

Abstract
Molecular dynamics simulations were carried out on Thermus thermophilus 70S ribosome with and without a nascent polypeptide inside the exit tunnel. Modeling of the polypeptide in the tunnel revealed two possible paths: one over Arg92 of L22 and one under (from the viewpoint of 50S on top of 30S). A strong interaction between L4 and Arg92 was observed without the polypeptide and when it passed over Arg92. However, when the polypeptide passed under, Arg92 repositioned to interact with Ade2059 of 23S rRNA. Using steered molecular dynamics the polypeptide could be pulled through the L4-L22 constriction when situated under Arg92, but did not move when over. These results suggest that the tunnel is closed by the Arg92-L4 interaction before elongation of the polypeptide and the tunnel leads the entering polypeptide from the peptidyl transferase center to the passage under Arg92, causing Arg92 to switch to an open position. It is possible, therefore, that Arg92 plays the role of a gate, opening and closing the tunnel at L4-L22. There is some disagreement over whether the tunnel is dynamic or rigid. At least within the timescale of our simulations conformational analysis showed that global motions mainly involve relative movement of the 50S and 30S subunits and seem not to affect the conformation of the tunnel.
INTRODUCTION
Ribosome is one of the supra-biomolecules used in the process of translating genetic information for the synthesis of polypeptides. The 70S ribosome from eubacteria is composed of a small (30S) and a large (50S) subunit. The 30S subunit decodes genetic information (1), and the 50S subunit is considered to be responsible for the formation of peptide bonds, and the elongation of the nascent polypeptide (2). The nascent polypeptide is generated at the peptidyl transferase center (PTC) of the 50S subunit, where a new amino acid from aminoacyl-tRNA is added to the carboxyl terminus of the growing polypeptide (3,4). The nascent polypeptide passes through a tunnel, which starts at the PTC and continues through the 50S subunit (4–6). The length of the tunnel is ∼100 Å and the diameter is between 10 and 20 Å. Crystal structures have shown that the tunnel is rather kinked, has a nonuniform diameter, and contains grooves and cavities (3–5).
As the nascent polypeptide passes through the tunnel and leaves the ribosome, it encounters three main environments: the entrance, the constriction, and the exit (7).
The entrance part of the tunnel is formed by the central loop of domain V in 23S rRNA, and can be targeted by several antibiotics such as chloramphenicol, clindamycin, and several members of the macrolide family such as erythromycin. They inhibit protein elongation by binding to the entrance and blocking the path of the nascent polypeptide (8,9).
At about one third of the tunnel length away from the PTC, the nascent peptide meets a narrow constriction. The wall of the tunnel at this region is composed mainly of ribosomal RNA (nucleotides of domain I through V of the 23S rRNA). However, the narrowest part of the tunnel is formed by the protruding loops of two ribosomal proteins L22 and L4, which approach the tunnel from opposite sides and intercalate between rRNA segments of the 23S rRNA (3–5). The tips of these protrusions form an ∼12 Å long and 15 Å wide segment of the exit tunnel (3,5). It is known that specific amino acid sequences in nascent polypeptides found in TnaC (the leader peptide of Escherichia coli tryptophanase operon) and SecM (a protein belonging to the secretion monitoring system) interact with the tunnel around the L4-L22 constriction and induce translation arrest (10–13). The x-ray crystallographic structure of Deinococcus radiodurans ribosome suggests that the conformational change of the extended loop of L22 could block the tunnel, and this might cause elongation arrest of these special sequences (14).
After the nascent polypeptide has passed through the L4-L22 constriction, the tunnel widens and branches out. Crystal structures have revealed that this widening of the tunnel is flanked by L23 and L29, which are positioned at the surface of the 50S (3,5). It is known that the ribosome-associating chaperone trigger factor in eubacteria specifically interacts with L23, and binds to nascent polypeptides to prevent their aggregation and assist their folding (15).
There is some disagreement over whether the tunnel is dynamic or rigid. Based on cryo-electron microscopy (cryo-EM) data, Gabashvili et al. suggested that the tunnel is dynamic (6). Voss et al. claim that the tunnel is rigid (16,17) as thermal fluctuations estimated from the Debye-Waller factors (B-factors) of atoms that form the wall of the tunnel are low (3), and structures of RNA surrounding the tunnel are basically identical in a variety of 70S ribosome crystallographic structures (3,4,18–22).
To understand the dynamics of the 70S ribosome, a number of computational studies have been carried out. Some have used elastic network models (23,24) and coarse-grained molecular dynamics (MD) simulations (25) to analyze the global motions. Of note is all-atom MD simulations with explicit water molecules (26–28) to analyze the translocation of tRNA through the 70S ribosome (26).
Here, all-atom MD simulations were carried out on a Thermus thermophilus 70S ribosome in water with and without a nascent polypeptide inside the tunnel. The purpose is to understand how the exit tunnel of ribosome regulates the passage of the polypeptide on an atomic level and to assess the role of global motions in the conformation of the exit tunnel. Simulations on this ∼1,900,000 atom system were carried out for a total time of 20 ns using massive parallel computers. The probable path of the nascent polypeptide through the L4-L22 constriction was determined, and the relationship between global motions of the whole 70S ribosome molecule and the conformation of the exit tunnel were analyzed.
MATERIALS AND METHODS
Modeling of T. thermophilus 70S ribosome
There have been several attempts to create all-atom models of the 70S ribosome (29,30). Here, we constructed an atomic model of the 70S ribosome and a nascent polypeptide inside the tunnel. The crystallographic structure of T. thermophilus 70S ribosome including mRNA and two tRNA molecules at the aminoacyl-tRNA (A) and peptidyl-tRNA (P) sites (Protein Data Bank (PDB) code: 1YL3 and 1YL4, resolution: 5.5 Å) (31) was used. The missing parts in 1YL3 were modeled by using T. thermophilus (PDB code: 487D, resolution: 7.5 Å) (32) and homologous molecules from Haloarcula marismortui (PDB code: 1YJW, resolution: 2.9 Å) (33), (PDB code: 1QVG, resolution: 2.9 Å) (34), and Deinococcus radiodurans (PDB code: 1NJM, resolution: 3.6 Å) (35) as templates. Modeling of the missing residues in 1YL3 was done by superposing 1YL3 and one of the aforementioned structures that contained the missing residues on regions that flank them. Details are given in Table 1. The missing nucleotides 2157–2173 of the 23S ribosomal RNA in 1YL3 were modeled using InsightII.
TABLE 1.
Modeling of T. thermophilus 70S ribosome
| Modeled residues in 1YL3 | Modeled using PDB | Modeled by fitting to residues |
|---|---|---|
| 64–66 of L1 | 487D | 5–63 and 67–228 of L1 |
| 1–37, 113–170 and 321–337 of L3 | 1YJW | 36–49, 61–67, 69–113 and 172–309 of L3 |
| 70–78 and 121–137 of L4 | 1YJW | 1–69 and 138–246 of L4 |
| 157–174 of L5 | 1QVG | 10–29 and 35–156 of L5* |
| 111–130 of L13 | 1YJW | 4–110 and 131–140 of L13 |
| 9-50 of L15 | 1YJW | 51–145 of L15 |
| 130–186 of L18 | 1YJW | 9–129 of L18 |
| 178–223 of L25 | 1NJM | 1–177 of L25† |
Missing residues 30–34 of L5 in 1YL3 were modeled using the Biopolymer module in InsightII (Accelyrs, San Diego, CA).
Missing atoms along the modeled Cα trace were modeled using InsightII.
Building a nascent polypeptide chain inside the 70S ribosome
As the atomic structure of a nascent polypeptide inside the exit tunnel has not yet been determined by x-ray crystallography, we modeled possible conformations of a polyalanine inside the tunnel. A nascent polypeptide chain was built in the exit tunnel from the outside to the PTC using two procedures, a path finding procedure and a polypeptide building procedure.
Path finding procedure
Possible paths for the polypeptide were determined using the full-atom model of ribosome. The basic idea was to simulate moving a sphere from the PTC to the exterior of the ribosome. The following five-step procedure was used. 1), A sphere with a diameter of 6.0 Å was placed at the PTC, specifically at an arbitrarily chosen “vacant point” 2.0 Å away from the N6 atom of Ade76 of tRNA. This point was called the origin. A “vacant point” is where the sphere could be placed without overlapping any atom (with van der Waal's radius) of the ribosome. 2), A vacant point 1.0 Å away from the origin was randomly chosen. We name this point A. 3), A point 1.0 Å away from point A was randomly chosen and checked for being a vacant point. We named this point B. 4), The distances between point B and the origin (= dist1), and between point A and the origin (= dist2) were calculated. If dist1 > dist2, then we accepted point B as the new point A and placed a new sphere there. If dist1 ≤ dist2, then the acceptance of point B as the new point A was determined by the following criterion:
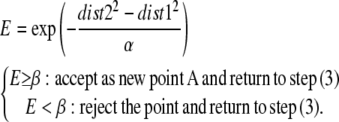 |
(1) |
α was a constant set so that E ranged between 0 and 1, and β was a random number between 0 and 1 generated each time the evaluation was carried out. If dist1 was >100 Å, namely far greater than the radius of the ribosome, the procedure was terminated. By repeating this procedure, we obtained 1,000 different paths. For the current study, we selected 10 paths that ran through the exit tunnel proposed by Voss et al. (16), as representatives.
Polypeptide building procedure
The peptide was built on each path using empirical distributions of bond lengths and bond angles found in nonhomologous proteins with known three-dimensional (3D) structures from the PDB. From 3D structures of proteins whose amino acid sequence identities are <50% among one another, we extracted the values of the following and built tables: i), distance between ith Cα and i + 1th Cα; ii), distance between ith Cα and C; iii), distance between ith Cα and i + 1th N; iv), distance between ith C and i + 1th N; v), coordinate of i + 1th C and i + 2th N, based on the local coordinate spanned by ith C, i + 1th N and i + 1th Cα; vi), coordinate of i + 1th Cα, based on the local coordinate spanned by ith Cα, ith C and i + 1th N; and vii), coordinate of i + 1th Cβ and ith O, based on the local coordinate spanned by ith N, Cα, and C. The peptide was built using the following steps: 1), the first Cα atom was placed on the path at the terminus furthest from the PTC; 2), the C atom was placed at a distance, randomly chosen from Table (ii), from the first Cα atom, and the Cα atom for the second residue was placed at a distance, randomly chosen from Table (i), from the first Cα atom; 3), the N atom for the second residue was placed in the plane spanned by the first Cα, the first C and the second Cα, and with a distance between Cα and N atoms chosen randomly from Table (iii); if the distance between the N atom and the C atom was not found in Table (iv), then we discarded the coordinate of the C atom and went back to step 2; and 4), the C atom for the second residue and the N atom for the third residue were placed using Table (v), and the Cα atom of the third residue was placed using Table (vi). If the Cα atom of the third residue was not located on the path, we discarded the C, N, and Cα atoms, and placed these atoms anew. By applying step 4 repeatedly, we could extend the polypeptide backbone until the C-terminal Cα atom located within the 9.0 Å distance from N6 atom of Ade76 of tRNA at the P-site. Once the whole polypeptide backbone was built (without O atoms), we finally placed Cβ and O atoms for all residues based on Table (vii) and finished the procedure. This resulted in 10 polyalanines, one for each path.
We removed some N-terminal amino acids to set the length of each chain to 36 residues. The constructed polyalanines were all in an extended conformation with ∼3.5 Å per amino acid. Thus, the length of the modeled polyalanine is ∼100 Å and this is long enough for it to fit along the path from the PTC to the distal end of the tunnel. Analysis of the polyalanine conformations revealed that they followed two distinctive and well defined paths, one over Arg92 of L22, the other under Arg92 of L22 as shown in Fig. 1. To define “over” and “under” the ribosome is oriented so that 50S is on top of 30S. They will be referred to as “case-over” and “case-under” from here on. Of the 10 polyalanines, 6 were positioned over Arg92 and 4 were positioned under.
FIGURE 1.

Two of the modeled polyalanines inside the exit tunnel. Ribosomal proteins L4 and L22 are located at the back and front, respectively. L4 and L22 (wire-ribbon models) are shown in dark khaki and coral, respectively. From the PTC, two nascent polyalanines (space-filling models) pass through the exit tunnel in the large subunit, 50S, passing over and under Arg92 of L22. They are shown in blue and red, respectively. Ade76 of tRNA at the P-site is depicted in space-filling model. For clarity other molecules in the 70S ribosome have not been included.
To optimize the conformations, a simulated annealing (SA) was carried out for each polyalanine in vacuum. The SA was carried out using AMBER 8.0 (36) with the force-field of Cornell et al. (37), included in the parm98.dat parameter set. To reduce the computation time, ribosome molecules that were >30 Å from the tunnel, (ribosomal proteins L1, L5, L6, L7, L12, L9, L11, L13, L14, L15, L16, L18, L19, L25, L30, L27, L31, L33, L35, L36, 16S, mRNA, and the 30S subunit) were removed from the system. The total number of atoms in the system (polyalanine, 5S rRNA, 23S rRNA, L2, L3, L4, L15, L16, L22, L23, L24, L29, L17, L20, L21, L32, L34, tRNA at the A-site, and tRNA at the P-site) was 130,147. A distance-dependent dielectric constant of 4.0 r was used, and nonbonded interactions were evaluated with a cut-off radius of 14 Å. A time step of 0.5 fs was used throughout the SA. Except for the modeled polyalanine, all the atoms in the system were restrained by a strong harmonic force constant of 10 kcal/mol Å2. The atoms of the polyalanine, except for the Cα of the C-terminal, were free to move. The Cα atom at the C-terminal near the PTC was restrained by a force constant of 1.0 kcal/mol Å2 to keep the C-terminal near the PTC and to mimic a situation where the polyalanine was elongating from the PTC. The system was heated from 0 to 800 K during the first 10 ps and was then equilibrated for 20 ps. The system was then gradually cooled for 70 ps from 800 K to 350 K. The SA was repeated 10 times for each of the 10 models, and the resulting coordinate sets were stored as possible conformations of the polyalanine at local minimum energy regions. Each of the 100 conformations was minimized for 1,000 steps using steepest descent followed by 5,000 steps of conjugate gradient without constraining the polyalanine.
Despite this procedure, the 100 polyalanine chains still followed two distinct paths through the L4-–L22 constriction, one over Arg92 (case-over), the other under Arg92 (case-under). As a representative of the 60 chains following the case-over path the minimum energy (energy was defined as the total of the internal energy of the polyalanine and the interaction energy between the polyalanine and the ribosome) chain was selected, likewise for the 40 chains of case-under. The distance between the Cα atoms of the N-terminal and C-terminal of these two models decreased during the SA from an initial distance of 90 Å and 88 Å to 57 Å and 41 Å, respectively. By adding the parts of the 70S ribosome that had been removed, two structural models of the 70S ribosome with a polyalanine inside the tunnel were constructed. Finally, each of the assembled structures was minimized to alleviate the stress at the interfaces between the constructed optimal structure and the parts of the 70S ribosome that had been removed.
Free MD simulation of the 70S ribosome
In addition to the two models of the 70S ribosome with a polyalanine inside the exit tunnel, a model of the 70S ribosome without a polyalanine was constructed to analyze the influence of the polyalanine on the dynamics of the 70S ribosome. These three models were independently placed in a rectangular box 286 Å × 286 Å × 263 Å. In this box, all the atoms of the 70S ribosome were >15 Å from the edge of the box. A large buffer size of 15 Å was used to avoid possible artificial interactions with the periodic image even if large conformational changes were to occur. To neutralize the negative charges of the 70S ribosome, sodium ions were placed at positions with large negative electrostatic potential. Then 539,992 water molecules were added to surround the two systems with the polyalanine. For the system without the polyalanine 540,043 water molecules were added. Each of the three systems comprised 1,878,425 atoms. For the two systems with a polyalanine, the number of atoms was as follows: 70S ribosome = 253,897, polyalanine = 363, sodium ions = 4,399, and water = 539,992 × 3. For the system without a polyalanine the number of atoms was: 70S ribosome = 253,897, sodium ions = 4,399, and water = 540,043 × 3.
Although metal ions such as magnesium or potassium are necessary for ribosomal function, they were not included in our study because: 1), magnesium and potassium ions are not given in the PDB files for ribosome, and it is not easy to correctly locate and equilibrate them inside and around ribosome in our rather short timescale MD simulations; in addition, it has been reported that potassium and chloride aggregate in biomolecular simulations using the AMBER force field (38), indicating that the simple inclusion of these ions may unnaturally affect the system; 2), our main intention is to observe the movement of the polypeptide inside the tunnel using steered MD (SMD) simulations (39); we assume that, in the SMD simulations in which the polypeptide is pulled through the tunnel, the movement of the polypeptide inside the tunnel is not significantly affected by the presence or absence of a particular type of ion; and 3), we assume that they do not significantly affect large-scale movements of the 70S ribosome.
Recent developments in computer technology have made all-atom molecular dynamics simulations of ribosome possible (26–28). In our study, to simulate the 70S ribosome in explicit water (using TIP3P model (40)), we used an in-house MD simulation program, called SCUBA (Simulation Codes for hUge Biomolecular Assembly) that is now being developed by the Japan Atomic Energy Agency and the University of Tokyo (41). The dielectric constant used was 1.0 and the van der Waals interactions were evaluated with a cut-off smoothly shifted to zero at 8 Å. The nonbonded list, including neighboring atoms within 8 Å, was updated every 50 fs. The particle-particle particle-mesh method (PPPM) was used for the electrostatic interactions (42). These interactions were split into short- and long-range contributions and the long-range potential was obtained by gridding the charges. The long-range contributions were evaluated with grid spacing of ∼1 Å. To utilize parallel computers using 128 CPUs efficiently, the size of the charge grid was chosen to be 28 = 256 for each dimension so that the fast Fourier transform could be applied to increase the speed of the calculation of the long-range contributions.
Steepest descent was carried out for 1,000 steps, followed by conjugate gradient for 9,000 steps. All heavy atoms of the 70S ribosome from T. thermophilus, tRNA at the A-site, tRNA at the P-site, mRNA, and the polyalanine were harmonically restrained using a force-constant of 10 kcal/mol Å2. The modeled parts of the 70S ribosome based on H. marismortui, the nonheavy atoms of the 70S ribosome and the nascent peptide, and all ions and water molecules were free to move. The average gradient of the potential energy at the final step of the minimization was 1.36 × 10−3 kcal/mol Å.
The MD simulation of the system was carried out for 3.5 ns at a constant pressure of one bar and a temperature of 350 K. The temperature of 350 K was chosen to match the optimal conditions for growth of T. thermophilus (43). The constant temperature and pressure algorithm developed by Martyna et al. (44) was used to control the temperature and pressure of the system. To integrate the equation of motion, the multi time step (MTS) algorithm developed by Zhou et al. (45) was used with a time step of 1 fs for short forces with the smooth cut-off of 8 Å and 2 fs for other long-range forces. Each system was first heated from 0 K to the 350 K within 500 ps during which the molecules and sodium ions were fixed with decreasing restraints and the water molecules were allowed to move. After the restraints were removed, the system was equilibrated for 1 ns with no restraint. During the equilibration the weights of barostat and thermostat were set at 105 ps2 · kcal/mol so that the relaxation times for the temperature and pressure control were both ∼1 ps. After equilibration, the weights were set at 109 ps2 · kcal/mol so that the relaxation times were 100 ps. These values make the coupling to the temperature and pressure baths weak enough to avoid any significant artificial effects on the atomic properties of the system. The 2-ns MD trajectory, from 1.5 ns to 3.5 ns, was analyzed. The coordinates of all the atoms including those of water and hydrogen atoms were collected every 1 ps for later analysis.
Steered MD simulations
Spontaneous movement of the polyalanine through the exit tunnel cannot be observed within several nanoseconds as the rate of elongation of the nascent polypeptide is ∼20 amino acids per second in vivo (46). To observe the movement of the polyalanine through the exit tunnel, we employed steered MD (SMD) simulations (39) by imposing a biased force on an atom of the polyalanine, to pull the nascent polyalanine along the tunnel. The purpose of applying SMD is to determine which of the two paths, case-over (over Arg92), or case-under (under Arg92), is the true path of the polyalanine.
During the SMD simulation, the pulling force exerted on the Cα atom was
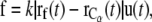 |
(2) |
where k is the force constant and 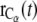 is the coordinate of the selected Cα atom at time t, the time elapsed from the beginning of the SMD simulation.
is the coordinate of the selected Cα atom at time t, the time elapsed from the beginning of the SMD simulation. 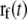 is the coordinate on which the center of the harmonic potential to produce the pulling force is located:
is the coordinate on which the center of the harmonic potential to produce the pulling force is located:
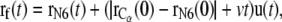 |
(3) |
where v is the constant velocity, and 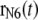 is the coordinate of the N6 atom of Ade76 of tRNA at the P-site.
is the coordinate of the N6 atom of Ade76 of tRNA at the P-site. 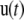 is the unit vector from the N6 atom in the direction of
is the unit vector from the N6 atom in the direction of 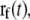 and is described as
and is described as
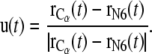 |
(4) |
In our SMD simulations, the value of v was set at 10 Å/ns (106 μm/s) to move the polyalanine along the tunnel slowly. This speed is significantly slower than the diffusion of the water molecules in the tunnel (see Results). The value of k was set at 10 pN/Å, corresponding to the root mean-square fluctuation, equal to 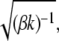 of 2.2 Å at 350 K. The force is sufficiently weak to allow the Cα atom to fluctuate in directions other than that of the applied force. This would enable the polyalanine to move along the curved tunnel naturally. The six rotational and translational motions of the ribosome in the simulation box were set to zero every 1 ps.
of 2.2 Å at 350 K. The force is sufficiently weak to allow the Cα atom to fluctuate in directions other than that of the applied force. This would enable the polyalanine to move along the curved tunnel naturally. The six rotational and translational motions of the ribosome in the simulation box were set to zero every 1 ps.
SMD simulations were carried out for 4 ns after the equilibration period for the free MD simulations. In our SMD simulations, a Cα atom of the polyalanine was pulled. To select the atom to be pulled, the Cα atom of L4 and the Cα atom of L22 closest to each other at the start of the SMD simulation were identified. The Cα atom to be pulled satisfied the following criteria: 1), it was >10 Å from both the Cα atoms on L4 and L22; 2), it was the closest Cα atom to the Cα atoms on L4 and L22; and 3), it was on the exit side of the line joining the Cα atoms of L4 and L22. The tunnel expands outward rapidly from the place around the selected Cα atoms, thus, a distance of 10 Å would prevent the L4-L22 constriction from being excessively disturbed when the selected Cα atom in the polyalanine was pulled. The selected Cα atom was pulled from the N6 atom of Ade76 of tRNA at the P-site in the direction of this selected Cα atom.
Visualization of the location of the L4-L22 constriction and the polyalanine in the tunnel
To visualize the mobile water around the L4-L22 constriction and the polyalanine in the tunnel, the diffusion of water molecules in the tunnel was calculated. To calculate the diffusion constant of the water molecules, the space of the system was divided into cells. The mean-square displacement of the ith water molecule at a cell coordinate, 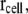 at the center of each cell can be calculated as
at the center of each cell can be calculated as
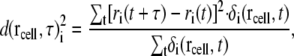 |
(5) |
where ri(t) is the coordinate of the ith water molecule at a given time t. The term within the square brackets is the square of the displacement of the ith water molecule during a time interval τ.  is 1 if the ith water molecule is within the cell at rcell at a given time t; otherwise, it is 0. The average mean-square displacement at a given cell coordinate is then
is 1 if the ith water molecule is within the cell at rcell at a given time t; otherwise, it is 0. The average mean-square displacement at a given cell coordinate is then
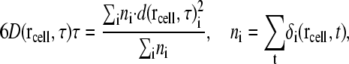 |
(6) |
where ni is the number of times the ith water molecule was found in the cell at rcell in the saved MD trajectories, and D is the diffusion constant of the water molecules. To calculate the diffusion constant, we set the length of each cell and the interval time τ at 0.5 Å and 100 ps respectively. The positions of the water molecules were measured by best-fitting the atoms of ribosome that are within 50 Å of the tip of the side chain of Arg92 of L22 with those of the initial structure.
PCA and domain motion analysis: influence on tunnel conformation
To investigate the influence of the global dynamics of the 70S ribosome on the dynamics of the tunnel, a principal component analysis (PCA) of the atomic fluctuations (47–50) was carried out to analyze the large-scale collective motions of the 70S ribosome for 2 ns for both case-over and case-under. The covariance matrix was calculated from the MD trajectory using the phosphorus atoms of all the ribosomal RNA and the two tRNA molecules, and the Cα atoms of all the ribosomal proteins. The order of the covariance matrix was 33,162. The covariance matrix was diagonalized to identify the most significant motions of the solute along the corresponding eigenvectors.
DynDom3D
DynDom (51,52) is able to determine dynamic domains, hinge axes, and hinge-bending residues from two protein structures that have different conformations. DynDom generates short segments of the amino-acid chains of these proteins by use of a sliding window and the calculation of the rotation vector associated with the rotation of these segments between the two structures. By treating the components of these rotation vectors as coordinates in a “rotation space”, segments that rotate together will have rotation points colocated, indicating possible rigid domains within the structure. Thus domains can be identified from the distribution of rotation points. The original DynDom, however, can only analyze single protein chains; it cannot analyze nucleotides or supra-molecules comprising two or more biomolecules. Consequently, we have used a new program, DynDom3D (manuscript in preparation), which was specifically developed to deal with supra-molecules. DynDom3D samples atoms using a sliding 3D block. Unlike the original DynDom, DynDom3D is blind to atomic bonding and atom type making it suitable for the analysis of multimeric complexes of any type of constituent molecule.
The input parameters of the DynDom3D program were set as follows: the maximum number of clusters was 5, the grid size for determining the rotation of the block was 7 Å, the block factor was 4, the occupancy was 0.3, and the minimum ratio of external to internal displacement was 0.4. The ratio of internal to external displacement determines the acceptance criterion for a given domain pair. This low value was required due to noise often seen from MD results. The minimum size for a dynamic domain was set to 10,000 atoms as we are not interested in local motions of small regions.
Computational resources
All the MD and SMD simulations covering a total time of 20 ns were carried out on 128 Itanium-2 processors of the Altix 3700 Bx2 supercomputer of Japan Atomic Energy Agency and 128 vector processors of the Earth Simulator of the Japan Agency for Marine-Earth Science and Technology. More than 300,000 CPU hours were used.
RESULTS
MD simulation with no polyalanine present
Structure of L4-L22 loops
A snapshot of a part of the structure around the L4-L22 constriction at 1 ns is shown in Fig. 2 A. L4 and L22 have extended loops around residues 56–85 in L4 and residues 80–99 in L22 respectively. The extended loop of L4 can be described as having two tips, one at residues 60–68, and the other at residues 72–79.
FIGURE 2.

Snapshot of the conformations around the L4-L22 constriction at 1ns of the free MD simulation. Residues 56–85 of L4 and 80–99 of L22 that form extended loops are shown in dark khaki and coral. They are depicted as a ribbon . Phe61, Gln67, Asp74 of L4, Arg92 of L22, Ade2059 of the 23S ribosomal RNA and Ade76 of tRNA at the P-site are shown in blue, cyan, red, yellow, deep pink and black, respectively. They are all depicted as wire models. The polyalanines are depicted as backbone models. (A) Case without the polyalanine (B) In case-under, Ala15 and Ala18–20 of the polyalanine are shown in red and blue, respectively. Other residues of the polyalanines are shown in black. (C) Superposition of the conformations in the case without the polyalanine and case-under. In the case without the polyalanine, L4 and L22 are shown in white, and residues are depicted as thin wire models. Color coding is the same as that used in (B and C). The view is from the PTC in the direction of the tunnel. The yellow and blue arrows show the large movements of Arg92 of L22 and Phe61 of L4, respectively. To keep the image clear, the polyalanines have not been included. (D) In case-over, Ala19 and Ala25–27 of the polyalanine are shown in red and blue, respectively. The figure was illustrated using Chimera (59).
Fig. 2 A shows that the extended loop of L22 and its Arg92 that protrudes into the tunnel extends toward L4. The side chain of Arg92 blocks the tunnel and separates it into two passages, one that passes over and one that passes under Arg92. Water-mediated interactions exist between Arg92 of L22 and Phe61 of L4. However, this water-mediated interaction is not very strong as the tip of the side chain of Arg92 often swayed between Gln67 and Asp94, and interacted with them via other bridging water molecules.
Fig. 3 A shows a snapshot of a part of the L4-L22 constriction at 1 ns, in the large tunnel where water molecules have diffusion constants >1.67 × 10−5 cm2/s (meaning that after 100 ps, the water molecules had moved an average of 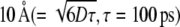 ). This value of 1.67 × 10−5 cm2/s was ∼25% of the diffusion constant of the bulk water at 350 K. The diameter of the high diffusion region over and under the side chain of Arg92 of L22 was >6 Å, large enough for the modeled polyalanine to pass over or under the side chain of Arg92.
). This value of 1.67 × 10−5 cm2/s was ∼25% of the diffusion constant of the bulk water at 350 K. The diameter of the high diffusion region over and under the side chain of Arg92 of L22 was >6 Å, large enough for the modeled polyalanine to pass over or under the side chain of Arg92.
FIGURE 3.

Location of the L4-L22 constriction and the polyalanine in the tunnel. Regions of the high diffusion constant of water molecules (>1.67 × 10−5 cm2/s) in the exit tunnel are represented. L4 and L22 are depicted as ribbon model. The diffusion constant was calculated as in Eq. 6. The polyalanine is shown in green and depicted in backbone model. (A) Case without the polyalanine. (B and C) cases over and under. The figure was illustrated using Chimera (59).
MD simulation with polyalanine under loop (case-under)
Structure of L4-L22 loops
A snapshot of a part of the L4-L22 constriction and the polyalanine at 1 ns is shown in Fig. 2 B. The interactions between Phe61, Gln67, and Asp74 of L4 and Arg92 of L22, which were observed in the case without the polyalanine, were not observed. Interestingly, the tip of the side chain of Arg92 moved ∼8 Å from its initial position, and repositioned to interact with Ade2059 of 23S rRNA during equilibration. As Ade2059 and the neighboring nucleotide Ade2058 are well known to be important for interacting with some antibiotics such as erythromycin (8,53), this may imply that Arg92 of L22 also plays an important role in determining the way these antibiotics bind to the entrance of the tunnel. As Arg92 of L22 moved from its initial position, the side chain of Phe61of L4 also flipped to widen the tunnel and interacted with residues 20–25 of the polyalanine, as shown in Fig. 2 C.
Steered MD
Following the procedure in Materials and Methods, the Cα atom of residue 15 was selected to be pulled. Residue 15 is shown in red, in Fig. 2 B.
The amplitude of the pulling force was monitored at each time step and is shown in Fig. 4 A. Fig. 4 A shows that the pulling force gradually increases up to ∼250 pN. This is comparable to forces used in atomic force microscopy experiments.
FIGURE 4.

Results of SMD. (A) The amplitudes of the pulling force are plotted against the time. The unit of the force is pN. The straight and broken lines represent in case-over and case-under, respectively. (B and C) Distances moved along the exit tunnel are plotted against the time. The vertical axis shows the distance of the four Cα atoms of the polyalanine measured by projecting their positions onto the line passing the N6 atom of Ade76 of tRNA at the P-site and the middle point of the line joining the Cα atom of Asp74 of L4 and that of Arg92 of L22 at the starting time of the SMD simulation. The origin of the vertical axis is the middle point. The units of the distance and time are Å and ps, respectively. (B) Case-under, distance moved along the exit tunnel for the pulled Cα atom of residue 15 and the Cα atoms of residues 18–20 of the polyalanine are plotted against the time with broken and straight lines, respectively. Residue 15 and residues 18–20 are shown in red and blue in Fig. 2 B, respectively. (C) Case-over, distance moved along the exit tunnel for the pulled Cα atom of residue 19 and the Cα atoms of residues 25–27 of the polyalanine are plotted against the time with broken and straight lines, respectively. Residue 19 and residues 25–27 are shown in red and blue in Fig. 2 D, respectively.
The movement of the polyalanine from its initial position in the direction of the exit tunnel is shown in Fig. 4 B. To show the movement of the polyalanine around the L4-L22 constriction clearly, the movements of only the pulled Cα atom of residue 15 and three Cα atoms of residues 18–20 are shown in Fig. 4 B. Representative points of these three residues are shown in blue in wire model in Fig. 2 B. The direction of the movement along the exit tunnel was from the N6 atom of Ade76 of tRNA at the P-site to the middle point of the line joining the Cα atom of Asp74 of L4 and that of Arg92 of L22. Displacements were measured from the middle point.
Fig. 4 B shows that in case-under, the pulled Cα atom started moving as soon as the force was applied. The average displacement of the Cα atoms of residues 18–20 around the L4-L22 constriction along the tunnel during 0–3 ns was 5.2 Å. As a result, two Cα atoms of residues 19 and 20 crossed the line between the Cα atoms of Asp74 of L4 and Arg92 of L22 within 3 ns. This indicates that the polyalanine could move through the L4-L22 constriction.
Fig. 3 B shows a snapshot of a part of the L4-L22 constriction and polyalanine at 3 ns of the SMD simulation, in the high diffusion region for water molecules with a diffusion constant of at least 1.67 × 10−5 cm2/s. The high diffusion region spans the L4-L22 constriction connecting the PTC and the exit.
MD simulation with polyalanine over loop (case-over)
Structure of L4-L22 loops
A snapshot of a part of the L4-L22 constriction and the polyalanine at 1 ns is shown in Fig. 2 D. In contrast to case-under, strong interactions among Gln67, Phe61, and Asp74 at the tips of L4, and Arg92 of L22 were maintained during the MD simulations.
Steered MD
Following the procedure in Materials and Methods, the Cα atom of residue 19 was selected to be pulled. Residue 19 is shown in red, in Fig. 2 D.
The amplitude of the pulling force was monitored at each time step and is shown in Fig. 4 A. Fig. 4 A shows that the pulling force gradually increases up to ∼250 pN. The increment of the pulling force in case-over is larger than that in case-under. This indicates that the pulled atom in case-under moved more easily than that in case-over.
The movement of the polyalanine from its initial position in the direction of the exit tunnel is shown in Fig. 4 C. To show the movement of the polyalanine around the L4-L22 constriction clearly, the movements of only the pulled Cα atom of residue 19 and three Cα atoms of residues 25–27 are shown in Fig. 4 C. Representative points of these three residues are shown in blue in wire model in Fig. 2 D. Fig. 4 C shows that all the residues basically stayed in the same positions during 0–1 ns, indicating that the structure of the polyalanine around residues 19–27 was stable. The pulled Cα atom then started to move at ∼1 ns. Interestingly, as the pulled Cα atom moved rapidly in the direction of the exit at ∼1.2 ns, residues 25–27 around the L4-L22 constriction moved in the opposite direction as if they resisted moving with the pulled Cα atom. This indicates that the structure between the pulled Cα atom and residue 25 was stretched due to the resistance of residues 25–27, and residues 25–27 went backward to release the stress accumulated at the L4-L22 constriction due to its having been dragged along with residues 25–27. This phenomenon was observed again at ∼3 ns. As a result, the average displacement of the Cα atoms of residues 25–27 along the tunnel during 0–3 ns was negative (−1.4 Å). These results indicate that the polyalanine was not able to move through the L4-L22 constriction at all.
These results contrast with those of case-under where the polyalanine moved 5–10 Å with a lower average force. Consequently, out of the two paths determined by modeling, case-under seems the likely path for elongation of the polyalanine. It is possible that case-under represents an open-gate conformation, and case-over a closed-gate conformation (see Discussion).
Fig. 3 C shows a snapshot of a part of the structure around the L4-L22 constriction and the polyalanine at 3 ns of the SMD simulation in a high diffusion region for water molecules with a diffusion constant of at least 1.67 × 10−5 cm2/s. The high diffusion region in case-over is smaller than in case-under, and it does not form a connected region. (They are connected under the threshold of 1.20 × 10−5 cm2/s.) This might be because the L4-L22 constriction is closed, hindering the movement of water molecules.
Global motions in the ribosome and effect on tunnel conformation
It has been suggested that the conformational change of the 70S ribosome may be accompanied by changes in tunnel conformation (6), such as would be the case for a peristaltic pump facilitating the movement of the polyalanine along the tunnel. However, Voss et al. (16) and Stietz (17) have concluded that the tunnel is rigid.
We carried out principal component analysis on the free MD simulations and a subsequent conformational analysis to determine whether global motions affect the tunnel conformation. It has been shown that a large portion of the overall fluctuations of a macromolecule can often be accounted for by a small number of eigenvectors having the largest eigenvalues and that these modes are sometimes related to the function of the macromolecule (54). In both case-under and case-over, fluctuation in first principal component eigenvector (PC1) was significantly larger than in the other principal components. The ratio of the first eigenvalue to the second eigenvalue was >3 (data not shown). However, due to the size and complexity of the 70S ribosome it is difficult to interpret this conformational change, even when it is restricted to a single principal component. Therefore, we used domain movement analysis software, DynDom3D (see Materials and Methods), to analyze the movement along PC1.
Polyalanine present (case-under)
Fig. 5 A shows the dynamic domains of the 70S ribosome of the first PC in case-under. DynDom3D classified the dynamic domains into three parts: domains I and III included a large part of 50S and the head part of 50S, respectively. Domain II comprised a large part of 30S. The axis of domains I and II was located at the boundary between 50S and 30S and passed near to L1 in domain I, L2, S11, S13 in domain II, and L5 in domain III. This indicates that when the space between 50S and 30S at the A-site opens, the space between 50S and 30S at the E-site closes. This opening and closing motion would facilitate the incorporation and emission of the tRNA molecules. Domain III rotates against domain I as well as domain II. Domain III included most of L5, L18, L21, L25, L27, L30, and 5S RNA of 50S. The axis of domains I and III passed nearby L27 in domain III and L22 and L34 in domain I. This motion seems to be similar to the motion of the 70S ribosome of mode 5 analyzed by elastic network model (24). (Animation of this mode is available at http://ribosome.bb.iastate.edu/70SnKmode.)
FIGURE 5.

Dynamic domains analyzed by DynDom3D. Each dynamic domain is colored. The regions that were not assigned to a dynamic domain are shown in green. tRNA at the P-site and the other molecules are depicted as space-filling and wire models. (A) The first mode in case-under: the opening and closing motion of 50S and 30S. The dynamic domains are composed of domain I (a large part of 50S including L7/L12 stalk and the body-part (the T-loop, T-stem, D-loop, and D-stem) of tRNA at the A-site and the P-site shown in blue), domain II (a large part of 30S, L1 stalk, and the anticodon stem and anticodon loop of tRNA at the A-site and the P-site shown in red), domain III (L5, L18, L21, L25, L27, L30, and 5S RNA of 50S, and nucleotides Gua15–Gua22 and Ura50–Ade64 of tRNA at the P-site shown in yellow). The axis for domains I and II is depicted as an arrow in blue with a tip in red, whereas the axis for domains I and III is depicted as an arrow in blue with a tip in yellow. (B) The first mode in case-over: ratchet-like movement of 50S and 30S. The dynamic domains are composed of domain I (a large part of 50S including L7/L12 stalk and region of the body-part of tRNA molecules at the P-site and the A-site shown in blue), domain II (a large part of 30S, L1 stalk, and the anticodon stem and anticodon loop of tRNA at the A-site and the P-site shown in red). The axis for domain I and domain II is depicted as an arrow in blue with a tip in red.
Polyalanine present (case-over)
Fig. 5 B shows the dynamic domains of the 70S ribosome of the first PC in case-over. DynDom3D classified the dynamic domains into two parts: domains I and II. Domains I and II included a large part of 50S and 30S, respectively. The axis of domains I and II passed near S12 in 30S, the acceptor stem and anticodon loop of tRNA at the A-site, the intersubunit bridges B2a, B3 at the boundary between 50S and 30S, the L4-L22 constriction, and L24 in 50S. B2a is composed of helix44 of 16 rRNA (h44) and helix69 of 23S and 5S rRNA (H69), and B3 is composed of h44 and H71 (hereafter, “h” will be used for the helices of 16S rRNA and “H” will be used for those of 23S and 5S rRNA.) The movement analyzed by DynDom3D seems to correspond with a so-called ratchet-like motion of the 30S and 50S subunits. This movement was observed by cryo-EM as a functional movement between the free state and the binding state of elongation factor G (55). This ratchet-like movement has also been observed by elastic network models (23,24), and a coarse-grained MD simulation (25).
The differences in conformation of the polyalanine and the L4-L22 constriction between case-under and case-over may not be large enough to influence the global dynamics of the system. It is likely therefore that the difference between PC1 in these two cases is due to a lack of sampling in this very large system within a 2-ns time period. However, the movements seen in PC1 are functionally plausible in that one corresponds to the ratchet-like movement (55) and the other to an open and closing movement between 50S and 30S.
In both cases domain I includes the L7/L12 stalk that comprises L7/L12, L11, and H42-44 in 23S rRNA. The L7/L12 stalk is considered to interact with initiation, elongation and termination factors (56). In both cases domain II includes the L1 stalk that comprises L1 and H76–H78. This is consistent with cryo-EM studies (57) and a coarse-grained MD simulation (25) that have shown that the rotation of 30S is accompanied by the movement of the L1 stalk, The movement of the L1 stalk is also considered to be involved in the release of tRNA at the E-site and to facilitate the translational movement of tRNA from the P-site to the E-site (22).
Interestingly in case-under, the tRNA at the P-site spans all three domains, and in case-over it spans both domains. This suggests that translocation of tRNA is related to these global movements. However, in both case-under and case-over the tunnel is situated within a single domain (domain I) spanning a large part of 50S that indicates that there is no twist or peristaltic motion in the tunnel. The fact that both of the axes of domains I and III in case-under and the axis of domains I and II in case-over pass near the region of the tunnel implies that the motion of the tunnel is small. Indeed, the mass-averaged root mean square of the atomic fluctuations (RMSF) of the heavy atoms within 10 Å of the polypeptide were very small, 1.0 Å and 0.96 Å and in case-under and case-over, respectively. Therefore, our results do not support the case for a peristaltic pump facilitating the movement of the polypeptide along the tunnel but favor the interpretation that the tunnel is rigid (16,17). However, our results cannot rule out the possibility that a large conformational change of the tunnel could occur during the entire reaction cycle that is not covered by our simulations.
DISCUSSION
Comparison with structures determined by x-ray crystallography
From our results case-under clearly represents the polyalanine in the state of elongation. What could case-over represent? It is possible that it is a state of elongation arrest as the polyalanine is unable to move through the L4-L22 constriction possibly due to the Arg92 of L22 interacting strongly with L4. This could be similar to the elongation arrest of a polypeptide with special sequences such as SecM or TnaC, where it has been suggested that two arginines on L22 block the tunnel in D. radiodurans ribosome (14).
In D. radiodurans ribosome, a large conformational change in the L4-L22 constriction between its open and closed states was actually observed by x-ray crystallography. Structural studies of D. radiodurans 50S (D50S) by Berisio et al. (14) have shown that a macrolide antibiotic, troleandomycin (TAO), binds to Ade2041 (that corresponds to Ade2058 in T. thermophile 70S ribosome (T70S) in our study) of the RNA wall of the exit tunnel and flips the β-hairpin of L22 from a region around Ade2041 to block the tunnel. They also speculated that the SecM and TnaC sequences may act like TAO and induce the extended loop of L22 to block the tunnel to prevent elongation. In D50S, Arg109 and Arg111 of L22 (that correspond to Arg88 and Arg90 of L22 in T70S in our study) are considered to form a “double hook” that switches between the open and closed states of the tunnel (14).
Taking this into account it is possible that in T70S the side chain of Arg92 of L22 plays the role of a “switch” that opens and closes the exit tunnel at the L4-L22 constriction. If this mechanism can be applied to the double-hook of D50S, when an antibiotic such as TAO comes near Ade2059, the side chain of Arg92 of L22, interacting with Ade2059 during the elongation of a polypeptide through the tunnel, as in case-under, would flip from the open position to the closed position, as in case-over. This conformational change would block the tunnel and induce elongation arrest of the nascent polypeptide.
We infer that the basic mechanism of opening and closing the L4-L22 constriction in D. radiodurans and T. thermophile ribosome is similar. However, several differences in the conformational change of the L4-L22 constriction in D50S and T70S should be noted: 1), the native conformation without the polypeptide in the tunnel is open in D50S, but closed in T70S; and 2), the conformational change of the L4-L22 constriction between the open and closed states was only observed at a small region around Asp92 of L22 in T70S in our study, whereas in D50S a large region including residues 105–107 and 113–115 (that correspond to residues 84–86 and 92–94 in T70S) was involved in the conformational change of the L4-L22 constriction.
Influence of global motions of the 70S ribosome on the conformation of the tunnel
PCA showed that the main modes of motion are global motions mainly involving the relative movement of the 50S and 30S subunits. These motions may be important for the translocation of tRNA molecules between 50S and 30S, but did not show any conformational changes within the tunnel such as a peristaltic pump to facilitate the movement of the polypeptide.
How does the polypeptide leave the ribosome through the exit tunnel? It is unlikely that the newly added carboxyl terminus at the PTC simply pushes the preceding part of the polypeptide, because the flexible parts of the polypeptide would fold and clog up the tunnel. We speculate that the driving force for the nascent polypeptide to move along the tunnel may come from an external force derived not from the dynamics of the tunnel itself but from a static property of the tunnel such as a gradual decrease of the electrostatic potential inside the tunnel (58), or an entropic force that is derived from the flexibility of the polypeptide. Mobile water molecules surrounding the polypeptide may assist on its smooth movement through the tunnel.
CONCLUSION
Modeling of a nascent polypeptide in the tunnel of the 70S ribosome in the crystal form has shown that the extended loops of L4 and L22 partially hinder the passage of the polypeptide. With 50S on top and 30S underneath, modeling revealed two possible paths for the polypeptide: one over Arg92 of L22 and one under Arg92. Our simulations of the 70S ribosome revealed the likely path of nascent polypeptide in the exit tunnel to be under Arg92 of L22. Our results suggest that the tunnel is closed by the Arg92-L4 interaction before the reaction of elongation of the polypeptide and that the tunnel leads the entering nascent polypeptide from the PTC to the passage passing under Arg92, causing it to switch to an open position. Subsequently, the L4-L22 constriction would open the passage for the polypeptide to enable it to move along the tunnel. In particular, the conformational change of the side chain of Arg92 seems to play the role of a gate, opening and closing the exit tunnel at the L4-L22 constriction.
PCA showed that within the timescale of several nanoseconds, the main modes of motion are global motions mainly involving the relative movement of the 50S and 30S subunits. These motions may be important for the translocation of tRNA molecules between 50S and 30S, but do not show any conformational changes within the tunnel that facilitate the movement of the polypeptide such as would be the case for a peristaltic pump.
Acknowledgments
We are grateful to Dr. Kei Yura at Ochanomizu University for kindly modeling the structures of the nascent polypeptide in the 70S ribosome. We are also grateful to Guru Poornam at the University of East Anglia for helpful discussion regarding the DynDom3D software.
This research was supported by Strategic International Cooperative Program, Japan Science and Technology Agency (JST). The computations were done partially on the Earth Simulator of the Japan Agency for Marine-Earth Science and Technology (JAMSTEC). Dr. Hayward's contribution was partly supported by a Biotechnology and Biological Sciences Research Council (UK) grant (BB/C004124/1).
Editor: Angel E. Garcia.
References
- 1.Ogle, J. M., D. E. Brodersen, W. M. Clemens Jr., M. J. Tarry, A. P. Carter, and V. Ramakrishnan. 2001. Recognition of cognate transfer RNA by the 30S ribosomal subunit. Science. 292:897–902. [DOI] [PubMed] [Google Scholar]
- 2.Muth, G. W., L. Ortoleva-Donnelly, and S. A. Strobel. 2000. A single adenosine with a neutral pKa in the ribosomal peptidyl transferase center. Science. 289:947–950. [DOI] [PubMed] [Google Scholar]
- 3.Ban, N., P. Nissen, J. Hansen, P. B. Moore, and T. A. Steitz. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science. 289:905–920. [DOI] [PubMed] [Google Scholar]
- 4.Harms, J., F. Schluenzen, R. Zarivach, A. Bashan, S. Gat, I. Agmon, H. Bartels, F. Franceschi, and A. Yonath. 2001. High resolution structure of the large ribosomal subunit from a mesophilic Eubacterium. Cell. 107:679–688. [DOI] [PubMed] [Google Scholar]
- 5.Nissen, P., J. Hansen, N. Ban, P. B. Moore, and T. A. Steitz. 2000. The structural basis of ribosome activity in peptide bond synthesis. Science. 289:920–930. [DOI] [PubMed] [Google Scholar]
- 6.Gabashvili, I. S., S. T. Gregory, M. Valle, R. Grassucci, M. Worbs, M. C. Wahl, A. E. Dahlberg, and J. Frank. 2001. The polypeptide tunnel system in the ribosome and its gating in erythromycin resistance mutants of L4 and L22. Mol. Cell. 8:181–188. [DOI] [PubMed] [Google Scholar]
- 7.Jenni, S., and N. Ban. 2003. The chemistry of protein synthesis and voyage through the ribosomal tunnel. Curr. Opin. Struct. Biol. 13:212–219. [DOI] [PubMed] [Google Scholar]
- 8.Schlünzen, F., R. Zarivach, J. Harms, A. Bashan, A. Tocilj, R. Albrecht, A. Yonath, and F. Franceschi. 2001. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 413:814–821. [DOI] [PubMed] [Google Scholar]
- 9.Hansen, J. L., P. B. Moore, and T. A. Steitz. 2003. Structures of five antibiotics bound at the peptidyl transferase center of the large ribosomal subunit. J. Mol. Biol. 330:1061–1075. [DOI] [PubMed] [Google Scholar]
- 10.Nakatogawa, H., and K. Ito. 2002. The ribosomal exit tunnel functions as a discriminating gate. Cell. 108:629–636. [DOI] [PubMed] [Google Scholar]
- 11.Gong, F., and C. Yanofsky. 2002. Instruction of translating ribosome by nascent peptide. Science. 297:1864–1867. [DOI] [PubMed] [Google Scholar]
- 12.Tenson, T., and M. Ehrenberg. 2002. Regulatory nascent peptides in the ribosomal tunnel. Cell. 108:591–594. [DOI] [PubMed] [Google Scholar]
- 13.Cruz-Vera, L. R., S. Rajagopal, C. Squires, C. Yanofsky. 2005. Features of ribosome-peptidyl-tRNA interactions essential for tryptophan induction of TNA operon expression. Mol. Cell. 19:333–343. [DOI] [PubMed] [Google Scholar]
- 14.Berisio, R., F. Schluenzen, J. Harms, A. Bashan, T. Auerbach, D. Baram, and A. Yonath. 2003. Structural insight into the role of the ribosomal tunnel in cellular regulation. Nat. Struct. Biol. 10:366–370. [DOI] [PubMed] [Google Scholar]
- 15.Kramer, G., T. Rauch, W. Rist, S. Vorderwulbecke, H. Patzelt, A. Schulze-Specking, N. Ban, E. Deuerling, and B. Bukau. 2002. L23 protein functions as a chaperone docking site on the ribosome. Nature. 419:171–174. [DOI] [PubMed] [Google Scholar]
- 16.Voss, N. R., M. Gerstein, T. A. Steitz, and P. B. Moore. 2006. The geometry of the ribosomal polypeptide exit tunnel. J. Mol. Biol. 360:893–906. [DOI] [PubMed] [Google Scholar]
- 17.Steitz, T. A. 2008. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 9:242–253. [DOI] [PubMed] [Google Scholar]
- 18.Yusupov, M. M., G. Z. Yusupova, A. Baucom, K. Lieberman, T. N. Earnest, J. H. Cate, and H. F. Noller. 2001. Crystal structure of the ribosome at 5.5 Å resolution. Science. 292:883–896. [DOI] [PubMed] [Google Scholar]
- 19.Klein, D. J., P. B. Moore, and T. A. Steitz. 2004. The roles of ribosomal proteins in the structure, assembly and evolution of the large ribosomal subunit. J. Mol. Biol. 340:141–177. [DOI] [PubMed] [Google Scholar]
- 20.Schuwirth, B. S., M. A. Borovinskaya, C. W. Hau, W. Zhang, A. Vila-Sanjurjo, J. M. Holton, and C. J. H. Doudna. 2005. Structure of the bacterial ribosome at 3.5 Å resolution. Science. 310:827–834. [DOI] [PubMed] [Google Scholar]
- 21.Selmer, M., C. M. Dunham, F. V. Murphy IV, A. Weixlbaumer, S. Petry, A. C. Kelley, J. R. Weir, and V. Ramakrishnan. 2006. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 313:1935–1942. [DOI] [PubMed] [Google Scholar]
- 22.Korostelev, A., S. Trakhanov, M. Laurberg, and H. F. Noller. 2006. Crystal structure of a 70S ribosome-tRNA complex reveals functional interactions and rearrangements. Cell. 126:1065–1077. [DOI] [PubMed] [Google Scholar]
- 23.Tama, F., M. Valle, J. Frank, and C. L. Brooks III. 2003. Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA. 100:9319–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang, Y. M., A. J. Rader, I. Bahar, and R. L. Jernigan. 2004. Global ribosome motions revealed with elastic network model. J. Struct. Biol. 147:302–314. [DOI] [PubMed] [Google Scholar]
- 25.Trylska, J., V. Tozzini, and J. A. McCammon. 2005. Exploring global motions and correlations in the ribosome. Biophys. J. 89:1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanbonmatsu, K. Y., S. Joseph, and C. S. Tung. 2005. Simulating movement of tRNA into the ribosome during decoding. Proc. Natl. Acad. Sci. USA. 102:15854–15859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanbonmatsu, K. Y. 2006. Energy landscape of the ribosomal decoding center. Biochimie. 88:1053–1059. [DOI] [PubMed] [Google Scholar]
- 28.Sanbonmatsu, K. Y., and C. S. Tung. 2007. High performance computing in biology: multimillion atom simulations of nanoscale systems. J. Struct. Biol. 157:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tung, C. S., S. Joseph, and K. Y. Sanbonmatsu. 2002. All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nat. Struct. Biol. 9:750–755. [DOI] [PubMed] [Google Scholar]
- 30.Tung, C. S., and K. Y. Sanbonmatsu. 2004. Atomic model of the Thermus thermophilus 70S ribosome developed in silico. Biophys. J. 87:2714–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenner, L., P. Romby, B. Rees, C. Schulze-Briese, M. Springer, C. Ehresmann, B. Ehresmann, D. Moras, G. Yusupova, and M. Yusupov. 2005. Translational operator of mRNA on the ribosome: how repressor proteins exclude ribosome binding. Science. 308:120–123. [DOI] [PubMed] [Google Scholar]
- 32.Mueller, F., I. Sommer, P. Baranov, R. Matadeen, M. Stoldt, J. Wöhnert, M. Görlach, M. van-Heel, and R. Brimacombe. 2000. The 3D arrangement of the 23 S and 5 S rRNA in the Escherichia coli 50 S ribosomal subunit based on a cryo-electron microscopic reconstruction at 7.5 Å resolution. J. Mol. Biol. 298:35–59. [DOI] [PubMed] [Google Scholar]
- 33.Tu, D., G. Blaha, P. B. Moore, and T. A. Steitz. 2005. Structures of MLSbK antibiotics bound to the mutated large ribosomal subunits provide a structural explanation of resistance. Cell. 121:257–270. [DOI] [PubMed] [Google Scholar]
- 34.Schmeing, T. M., P. B. Moore, and T. A. Steitz. 2003. Structures of deacylated tRNA mimics bound to the E site of the large ribosomal subunit. RNA. 9:1345–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bashan, A., I. Agmon, R. Zarivach, F. Schluenzen, J. Harms, R. Berisio, H. Bartels, F. Franceschi, T. Auerbach, H. A. Hansen, E. Kossoy, M. Kessler, and A. Yonath. 2003. Structural basis of the ribosomal machinery for peptide bond formation, translocation, and nascent chain progression. Mol. Cell. 11:91–102. [DOI] [PubMed] [Google Scholar]
- 36.Pearlman, D. A., D. A. Case, J. W. Caldwell, W. S. Ross, I. Cheatham, T. E. S. DeBolt, D. Ferguson, G. Seibel, and P. A. Kollman. 1995. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput. Phys. Commun. 91:1–41. [Google Scholar]
- 37.Cheatham, T. E., P. Cieplak, and P. A. Kollman. 1999. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16:845–862. [DOI] [PubMed] [Google Scholar]
- 38.Auffinger, P., T. E. Cheatham III, and A. C. Vaiana. 2007. Spontaneous formation of KCl aggregates in biomolecular simulations: a force field issue? J. Chem. Theory Comput. 3:1851–1859. [DOI] [PubMed] [Google Scholar]
- 39.Isralewitz, B., M. Gao, and K. Schulten. 2001. Steered molecular dynamics and mechanical functions. Curr. Opin. Struct. Biol. 11:224–230. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 41.Ishida, H., M. Higuchi, Y. Yonetani, T. Kano, Y. Joti, A. Kitao, and N. Go. 2006. Analysis of the function of a large-scale supra-biomolecule system by molecular dynamics simulation system, SCUBA (Simulation Codes for hUge Biomolecular Assembly). Annual Report of the Earth Simulator Center April 2005–March 2006. 237–240.
- 42.Hockney, R. W., and J. W. Eastwood. 1981. Computer Simulation Using Particles. McGraw-Hill, New York.
- 43.Oshima, T., and K. Imahori. 1974. Description of Thermus thermophilus (Yoshida and Oshima) comb. nov., a nonsporulating thermophilic bacterium from a Japanese thermal spa. Int. J. Syst. Bacteriol. 24:102–112. [Google Scholar]
- 44.Martyna, G. J., M. E. Tuckerman, D. J. Tobias, and M. L. Klein. 1996. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 87:1117–1157. [Google Scholar]
- 45.Zhou, R., E. Harder, H. Xu, and B. J. Berne. 1996. Efficient multiple time step method for use with Ewald and particle mesh Ewald for large biomolecular systems. J. Chem. Phys. 87:1117–1157. [Google Scholar]
- 46.Kjeldgaard, N. O., and K. Gaussing. 1974. Regulation of biosynthesis of ribosomes. In Ribosomes. M. Nomura, et al., editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 369–392.
- 47.Ichiye, T., and M. Karplus. 1991. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 11:205–217. [DOI] [PubMed] [Google Scholar]
- 48.Kitao, A., F. Hirata, and N. Go. 1991. The effects of solvent on the conformation and the collective motions of protein: normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem. Phys. 158:447–472. [Google Scholar]
- 49.García, A. E. 1992. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 68:2696–2699. [DOI] [PubMed] [Google Scholar]
- 50.Amadei, A., A. B. Linssen, and H. J. Berendsen. 1993. Essential dynamics of proteins. Proteins. 17:412–425. [DOI] [PubMed] [Google Scholar]
- 51.Hayward, S., and H. J. C. Berendsen. 1998. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins. 30:144–154. [PubMed] [Google Scholar]
- 52.Hayward, S., and R. A. Lee. 2002. Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J. Mol. Graph. Model. 21:181–183. [DOI] [PubMed] [Google Scholar]
- 53.Tenson, T., M. Lovmar, and M. Ehrenberg. 2003. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 330:1005–1014. [DOI] [PubMed] [Google Scholar]
- 54.Berendsen, H. J. C., and S. Hayward. 2000. Collective protein dynamics in relation to function. Curr. Opin. Struct. Biol. 10:165–169. [DOI] [PubMed] [Google Scholar]
- 55.Frank, J., and R. K. Agrawal. 2000. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature. 406:318–322. [DOI] [PubMed] [Google Scholar]
- 56.Helgstrand, M., C. S. Mandava, F. A. A. Mulder, A. Liljas, S. Snayal, and M. Akke. 2007. The ribosomal stalk binds to translation factors IF2, EF-Tu, EF-G and RF3 via a conserved region of the L12 C-terminal domain. J. Mol. Biol. 365:468–479. [DOI] [PubMed] [Google Scholar]
- 57.Valle, M., A. Zavialov, J. Sengupta, U. Rawat, M. Ehrenberg, and J. Frank. 2003. Locking and unlocking of ribosomal motions. Cell. 114:123–134. [DOI] [PubMed] [Google Scholar]
- 58.Lu, J., W. R. Kobertz, and C. Deutsch. 2007. Mapping the electrostatic potential within the ribosomal exit tunnel. J. Mol. Biol. 371:1378–1391. [DOI] [PubMed] [Google Scholar]
- 59.Pettersen, E. F., T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng, and T. E. Ferrin. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612. [DOI] [PubMed] [Google Scholar]