flux is through the small ubiquitous protein calmodulin (CaM), which cooperatively binds Ca2+ and transduces the signal to a family of over 100 CaM-binding proteins (2). It has been estimated that the total concentration of CaM-binding proteins is ∼2-fold higher than the total CaM concentration in cells (3,4). The formation of CaM complexes with target proteins occurs throughout the cell as there are cytosolic, nuclear, and membrane proteins that can bind CaM in either its Ca2+-bound or Ca2+-free configurations (5).
As such, interactions of CaM with its target proteins affect its mobility through the cytoplasm (6,7). A particular example of this problem, approached by simulations, suggests that a CaM target protein, neurogranin, has the potential to regulate the encounter rate between Ca2+-saturated CaM and its downstream targets during Ca2+ transients (8). In other examples, a regulator of CaM signaling when phosphorylated can increase its affinity for CaM acting as a competitive inhibitor of other CaM-activated enzymes (9). The general concept is that CaM has been thought of as limiting in its role as a messenger (4). Thus, the model put forward is that CaM is sequestered by its targets in basal conditions and the Ca2+ signals activate downstream targets to which CaM is already bound or by redistributing it to nearby targets with higher affinity for the Ca2+-saturated form of CaM.
One of the major targets in cells for Ca2+/CaM is Ca2+/CaM-dependent protein kinase II (CaMKII), a Ser/Thr directed protein kinase abundant in brain and other tissues (10). CaMKII is a 12-subunit enzyme that was shown to increase its affinity to CaM following autophosphorylation, a phenomenon called CaM trapping (11,12). This suggests that CaMKII may have a role in modulating the pool of available CaM inside cells. Eventually, activation of CaMKII can lead to physiological changes in the strength of synaptic connections (13,14) and modulate the force/contraction relationship in cardiac tissues, among many other functions (15–17).
Regardless of the amount of information that has been acquired over the years on Ca2+ signaling through CaM, the question of how CaM diffuses throughout the cell to regulate specific pathways is still open. When direct observations of CaM mobility and availability have been undertaken, some results are contradictory mainly because of the lack of spatial resolution, the methodology employed, the cell model used, and the reporter molecule (3,4,6,7,18–21). Our approach is to combine the novel fluorescent spectroscopy (FCS) techniques, raster image correlation spectroscopy (RICS) (22–26), and number and brightness (N&B) analysis (27). Together, these techniques provide a methodology to investigate the global diffusive behavior of CaM and determine how CaM interacts with binding targets throughout the cell.
Using these methodologies, our data show that in human embryonic kidney 293 (HEK293) cells, CaM diffuses at a rate slower than expected on the basis of the molecular weight and cytoplasmic viscosity (10 μm2/s), whereas enhanced green fluorescent protein (eGFP) diffuses at ∼20 μm2/s. In the nucleus, CaM has a brightness corresponding to the monomeric form of eGFP and is fast diffusing (80%). In the cytoplasm, only 60% of the molecules diffuse at the rate of 10 μm2/s. Combining the data from RICS and N&B, we found regions of the cell where only one molecule of CaM is interacting with a putative target and regions in which CaM is in complexes with higher binding stoichiometry. Coexpression of αCaMKII leads to slowed CaM diffusion and an increased number of CaM complexes, thus supporting the hypothesis that CaMKII can modulate CaM availability. In total, we conclude that there is little available CaM at rest or following an increase in Ca2+, supporting the idea that target binding sites must exceed the number of CaM molecules.
MATERIALS AND METHODS
Preparation of HEK cells
HEK293 cell were plated on poly-D-lysine-coated 35 mm tissue culture dishes 2 days before measurement, as previously described (28). Plasmids expressing eGFP, eGFP-CaM, and eGFP-CaMKII were transfected using Lipofectamine-2000 on the day of the experiment. In some experiments, cells were cotransfected with a plasmid expressing αCaMKII in the pCDNA-3 vector. After 3 h, cells were transferred into Hank's balanced salt solution (Invitrogen, Carlsbad, CA) containing 25 mM HEPES, pH 7.2, 2 mM CaCl2, and 0.8 mM MgCl2 before imaging. To increase or decrease the Ca2+, a 2 μM (final concentration) solution of ionomycin (Calbiochem, San Diego, CA) was added to the media. To reduce Ca2+, 10 mM EGTA replaced the 2 mM CaCl2 in the media of cells treated with 2 μM ionomycin.
Data acquisition with an LSM
Data were acquired using a commercial laser-scanning microscope (LSM) in an upright configuration (Zeiss LSM 510, Thornwood, NY). A 63× Apo water immersion (0.9 NA) infinity corrected lens was dipped directly into the imaging media at room temperature. The pinhole was set to 1 Airy unit. The 488 nm laser line was used with <4% of transmission power, and the eGFP signal was detected with a long-pass emission filter (505 nm cutoff). Laser output was initially adjusted to below the 4% level and never changed through the experiments reported in this work. Detector gain was set to 800 V whereas detector offset was set to 0.
Unless otherwise specified 256 × 256 images at 8 bits were collected at scan speed 5, which in the Zeiss LSM 510 corresponds to a pixel dwell time (τp) of 25.6 μs. A zoom setting of 10 with a pixel dimension (δr) of 0.052 μm was selected to oversample the point spread function (PSF). Under these conditions, the time to scan one line (τl) was 15.35 ms. A time series of 100 frames with no programmed delay between images was used to reduce statistical error. The excitation volume was calibrated using a dye solution of Alexa 488 (Molecular Probes, Eugene, OR). The diffusion coefficient (D) of this dye was set to 196 μm2/s (29) to determine the waist of the laser beam. ω0 = 0.32 ± 0.01 μm (Supplementary Material, Fig. S1 in Data S1). Images were processed and the data were fit using the SimFCS program (LFD, University of California at Irvine).
Raster image correlation spectroscopy
RICS (23) processing consists of calculating the two-dimensional (2D) spatial correlation function of each frame
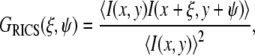 |
(1) |
where ξ,ψ are the spatial lag in the horizontal and vertical (x,y) dimensions, respectively.
The 2D autocorrelation function can be expressed in terms of two components, one is the scanning component (S(ξ,ψ)), which depends on the scanning parameters such as the size of the excitation volume and the pixel size, and the other component contains the information about the diffusion of the molecule
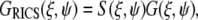 |
(2) |
where
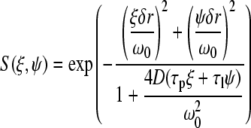 |
(3) |
and
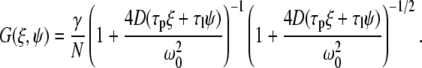 |
(4) |
In Eq. 4, γ is the illumination profile factor assumed to be 0.35 for a three-dimensional (3D) Gaussian using one-photon excitation, and N is the apparent number of molecules in the excitation profile.
To accurately deduce diffusion coefficients using RICS in live cells, one has to account for and subtract slow background motion (gross cellular motility) and movement of large visible vesicles or organelles that would give rise to fluctuations with an apparent diffusion of <0.002 μm2/s. In so doing, the contribution of the true immobile fraction is also filtered out. The subtraction algorithm consists of calculating the average intensity per pixel of a stack of images and subtracting pixel by pixel this image from the intensity of each image of the stack Ii(x,y). This subtraction could sometimes give a negative number. Thus, a number (a) equal to the average intensity of the stack is added to avoid this artifact. Mathematically, this correction can be written as
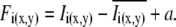 |
(5) |
The above equations describing the RICS 2D autocorrelation functions are valid for point particles. We discovered that a single component did not fit the data, and the need for a second component in Eq. 4 was required. We observed that the second component was in the range of 0.4 μm2/s for the eGFP transfected cells. The possible causes for such slow components include weak interactions with immobile obstacles, formation of large molecular complexes, and other intracellular features that were not removed by the subtraction algorithm.
Number and brightness analysis
The N&B analysis (30) considers each pixel of the image as a temporal data set. For each pixel the average and the variance is defined as follows:
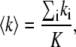 |
(6) |
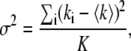 |
(7) |
where ki is the intensity at a given pixel and K is the number of frames analyzed. Using these definitions, the apparent brightness B and the apparent number of molecules N can be obtained using the following expressions as a function of the true brightness 〈ɛ〉 and the true number of particles n
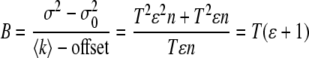 |
(8) |
and
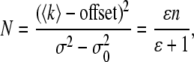 |
(9) |
where T is the average conversion factor between one photon detected and the number of digital levels produced by the analog electronics. The true brightness 〈ɛ〉 and the true number of particles n are thus dependent on the calibration of the instrument when using analog detectors (30), because one needs to consider the internal variance of the detector current, which is a linear function of the detector current and the readout term. Note that B depends only on the particle brightness and is independent of the number of particles. The calibration of the analog detector (30) for the Zeiss LSM 510 under the condition of our measurements (gain and offset) gave the value of T of 0.5 digital levels/photon. The value of the offset slightly varied from experiment to experiment but was always in the range of 1–1.5 digital levels, and 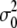 (the readout variance) was set to 0. As discussed in Digman et al. (27) a pixel value of B = 1 indicates that in that pixel there is only an immobile fraction. If in a pixel there is a ratio, R, of immobile and mobile fraction (the ratio is calculated from the relative contribution to the fluorescence intensity), then the value of the measured brightness ɛm in terms of the true brightness ɛ is given by
(the readout variance) was set to 0. As discussed in Digman et al. (27) a pixel value of B = 1 indicates that in that pixel there is only an immobile fraction. If in a pixel there is a ratio, R, of immobile and mobile fraction (the ratio is calculated from the relative contribution to the fluorescence intensity), then the value of the measured brightness ɛm in terms of the true brightness ɛ is given by
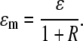 |
(10) |
Single-point two-photon fluorescent spectroscopy
Two-photon fluorescent spectroscopy (TPFCS) was achieved by using a mode-locked titan-sapphire laser tuned at 850 nm coupled to an inverted Olympus IX71 with a 60× water immersion objective (Olympus, Melville, NY). The sample was mounted on a No. 1 coverslip on top of the objective, and the fluorescent signal from the sample was collected by an avalanche photodiode and transmitted to a hardware correlator, as described elsewhere (31,32). After using a known dye to calibrate the system, 10 consecutive acquisitions of 10 s each were averaged and analyzed using a single component model of diffusion for the correlation function:
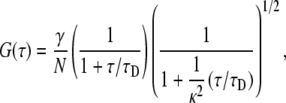 |
(11) |
where the diffusion coefficient for a two-photon system is obtained with 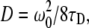 k is the structure parameter of the PSF, ω 0 is the waist of the PSF, N is the apparent number of molecules in the 3D Gaussian illumination volume, and γ is the illumination profile factor.
k is the structure parameter of the PSF, ω 0 is the waist of the PSF, N is the apparent number of molecules in the 3D Gaussian illumination volume, and γ is the illumination profile factor.
Immunocytochemistry
After RICS, HEK293 cells were fixed with a 4% solution of paraformaldehyde for 10 min and washed three times with a solution of 1× phosphate buffer saline. After permeabilization with 1% TX-100 in the presence of 5% bovine serum albumin, cells were incubated with a monoclonal-specific antibody to αCaMKII for 2 h. After several washes, cells were incubated with an Alexa-568 labeled goat anti-mouse secondary antibody for an additional 2 h before imaging on a Zeiss 510 confocal microscope with a 63× Apo water immersion (0.9 NA) infinity corrected lens with the zoom set to 2 and collecting 512 × 512 8-bit images. An eGFP signal was detected by excitation with the 488 nm laser line, whereas the Alexa-568 label was detected by excitation at 543 nm. A band-pass filter (505–530 nm) was used to discriminate the green signal, and a long-pass filter (<585 nm) was used for the red channel.
RESULTS
In vitro measurements of GFP constructs
The diffusion coefficients of green fluorescent protein (GFP), GFP-CaM, and GFP-CaMKII diffusing in 1× phosphate buffer saline were obtained by RICS and single-point TPFCS (see Materials and Methods for details on RICS and TPFCS). These data are presented in Table 1, where the standard deviation describes the day-to-day error in the measured diffusion coefficient of each of these proteins. eGFP tends to give a larger experimental error with RICS, but the error decreases as the size of the tracer is increased with a correspondingly slower diffusion coefficient. From single-point FCS, we know that at least a 1.6 difference in the radius of gyration is needed to differentiate without ambiguity the diffusion between two molecules (33). In our case, we know that CaM, a 16 kDa protein, is very flexible. The larger fluorescent tag eGFP is 27 kDa and has a rigid barrel shape and likely defines most of the mobility of eGFP-CaM, which exhibits only a slightly slower diffusion coefficient than eGFP. GFP-CaMKII gives almost a fourfold change in the diffusion coefficient when compared to GFP or GFP-CaM, consistent with its larger radius of gyration. Compared to GFP-CaM (42 kDa), GFP-CaMKII (970 kDa) is one of the largest soluble proteins found in the cytosol. Thus, it would correspond to one extreme for a protein tracer reporter for intracellular diffusion.
TABLE 1.
Comparison of means ± SD of diffusion coefficients as obtained by TPFCS and RICS in solution
| RICS
|
TPFCS
|
|
|---|---|---|
| D ± SD (μm2/s) | D ± SD (μm2/s) | |
| GFP | 88 ± 30 | 83 ± 14 |
| GFP-CaM | 79 ± 13 | 66 ± 4 |
| GFP-CaMKII | 25 ± 5 | 21 ± 0.3 |
Live cell measurements
Immunocytochemistry of HEK293 cells
To assess the distribution of each of the eGFP constructs used in this study, HEK293 cells were transfected with plasmids expressing eGFP, eGFP-CaM, eGFP-CaMKII, or the nonlabeled form of αCaMKII and were fixed, immunolabeled, and imaged on the LSM 510 (see Materials and Methods). Fig. 1 shows antibody labeling of αCaMKII in the red channel, and the green channel shows the expression of the various eGFP constructs. The final column shows image overlays of the two channels. It is clear from row A that there is no endogenous αCaMKII in HEK293 cells (absence of signal in the red channel). Note that eGFP is expressed throughout both the nucleus and cytoplasm of the cell (green channel) with some apparent enrichment in the nucleus. The bleed-through between channels is observed on row B, where no antibody treatment was added to HEK cells expressing eGFP-CaM. eGFP-CaM is distributed homogeneously within the cells with slightly lower expression in the nucleus. Strong immunostaining is evident when nonlabeled αCaMKII is expressed in the cells (row C) that disappear when the primary antibody is omitted (row D). αCaMKII is uniformly distributed in the cytoplasm but is largely excluded from the nucleus whether expressed without (row C) or with (row E) the eGFP tag. In cells expressing both eGFP-CaM and nonlabeled CaMKII (row C), GFP-CaM can be detected throughout the cytoplasm and the nucleus.
FIGURE 1.

Immunolabeling of transfected HEK293 cells. Red channel in most cases corresponds to the signal coming from Alexa-568 labeled goat anti-mouse secondary following labeling with a monoclonal antibody to αCaMKII. The green channel is eGFP or the eGFP construct signal. The overlay of the two channels is shown in the third column. (A) HEK293 cells expressing GFP; we see no evidence of endogenous αCaMKII, evidenced by the lack of signal in the red channel. (B) eGFP-CaM expressed on HEK293 cells when no antibody treatment was performed, showing little cross talk signal from the green channel into the red channel. eGFP-CaM is distributed homogeneously within the cells with slightly lower expression in the nucleus. (C) Coexpression of eGFP-CaM and the nonlabeled form of αCaMKII; we see similar results to those in B, plus we see signal from the red channel, indicating expression of αCaMKII in each cell that is also expressing eGFP-CaM. CaMKII is largely excluded from the nucleus, whereas eGFP and eGFP-CaM are not. The appearance of very bright spots possibly reflects recycling vesicles due to overexpression of eGFP-CaM. (D) Expression of eGFP-CaMKII, with the primary monoclonal antibody specific for αCaMKII was omitted from the staining protocol. Note the very weak signal in the red channel, which represents background fluorescence and/or some modest bleed-through of the green channel similar to row B. (E) eGFP-CaMKII expressed in HEK293 cells stained with monoclonal antibody specific to αCaMKII. Note that both signals overlay almost perfectly, as expected.
Diffusivity and availability of eGFP in HEK293 cells
We first characterized the diffusion, concentration, and brightness distribution of eGFP in HEK293 cells. eGFP represents a noninteracting molecule with the same molecular brightness as the molecule of interest, eGFP-CaM. From analyzing 16 cells, we obtained an average value for the diffusion coefficient and molecular brightness of intracellular eGFP in its monomeric form, and the numeric data are summarized in Table 2. As a representative example, Fig. 2 A shows a single confocal frame of a selected cell with the region of interest (ROI) identified as a red box. From that region, 100 frames were collected, as mentioned in Materials and Methods. Fig. 2 B shows the averaged intensity across the 100 frames (collected over 3 min), and by averaging, some of the features in the confocal image (Fig. 2 A) are blurred. After applying the N&B analysis, one can extract the spatial distribution of the number of molecules.
TABLE 2.
Diffusion coefficients, brightness, and concentrations obtained using RICS and N&B of eGFP constructs expressed in HEK293 cells
| Protein | Ca2+ | D ± SD (μm2/s) | % Fast Pool |
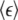 (Kc/m/s) (Kc/m/s) |
C (μM) |
|---|---|---|---|---|---|
| eGFP | 20 ± 5 | 90 ± 9 | 6.88 ± 1.5 | 1.5 ± 2.3 | |
| eGFP+nlCaMKII | 21 ± 8 | 86 ± 13 | 7.88 ± 2.9 | 1.75 ± 1.42 | |
| eGFP-CaMKII | 1.6 ± 0.9 | 74 ± 11 | 21.08 ± 11.05 | 2.16 ± 1.14 | |
| eGFP-CaM | 7.56 ± 5.6 | 2.1 ± 1.57 | |||
| eGFP-CaM+nlCaMKII | 10.65 ± 6.1 | 4.82 ± 2.63 | |||
| Nucleus | |||||
| eGFP-CaM | Basal | 10 ± 5 | 80 ± 19 | ||
| +Ca2+ | 10 ± 4 | 85 ± 16 | |||
| −Ca2+ | 10 ± 7 | 75 ± 12 | |||
| Cytoplasm | |||||
| eGFP-CaM | Basal | 11 ± 6 | 60 ± 17 | ||
| +Ca2+ | 11 ± 4 | 68 ± 17 | |||
| −Ca2+ | 10 ± 9 | 64 ± 26 | |||
| Nucleus | |||||
| eGFP-CaM+nlCaMKII | Basal | 7 ± 5 | 52 ± 28 | ||
| +Ca2+ | 12 ± 2 | 58 ± 23 | |||
| −Ca2+ | 6 ± 2 | 67 ± 11 | |||
| Cytoplasm | |||||
| eGFP-CaM+nlCaMKII | Basal | 7 ± 4 | 29 ± 27 | ||
| +Ca2+ | 8 ± 1 | 67 ± 10 | |||
| −Ca2+ | 4 ± 3 | 34 ± 10 |
Data are tabulated from different cellular compartments for RICS and after elevation Ca2+ (+Ca2+) or reduction Ca2+ (−Ca2+) as described in Materials and Methods. nlCaMKII refers to nonlabeled CaMKII. Values are means ± SD of at least six different cells measured for each condition. The average molecular brightness 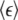 is calculated from Eq. 8 then divided by the pixel dwell time (25.6 μs) giving units of kilocounts per molecule per second (Kc/m/s).
is calculated from Eq. 8 then divided by the pixel dwell time (25.6 μs) giving units of kilocounts per molecule per second (Kc/m/s).
FIGURE 2.

RICS and N&B analysis of eGFP in HEK293 cells. (A) A confocal slice through a HEK293 cell expressing eGFP. (B) The average intensity of 100 frames of the ROI identified in A with a red box is shown with the pseudocolor scale from 0 to 255. (C) The concentration map obtained from the N&B analysis by converting the number of molecules from the average intensity as compared with a standard curve similar to the one found in the Supplementary Material (Data S1). (D) The spatial map of the brightness “B” that represents the variance of each pixel divided by the average intensity of each pixel, calculated from the 100 frames. (E) Brightness histogram of the data shown in D. The total number of pixels (y axis) are plotted against B (x axis). A green Gaussian profile is overlaid on the histogram whose peak represents the molecular brightness of monomeric eGFP. (F) The RICS analysis of the ROI shown in B. (G) The fit of the autocorrelation with a two-component model for this cytoplasmic region of the cell gives values of 20.1 μm2/s and 0.39 μm2/s.
The number of molecules can be converted into concentration using a standard curve similar to the one shown in Fig. S1 D (Data S1). Thus, panel C shows a spatial map that represents the concentration of eGFP in this region of the cell. As a result of the N&B analysis, one can also obtain a spatial map of the brightness of eGFP, shown in panel D. The apparent brightness “B” is obtained by calculating the variance over the intensity at each pixel over the 100 frames. From B the average molecular brightness 〈ɛ〉 can be extracted by considering the pixel dwell time, as reported in Table 2. In principle, integer times of molecular brightness represent a molecule with a single or multiple copies of the same fluorescent tag. We refer to the latter as complexes. These complexes appear when there is an association with increased binding stoichiometry.
In each pixel, we could have “monomeric” (with one eGFP) and “multimeric” (with more than one eGFP) complexes; thus we can only determine an average number of eGFPs (or eGFP multimers) forming complexes per pixel. For example from the average apparent brightness (B = 1.19) of all HEK cells expressing eGFP, a value of B = 1 corresponds to the immobile fraction and 0.19 is the actual contribution from the molecule. Multiples of this number (0.19) would represent eGFP forming complexes. It is clear from panel (D) that the molecular brightness of eGFP is distributed uniformly. This is quantitatively shown in the histogram of the brightness in Fig. 2 E, where the number of pixels is plotted against their brightness (variance/intensity). We superimposed on the histogram a Gaussian distribution (green line) from which the value of the peak molecular brightness is obtained. In this case the standard deviation of the distribution reflects the statistical noise of this measurement. The distribution for eGFP in HEK293 cells can be well described by a single Gaussian curve. The average molecular brightness, 〈ɛ〉, of GFP inside cells was found to be 6.88 ± 1.5 Kc/m/s (see Table 2).
Some of the artifacts that disrupt the histogram and thus the N&B analysis are the appearance of bright or dark vesicles because they increase the variance and appear as places with high B, more evident when the other eGFP constructs described below were expressed. However, they usually give a low background on the histogram with few pixels associated with these artifacts; thus they can be avoided when interpreting the data. Movement of the plasma and intracellular membranes also appear as regions with high brightness, but with low counts that can be observed directly on the screen and avoided for interpretation. Regardless of these possible artifacts, eGFP was shown to be homogeneously distributed in the nucleus and cytoplasm and served as a reproducible control with a molecular brightness of ∼7 Kc/m/s to compare against other eGFP-constructs.
Image correlation provides the mobility of the tracer molecule using the same time series of images that was used for the N&B analysis. To obtain the diffusion coefficient of smaller regions than the whole image shown in Fig. 2 B, a smaller ROI, indicated as a red box in Fig. 2 B, was used to calculate the image correlation (Fig. 2 F). Then, we used a version of Eq. 4 containing two diffusive components to fit the data. D1 represents the diffusion coefficient of the eGFP (20.1 μm2/s), and D2 was required to properly fit the data, with a value of ∼0.4 μm2/s, as described in Materials and Methods. A one-component model produced a poor fit, evident in the residuals, with a D slower than the first component in the two-component model. Careful analysis of the diffusion of eGFP over many cells and in different subcellular regions allowed us to determine that the average diffusion coefficient of eGFP was ∼20 μm2/s (see Table 2). From this diffusion coefficient, assuming eGFP is noninteracting, we deduce that the apparent viscosity of the cytoplasm is ∼4-fold that of buffer. As with the N&B analysis, we noticed that diffusion rates recovered by RICS could be distorted in specific regions of the image due to dark or bright moving vesicles and organelles, like mitochondria. For the average values reported in Table 2, the ROIs were carefully selected in an attempt to minimize these artifacts.
Diffusivity and availability of eGFP-CaMKII in HEK293 cells
We expressed eGFP-CaMKII in HEK293 cells as a tool to examine the diffusion of a large cytosolic protein complex with the potential to interact with CaM. Also, CaMKII assembles into a dodecameric complex (34) and has the theoretical potential to produce a molecule with 12 times the brightness of eGFP.
We present a typical example of data analyzed from an eGFP-CaMKII expressing HEK293 cell using the same layout as in Fig. 2. Fig. 3 A represents a confocal scan of the selected cell and Fig. 3 B represents the average intensity of the scanned region. Fig. 3 C shows the concentration map that clearly shows that eGFP-CaMKII is not expressed in the nuclear region. The concentration reported in Fig. 3 C is that of eGFP as determined for Fig. 2. The B-map (Fig. 3 D) shows that some values are ≤1, which correspond to the background counts and are areas where eGFP-CaMKII is not present, such as inside the nucleus. In the cytoplasm there is a large distribution of brightness ranging from one eGFP to complexes that contain five to eight copies of eGFP. This is expected because CaMKII is a multisubunit enzyme. This is clearly evident by the broadening in the histogram (Fig. 3 E), where complexes are responsible for the significant second Gaussian component (blue profile). This oligomeric complex of CaMKII expressed in the HEK293 cells has on average at least three fluorescent eGFP-CaMKII subunits per molecule. The heterogeneity in the second component presumably reflects the stochastic assembly of eGFP-CaMKII subunits with the endogenous unlabeled subunits in the HEK293 cells and/or incomplete maturation to produce the fluorescent state of the eGFP molecule. Since αCaMKII was not observed in untransfected HEK293 cells (see Fig. 1), eGFP-CaMKII subunits most likely are associated with one, or a combination, of the other three mammalian isoforms of CaMKII (β, γ, or δ) (35).
FIGURE 3.

RICS and N&B analysis of eGFP-CaMKII in HEK cells. (A) A confocal slice through HEK293 cells expressing eGFP-CaMKII. (B) The average intensity of 100 frames of the ROI identified in A is shown with the pseudocolor scale from 0 to 255. (C) The concentration map obtained from the N&B analysis. (D) The spatial map of the brightness “B”. (E) Brightness histograms of the data shown in D; the total number of pixels (y axis) are plotted against B (xaxis). The green Gaussian represents the contribution with the molecular brightness of eGFP, and the blue Gaussian represents the contribution of molecular complexes with an average of ∼3 eGFP-CaMKII subunits presumably assembled into the holoenzyme complex. (F) The RICS analysis of the ROI shown in B. (G) The fit of the autocorrelation with a two-component model for this cytoplasmic region of the cell gives values of 2.6 μm2/s and 0.14 μm2/s.
The diffusion analysis of eGFP-CaMKII by RICS of a small ROI indicated in Fig. 3 B gives the correlation analysis shown in Fig. 3 F. This spatial and temporal correlation was fit with two diffusion components, where we find that the faster component diffuses at a rate of 3.6 μm2/s and the slower component at a rate of 0.4 μm2/s, as done for the eGFP case. The component diffusing at 3.6 μm2/s is ∼10-fold slower than eGFP-CaMKII diffusion in buffer (Table 1), which implies that CaMKII is associated with other molecules or with itself or that it is hindered in its diffusion within the cytosolic matrix by its size.
Diffusivity and availability of eGFP-CaM in HEK293 cells
Our primary objective in this study was to establish the mobility and availability of CaM inside cells. Because of the numerous binding partners for CaM inside cells, it was expected that the mobility and distribution of eGFP-CaM would be complex. RICS analysis of eGFP-CaM expressed in HEK293 cells in basal conditions of Ca2+ is shown in Fig. 4. A confocal slice is shown of the selected cell in Fig. 4 A; note that the fluorescence is relatively uniform in the cytoplasm with the nucleus exhibiting less fluorescence. Then a smaller ROI was selected with the zoom tool (red box in Fig. 4 A) and 100 frames were collected (the average intensity is shown in Fig. 4 B, which includes an area of the nucleus and cytoplasm). Then RICS was applied to the ROI shown in Fig. 4 B, and after removing the immobile fraction we produced the spatial autocorrelation shown in Fig. 2 C. A two-component diffusion model was required to properly fit the data (Fig. 2 D). For this particular cell, the two components of diffusion of eGFP-CaM in the cytoplasm were 13.2 μm2/s and 0.036 μm2/s.
FIGURE 4.

RICS analysis of eGFP-CaM in HEK cells. (A) A confocal slice through a HEK293 cell expressing eGFP-CaM. (B) The average intensity of 100 frames of the ROI identified in A is shown with the pseudocolor scale from 0 to 255. (C) The spatial autocorrelation of the intensity data from the red box shown in B. The profile of the correlation function is indicative of multiple diffusive components. (D) The fit of the autocorrelation with a two-component model for this cytoplasmic region of the cell gives values of 13.2 μm2/s and 0.036 μm2/s. (E) and (F) The spatial map of the two components scaled to be centered on the diffusion of each component (D1 and D2) in μm2/s. A mask of some of the intracellular boundaries is overlaid as topographical maps and a guide where diffusion takes place. Diffusion maps were obtained by scanning a 32 × 32 pixel sequentially over the whole data shown on B with a step of 16 pixels. The fitted values were smoothed for visualization.
A diffusion map of the whole image shown in Fig. 4 B can be constructed by sequentially selecting a 32 × 32 pixel box spaced by 16 pixel steps. Each box is correlated and fitted. For the whole map, both in the nucleus and in the cytoplasm, the average diffusion of eGFP-CaM was ∼10 μm2/s (Fig. 4 E), which was consistent when comparisons were made between cells (See Table 2). As before, the fits required a second, slower component for diffusion in the range of 0.7–0.02 μm2/s (Fig. 4 F). However, the N&B analysis (see below) implies that eGFP-CaM forms molecular complexes containing more than one eGFP-CaM. For this reason the second component was allowed to float to capture these dynamics. The hot spots on Fig. 4 F represent a very dynamic region of these types of objects.
From the map shown in Fig. 4 E, we observed that eGFP-CaM diffuses faster, possibly noninteracting, at the top center of the image which corresponds to an area inside the nucleus. The black lines are a mask of some of the major cellular features evident from the image displayed in Fig. 4 B. In the cytoplasm the diffusion can slow to values of ∼4 μm2/s, indicating regions where eGFP-CaM most likely is moving as a complex with target proteins. However, the middle portion of the map (average behavior) shows diffusion at a rate close to 10 μm2/s, which is about half of that expected for diffusion considering the local viscosity as measured by eGFP. It is important to note that the spatial resolution observed in these maps is poor due to the 32 × 32 pixel window that is sequentially analyzed and further averaged with neighboring data. One technical issue to consider is that the 32 × 32 autocorrelation image is fitted with fewer points that reduce the statistical confidence of the fit. This confidence level increases if larger regions are selected, e.g., a 64 × 64 or 128 × 128 box. The spatial resolution of RICS is thus limited by the necessity of using a rather large area for the analysis. In summary, the overall diffusive behavior of eGFP-CaM across many cells showed that the average fraction of the faster diffusing component (∼10 μm2/s) was ∼80% in the nucleus and 60% in the cytoplasm; see Table 2 for a summary of these data. The immobile pool was subtracted for these analysis; thus, the percentages report the fraction of eGFP-CaM that diffuses at rates >0.002 μm2/s.
The N&B analysis of the cell presented in Fig. 4 is shown in Fig. 5. In Fig. 5 A the concentration distribution of eGFP-CaM is shown below a mask constructed from Fig. 4 B. The concentration histogram of this image is presented in Fig. 5 B. The B-map (brightness map) and its histogram are shown in Fig. 5, C and D, respectively. From the B-map we can see that in the nucleus and in large regions of the cytosol eGFP-CaM has the brightness of a single eGFP. This is evident in Fig. 5 E, where a binary map of the pixels showing the brightness of the monomeric eGFP is shown in red. This threshold value is selected by overlaying a Gaussian profile (green Gaussian) on top of the B-histogram (Fig. 5 D) and setting the threshold for the image to the peak of the profile. The blue Gaussian corresponds to a threshold value that would represent the “dimeric” state of eGFP-CaM, likely representing molecules with two bound CaM molecules; the map of such a selection is shown in Fig. 5 F as a binary map. From here, we see that almost no such complexes exist in the nuclear region, but in the cytoplasm we find that complexes are found near the perinuclear region and close to borders of other membranes. Even complexes with higher brightness are found (the additional pixels in Fig. 5 D at 1.4 variance/intensity and beyond), but they do not contribute significantly to the overall concentration of eGFP-CaM.
FIGURE 5.

N&B analysis of eGFP-CaM transfected HEK293 cell. (A) The concentration map of eGFP-CaM of the same cell shown in Fig. 4. (B) The concentration histogram as the total number of pixels (y axis) plotted against concentration in μM (x axis). (C) The brightness map as the variance/intensity is scaled over the range from 0.8 to 2.0. (D) Brightness histograms of the data shown in (C); the total number of pixels (y axis) are plotted against B (x axis). On top of the B-histogram, the green Gaussian represents the contribution of a single eGFP, and the blue Gaussian represents the pixels representing two eGFP-CaMs per complex. These pixels are shown on red as a binary map on E and F, respectively.
Some of the larger bright objects, with values for B ∼ 2, correspond to vesicles (1–2 μm diameter) filled with fluorescent protein, but those regions can be easily identified and ignored because they have abnormally large fluorescent intensity. By performing a careful selection of brightness and intensity in regions devoid of vesicles, we conclude that eGFP-CaM is found mainly in complexes with only one CaM molecule per complex in the nucleus; but in the cytoplasm, eGFP-CaM is also found in complexes with many copies.
Data from some of these cells were collected after elevation of Ca2+ with ionomycin and some after removal of Ca2+ with EGTA and ionomycin. However, no significant difference was observed between these treatments and cells at rest. We attribute this somewhat surprising result to the fact that the data were collected at steady sate following each treatment and not during the transition periods of increased or decreased Ca2+. These data are summarized in Table 2, which contains data on the molecular brightness of eGFP-CaM, the concentration of expressed protein, and the diffusion coefficient for different regions.
Cotransfection of GFP-CaM with nonlabeled αCaMKII in HEK293 cells
To examine the influence of CaM-binding targets on the brightness and mobility of eGFP-CaM, similar experiments to those just described were done but now coexpressing eGFP-CaM and nonlabeled αCaMKII in HEK293 cells. An example of single-cell analysis is presented in Fig. 6 where rows I and II represent the analysis of the same cell under basal conditions and at elevated Ca2+, respectively. Similar images from a second cell are presented in Fig. S3 (Data S1), but in that case we changed from basal to lower Ca2+ using ionomycin in the presence of the Ca2+-chelator EGTA. Column A represents the selected cell, and column B is the ROI from which the N&B analysis was done. The B-map and its histogram are shown in columns C and D, respectively. The brightness map (column C) shows that in a substantial fraction of pixels, eGFP-CaM exists as a monomer, particularly in the nuclear region, consistent with the data described above (Fig. 5).
FIGURE 6.

N&B analysis of eGFP-CaM coexpressed with nonlabeled CaMKII. Transfected HEK293 cells in basal conditions row I and with a media exchange for high Ca2+ (II). (A) A pseudocolor intensity profile of the whole cell. (B) The ROI from which RICS and N&B were determined. (C) The spatial map of the molecular brightness of 100 frames after subtraction of the slowly moving component (see Materials and Methods). (D) B-histogram where two Gaussian distributions are shown (green and blue lines). The total number of pixels (y axis) is plotted against B (x axis) scaled from 0.8 to 2. Note the long and asymmetric profile of the second (blue) component, which indicates a family of diffusing molecules with molecular brightness 3–12 times that of eGFP.
Since αCaMKII is largely excluded from the nucleus, we did not anticipate significant nuclear changes in eGFP-CaM behavior. However, in the cytoplasm enriched with αCaMKII (see Fig. 1 C), there is a significant fraction that binds at a stoichiometry of ∼3–6 eGFP-CaMs per moving molecule in regions that we are confident are not contaminated by vesicle artifacts. Those regions contaminated with artifacts are at the edge of the cell on the right and the large bright vesicles. The complexes of 3–6 eGFP-CaM molecules are likely due to binding to αCaMKII. This is in agreement with the reduction of the overall diffusion coefficient from 10 μm2/s to 7 μm2/s obtained by RICS in these same cells. Note again that the RICS value of 7 μm2/s represents the average diffusive behavior over a 32 × 32 pixel box. One common observation we noted is that in cells that are brighter (express increasing amounts of eGFP-CaM), as in the cells presented in Fig. 6 C, the perinuclear region increases in brightness. Interestingly, these complexes disappear after increasing Ca2+ (compare rows I and II column C where black arrows are present). This is one of the only consistent differences we found in manipulating Ca2+ inside the cells.
In summary, if we consider only the average molecular brightness per cell (Table 2), complexes were mostly found in cells that expressed eGFP-CaMKII, which was anticipated because αCaMKII is a multimeric protein. But, higher binding stoichiometry was also abundant in the cells expressing both eGFP-CaM and the nonlabeled form of αCaMKII, presumably because αCaMKII is binding to multiple copies of eGFP-CaM. These results from N&B analysis are consistent with the results using RICS, which shows that in these same cells, on average, we find slower diffusion coefficients (Table 2). Thus, we conclude that eGFP-CaM targets, such as αCaMKII, have a significant potential to alter the diffusivity and available pool of CaM.
DISCUSSION
We show that the combination of RICS and N&B analyses is a powerful technical combination to investigate the mobility and interaction of proteins inside cells. Applying these techniques led us to the conclusion that a significant fraction of CaM is binding to other molecules inside cells both at rest and at elevated Ca2+. We also examined the mobility and state of aggregation of eGFP (∼27 kDa) and eGFP-CaMKII (∼970 kDa). These two proteins are at the extremes of a broad range of sizes found in cells. eGFP is a noninteracting protein that gives a limiting high value for the diffusion of a small freely mobile protein. We found that eGFP in cells diffused at ∼20 μm2/s, ∼4-fold slower than in buffer and consistent with previous results (36–38). Also, eGFP was uniformly distributed and existed with the brightness of a single eGFP throughout the cytoplasm. For eGFP-CaMKII, hindered diffusion was found, typical of large proteins in the cytosol. eGFP-CaMKII exhibited an average diffusion coefficient of ∼1.6 μm2/s (Table 2), a factor∼13-fold slower than in buffer and consistent with our earlier studies (6). Furthermore, we observed that eGFP-CaMKII was partially immobilized in the cell (∼26% immobile fraction), presumably due to interactions with other cellular components or possibly itself (28), as suggested by the presence of large brighter complexes.
Our primary objective in this study was to establish the mobility and availability of CaM inside cells. The apparent viscosity of the cytoplasm of HEK cells was determined to be ∼4 times higher than that in buffer when eGFP was used as a tracer. The diffusion coefficient of eGFP-CaM in cells (∼10 μm2/s) was on average a factor of 2 slower than for eGFP in cells, and this fast mobile fraction represented ∼80% of CaM in the nucleus and ∼60% in the cytosol in basal conditions (Table 2). With a careful selection of regions to avoid artifacts, the slow diffusive component represented the motion of eGFP-CaM most likely bound in large complexes. In solution, there was no significant difference between the diffusion of eGFP and eGFP-CaM. Therefore, the value found for the diffusion of eGFP-CaM implies that either eGFP-CaM is bound to other proteins or that eGFP-CaM is weakly interacting with immobile structures, giving the appearance of slowed diffusive motion.
To distinguish between these two possibilities, we used the information from the N&B analysis. We observed from the B-map in Fig. 5 C that over the entire cell, ∼80% of the pixels had the brightness of a single eGFP-CaM (Fig. 5 D) and that the remainder of pixels report brightness corresponding to more than one eGFP-CaM per molecular complex (histogram Fig. 5 D). However, in the nucleus, >90% of eGFP-CaM was found as “monomers”, whereas in the cytoplasm ∼50% of the eGFP-CaM was monomers (Fig. 5 E). The rest of the pixels show increased brightness, mostly as apparent dimers, presumably from complexes formed with two eGFP-CaMs per target protein. Furthermore, there is a spatial separation between pixels with low and high brightness. Since pixels with large brightness must correspond to CaM-forming complexes, we must look at the map of the number of molecules to properly evaluate the availability of CaM. Using the values of the concentration map, we determined that most of the CaM molecules are part of molecular complexes.
In addition, a transient interaction with immobile structures is unlikely to give plots with high brightness. We reached this conclusion by considering the diffusion analysis and N&B. From the N&B, we obtained concentrations of eGFP-CaM in the μM regime; only very high affinity targets (kD ∼ nM or below) would be likely to bind and stay as a complex during the time frame of the fluctuations (∼4 s between frames). Transient interactions, although potentially slowing the diffusion, would not give a high brightness because higher variance over the acquisition time is required to resolve complexes. However, we acknowledge that there are CaM-binding partners with 1:1 stoichiometry that would also slow significantly the diffusion of eGFP-CaM and from diffusion analysis they cannot be resolved. Nevertheless, the finding of variable stoichiometry of CaM with binding partners leads to a new dimension on the regulatory function of CaM.
We also showed that introducing a target for the binding of CaM (αCaMKII) significantly influenced the mobility and availability of eGFP-CaM in the cells. This is evident in both the analysis of molecular brightness and diffusion analyzed with RICS (Table 2). Similar influences on CaM availability have been seen in a number of other examples where CaM-binding proteins were introduced into cells. Expression of nitric oxide synthase (21) or CaMKII (6) in HEK293 cells were both shown to significantly affect the availability of CaM.
The appearance of molecular complexes indicates that there are targets with multiple binding sites for CaM. In the context of what is known about CaM biochemistry, there is little available information about the possible function of multiple CaMs bound to specific targets. Nevertheless, there are many examples where single protein targets contain multiple binding sites for CaM. A few examples include the L-type calcium channel, which contains three CaM-binding domains (39): CaMKII or other kinases (40) and members of the myosin family of actin binding proteins (41). In the particular case of CaMKII, multiple CaMs are required for the enzyme to reach an autophosphorylated state, placing a requirement for multiple CaMs bound to a single holoenzyme (42). Thus, the existence of many CaMs associated to a target was expected to some degree even in the absence of CaMKII. The functional role of the multiple CaM-binding sites for CaM targets is an active research field that is also being approached by numerical simulations.
A number of past studies have evaluated the intracellular diffusion and availability of CaM either directly (6,7,19) or indirectly (3,4,20,21) using genetically engineered biosensors. Fluorescent recovery after photobleaching of microinjected fluorescent CaM in smooth muscle cells demonstrated an immobile pool of 37% (7), and the diffusion was four times slower than an inert tracer of similar size (43). These values are quite consistent with those we determined in HEK293 cells. The use of fluorescence resonance energy transfer based CaM sensors has also revealed that CaM-binding targets are in excess of available CaM and that the formation of CaM-target complexes is limited by CaM availability (4,20,21). However, quite different results have also been published. Gough and Taylor (19) used fluorescence anisotropy to show that <10% of fluorescein-labeled CaM microinjected into 3T3 cells at rest was bound. This apparent discrepancy is most likely due to the way the fluorescent probe was introduced competing with the endogenous pool of CaM or by the cell type used for the study.
In total, the presented results suggest that the majority of CaM is bound to target proteins spatially differentiable at rest and following Ca2+ elevations, implying that CaM-dependent protein activation occurs through competition for a limited pool of available CaM. We have also shown that the combination of RICS and N&B applied to data collected on a standard confocal microscope can provide enormous insight into the spatial map of the diffusion and formation of CaM-target complexes. This technology should be widely applicable to study the dynamics of protein mobility and protein interactions in the cytoplasm, nucleus (as shown here), or plasma membrane (44).
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Amélie Forest for technical assistance with cell culture and transfections.
This work was supported by a grant from the National Institute of Neurological Disorders and Stroke (NS26086) (M.N.W.) and the National Institutes of Health (S10-RR022531). M.N.W. also gratefully acknowledges an endowment from the William Wheless III Professorship. H.S. was supported by a training fellowship from the Keck Center Nanobiology Training Program of the Gulf Coast Consortia (National Institutes of Health grant No. 2 R90 DK071054-03S1). M.D. and E.G. acknowledge support from the Cell Migration Consortium U54 GM64346 (M.D. and E.G.), NIH-P41-RRO3155 (E.G.), and P50-GM076516 (E.G.).
Editor: Ian Parker.
References
- 1.Chin, D., and A. R. Means. 2000. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10:322–328. [DOI] [PubMed] [Google Scholar]
- 2.Clapham, D. E. 1995. Calcium signaling. Cell. 80:259–268. [DOI] [PubMed] [Google Scholar]
- 3.Persechini, A., and B. Cronk. 1999. The relationship between the free concentrations of Ca2+ and Ca2+-calmodulin in intact cells. J. Biol. Chem. 274:6827–6830. [DOI] [PubMed] [Google Scholar]
- 4.Persechini, A., and P. M. Stemmer. 2002. Calmodulin is a limiting factor in the cell. Trends Cardiovasc. Med. 12:32–37. [DOI] [PubMed] [Google Scholar]
- 5.Huang, C. Y., V. Chau, P. B. Chock, J. H. Wang, and R. K. Sharma. 1981. Mechanism of activation of cyclic nucleotide phosphodiesterase: requirement of the binding of four Ca2+ to calmodulin for activation. Proc. Natl. Acad. Sci. USA. 78:871–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim, S. A., K. G. Heinze, M. N. Waxham, and P. Schwille. 2004. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc. Natl. Acad. Sci. USA. 101:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luby-Phelps, K., M. Hori, J. M. Phelps, and D. Won. 1995. Ca2+-regulated dynamic compartmentalization of calmodulin in living smooth muscle cells. J. Biol. Chem. 270:21532–21538. [DOI] [PubMed] [Google Scholar]
- 8.Kubota, Y., J. A. Putkey, and M. N. Waxham. 2007. Neurogranin controls the spatiotemporal pattern of postsynaptic Ca2+/CaM signaling. Biophys. J. 93:3848–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rakhilin, S. V., P. A. Olson, A. Nishi, N. N. Starkova, A. A. Fienberg, A. C. Nairn, D. J. Surmeier, and P. Greengard. 2004. A network of control mediated by regulator of calcium/calmodulin-dependent signaling. Science. 306:698–701. [DOI] [PubMed] [Google Scholar]
- 10.Hudmon, A., and H. Schulman. 2002. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annu. Rev. Biochem. 71:473–510. [DOI] [PubMed] [Google Scholar]
- 11.Meyer, T., P. I. Hanson, L. Stryer, and H. Schulman. 1992. Calmodulin trapping by calcium-calmodulin-dependent protein kinase. Science. 256:1199–1202. [DOI] [PubMed] [Google Scholar]
- 12.Putkey, J. A., and M. N. Waxham. 1996. A peptide model for calmodulin trapping by calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 271:29619–29623. [DOI] [PubMed] [Google Scholar]
- 13.Colbran, R. J. 2004. Protein phosphatases and calcium/calmodulin-dependent protein kinase II-dependent synaptic plasticity. J. Neurosci. 24:8404–8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lisman, J., H. Schulman, and H. Cline. 2002. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 3:175–190. [DOI] [PubMed] [Google Scholar]
- 15.Berridge, M. J., M. D. Bootman, and H. L. Roderick. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4:517–529. [DOI] [PubMed] [Google Scholar]
- 16.Pitt, G. S. 2007. Calmodulin and CaMKII as molecular switches for cardiac ion channels. Cardiovasc. Res. 73:641–647. [DOI] [PubMed] [Google Scholar]
- 17.Schulman, H., P. I. Hanson, and T. Meyer. 1992. Decoding calcium signals by multifunctional CaM kinase. Cell Calcium. 13:401–411. [DOI] [PubMed] [Google Scholar]
- 18.Black, D. J., Q. K. Tran, and A. Persechini. 2004. Monitoring the total available calmodulin concentration in intact cells over the physiological range in free Ca2+. Cell Calcium. 35:415–425. [DOI] [PubMed] [Google Scholar]
- 19.Gough, A. H., and D. L. Taylor. 1993. Fluorescence anisotropy imaging microscopy maps calmodulin binding during cellular contraction and locomotion. J. Cell Biol. 121:1095–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran, Q. K., D. J. Black, and A. Persechini. 2003. Intracellular coupling via limiting calmodulin. J. Biol. Chem. 278:24247–24250. [DOI] [PubMed] [Google Scholar]
- 21.Tran, Q. K., D. J. Black, and A. Persechini. 2005. Dominant affectors in the calmodulin network shape the time courses of target responses in the cell. Cell Calcium. 37:541–553. [DOI] [PubMed] [Google Scholar]
- 22.Costantino, S., J. W. Comeau, D. L. Kolin, and P. W. Wiseman. 2005. Accuracy and dynamic range of spatial image correlation and cross-correlation spectroscopy. Biophys. J. 89:1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Digman, M. A., C. M. Brown, P. Sengupta, P. W. Wiseman, A. R. Horwitz, and E. Gratton. 2005. Measuring fast dynamics in solutions and cells with a laser scanning microscope. Biophys. J. 89:1317–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Digman, M. A., P. Sengupta, P. W. Wiseman, C. M. Brown, A. R. Horwitz, and E. Gratton. 2005. Fluctuation correlation spectroscopy with a laser-scanning microscope: exploiting the hidden time structure. Biophys. J. 88:L33–L36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolin, D. L., D. Ronis, and P. W. Wiseman. 2006. k-Space image correlation spectroscopy: a method for accurate transport measurements independent of fluorophore photophysics. Biophys. J. 91:3061–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wiseman, P. W., and N. O. Petersen. 1999. Image correlation spectroscopy. II. Optimization for ultrasensitive detection of preexisting platelet-derived growth factor-β receptor oligomers on intact cells. Biophys. J. 76:963–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Digman, M. A., R. Dalal, A. F. Horwitz, and E. Gratton. 2008. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 94:2320–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudmon, A., E. Lebel, H. Roy, A. Sik, H. Schulman, M. N. Waxham, and P. De Koninck. 2005. A mechanism for Ca2+/calmodulin-dependent protein kinase II clustering at synaptic and nonsynaptic sites based on self-association. J. Neurosci. 25:6971–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pristinski, D., V. Kozlovskaya, and S. A. Sukhishvili. 2005. Fluorescence correlation spectroscopy studies of diffusion of a weak polyelectrolyte in aqueous solutions. J. Chem. Phys. 122:14907. [DOI] [PubMed] [Google Scholar]
- 30.Dalal, R. B., M. A. Digman, A. F. Horwitz, V. Vetri, and E. Gratton. 2008. Determination of particle number and brightness using a laser scanning confocal microscope operating in the analog mode. Microsc. Res. Tech. 71:69–81. [DOI] [PubMed] [Google Scholar]
- 31.Iyer, V., M. Rossow, and M. N. Waxham. 2006. Peak two-photon molecular brightness of fluorophores is a robust measure of quantum efficiency and photostability. J. Opt. Soc. Am. B. 23:1420–1433. [Google Scholar]
- 32.Sanabria, H., Y. Kubota, and M. N. Waxham. 2007. Multiple diffusion mechanisms due to nanostructuring in crowded environments. Biophys. J. 92:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meseth, U., T. Wohland, R. Rigler, and H. Vogel. 1999. Resolution of fluorescence correlation measurements. Biophys. J. 76:1619–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kolodziej, S. J., A. Hudmon, M. N. Waxham, and J. K. Stoops. 2000. Three-dimensional reconstructions of calcium/calmodulin-dependent (CaM) kinase IIα and truncated CaM kinase IIα reveal a unique organization for its structural core and functional domains. J. Biol. Chem. 275:14354–14359. [DOI] [PubMed] [Google Scholar]
- 35.Gaertner, T. R., S. J. Kolodziej, D. Wang, R. Kobayashi, J. M. Koomen, J. K. Stoops, and M. N. Waxham. 2004. Comparative analyses of the three-dimensional structures and enzymatic properties of α, β, γ and δ isoforms of Ca2+-calmodulin-dependent protein kinase II. J. Biol. Chem. 279:12484–12494. [DOI] [PubMed] [Google Scholar]
- 36.Chen, Y., J. D. Muller, Q. Ruan, and E. Gratton. 2002. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luby-Phelps, K., F. Lanni, and D. L. Taylor. 1988. The submicroscopic properties of cytoplasm as a determinant of cellular function. Annu. Rev. Biophys. Biophys. Chem. 17:369–396. [DOI] [PubMed] [Google Scholar]
- 38.Swaminathan, R., C. P. Hoang, and A. S. Verkman. 1997. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys. J. 72:1900–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang, W., D. B. Halling, D. J. Black, P. Pate, J. Z. Zhang, S. Pedersen, R. A. Altschuld, and S. L. Hamilton. 2003. Apocalmodulin and Ca2+ calmodulin-binding sites on the CaV1.2 channel. Biophys. J. 85:1538–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buschmeier, B., H. E. Meyer, and G. W. Mayr. 1987. Characterization of the calmodulin-binding sites of muscle phosphofructokinase and comparison with known calmodulin-binding domains. J. Biol. Chem. 262:9454–9462. [PubMed] [Google Scholar]
- 41.Trybus, K. M. 2008. Myosin V from head to tail. Cell. Mol. Life Sci. 65:1378–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanson, P. I., T. Meyer, L. Stryer, and H. Schulman. 1994. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 12:943–956. [DOI] [PubMed] [Google Scholar]
- 43.Luby-Phelps, K., F. Lanni, and D. L. Taylor. 1985. Behavior of a fluorescent analogue of calmodulin in living 3T3 cells. J. Cell Biol. 101:1245–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caiolfa, V. R., M. Zamai, G. Malengo, A. Andolfo, C. D. Madsen, J. Sutin, M. A. Digman, E. Gratton, F. Blasi, and N. Sidenius. 2007. Monomer dimer dynamics and distribution of GPI-anchored uPAR are determined by cell surface protein assemblies. J. Cell Biol. 179:1067–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.