

Abstract
Inactivation of voltage-gated Kv1 channels can be altered by Kvβ subunits, which block the ion-conducting pore to induce a rapid (‘N-type') inactivation. Here, we investigate the mechanisms and structural basis of Kvβ1.3 interaction with the pore domain of Kv1.5 channels. Inactivation induced by Kvβ1.3 was antagonized by intracellular PIP2. Mutations of R5 or T6 in Kvβ1.3 enhanced Kv1.5 inactivation and markedly reduced the effects of PIP2. R5C or T6C Kvβ1.3 also exhibited diminished binding of PIP2 compared with wild-type channels in an in vitro lipid-binding assay. Further, scanning mutagenesis of the N terminus of Kvβ1.3 revealed that mutations of L2 and A3 eliminated N-type inactivation. Double-mutant cycle analysis indicates that R5 interacts with A501 and T480 of Kv1.5, residues located deep within the pore of the channel. These interactions indicate that Kvβ1.3, in contrast to Kvβ1.1, assumes a hairpin structure to inactivate Kv1 channels. Taken together, our findings indicate that inactivation of Kv1.5 is mediated by an equilibrium binding of the N terminus of Kvβ1.3 between phosphoinositides (PIPs) and the inner pore region of the channel.
Keywords: ion channel, Kvβ, N-type inactivation, phospholipids, potassium
Introduction
Voltage-gated potassium (Kv) channels are essential for regulating resting membrane potential, repolarization of action potentials, pacemaking and neurotransmitter release. Kv channels are tetrameric complexes formed by coassembly of four identical or homologous α-subunits. Rapid N-type inactivation of Kv1 channels can result from binding of a single N-terminal hydrophobic, ‘inactivation ball' peptide of an α-subunit to the inner pore region of the channel complex (Hoshi et al, 1990). The inactivation ball of Shaker B (Kv1.1 of Drosophila) α-subunits is a random coil in aqueous solution (Lee et al, 1993), but forms a β-hairpin structure when exposed to a more hydrophobic environment (Lee et al, 1993; Fernandez-Ballester et al, 1995). There may be variation in how inactivation ball peptides interact with the inner pore as the NMR structures of the inactivation balls of Kv1.4 and Kv3.4 α-subunits are clearly different (Antz et al, 1997).
An alternative structural basis of N-type inactivation of Kv1 channels has been described. Fast inactivation can also be mediated by the N terminus of a Kvβ subunit (Rettig et al, 1994; Heinemann et al, 1996) that is tethered to the T1 domain of a Kv1 α-subunit. For example, Kvβ1, Kvβ2 and Kvβ3 subunits alter the activation and inactivation gating of Kv1.5 channels (Leicher et al, 1998). The inactivation of Kv1 channels is diversified by alternative splicing of the Kvβ1 gene, resulting in the isoforms Kvβ1.1, Kvβ1.2 and Kvβ1.3. The N terminus of Kvβ1 subunits was proposed to enter the pore of a Kv1 channel as an extended peptide (Zhou et al, 2001). In contrast, the N-terminal ball peptides of Kv α-subunits were proposed to form a compact hairpin structure that binds to the inner vestibule to occlude the pore (Antz et al, 1997; Antz and Fakler, 1998). As illustrated by comparison of the N-terminal regions of two Kvα and three Kvβ subunits in Figure 1A, there is no apparent sequence conservation for inactivation ball peptides.
Figure 1.

N-type inactivation of Kv1.5 by Kvβ1.3. (A) Alignment of the N termini of Kvβ isoforms and of N-type inactivating Kv3.4 and Shaker channels. (B) Kv1.5 currents during short and long voltage steps to +70 mV, illustrating slow time course of C-type inactivation. (C) Superimposed current traces in response to depolarizations applied in 10-mV increments to test potentials ranging from −70 to +70 mV for Kv1.5 alone, co-expressed with Kvβ1.3 or with a Kvβ1.3, which lacks the N-terminal amino acids 2–10.
Mutations in the N terminus of Kvβ or Kv1 subunits can prevent their ability to inactivate Kv channels. For example, deletion of 10 amino acids from the N terminus of Kvβ1.3 (Uebele et al, 1998) causes a loss of function as does the L7E mutation in Shaker B α-subunits (Hoshi et al, 1990). Cysteine residues at position 7 of Kvβ1.1 (Rettig et al, 1994), position 6 of Kv3.4 (Stephens and Robertson, 1995) or position 13 of Kv1.4 (Ruppersberg et al, 1991) confer a redox sensitivity to channel inactivation. The loss of function by L7E or L7R in Shaker B (Hoshi et al, 1990) can be mimicked by phosphorylation of Y8 that prevents formation of a functional hairpin structure (Encinar et al, 2002). In addition, N-type inactivation of Kv1.5/Kvβ1.3 channels is modulated by protein kinase C (Kwak et al, 1999) and inactivation of Kv1.1/Kvβ1.1 is antagonized by intracellular Ca2+ (Jow et al, 2004). However, the molecular mechanisms and structural basis of Kvα–Kvβ interactions that mediate these effects are poorly understood.
N-type inactivation of Kv3.4 alone or inactivation of Kv1.1 mediated by Kvβ1.1 are antagonized by PIP2 (Oliver et al, 2004). For Kv3.4, binding of PIP2 to residues R13 and K14 of the N terminus appears to mediate this effect (Oliver et al, 2004). Although all three Kvβ1 isoforms introduce N-type inactivation, they differ in inactivation kinetics, intracellular modulation and expression pattern. This diversity plus cellular regulation helps to tune K+ channels to serve specific function.
We recently identified residues in the pore region of Kv1.5 that interact with Kvβ1.3 (Decher et al, 2005). Blockade of Kv1.5 by drugs such as S0100176 and bupivacaine can be modified by Kvβ1.3. Accordingly, site-directed mutagenesis studies revealed that the binding sites for drugs and Kvβ1.3 partially overlap (Gonzalez et al, 2002; Decher et al, 2004, 2005). In the present study, we used a mutagenesis approach to identify the residues of Kvβ1.3 and Kv1.5 that interact with one another to mediate fast inactivation. We also examined the structural basis for inhibition of Kvβ1.3-mediated inactivation by PIP2. Taken together, our findings indicate that when dissociated from PIP2, the N terminus of Kvβ1.3 forms a hairpin structure and reaches deep into the central cavity of the Kv1.5 channel to cause inactivation. This binding mode of Kvβ1.3 differs from that found earlier for Kvβ1.1, indicating a Kvβ1 isoform-specific interaction in the pore cavity.
Results
Identification of residues important for Kv1.3 function using cysteine- and alanine-scanning mutagenesis
Wild-type (WT) Kv1.5 channels activate rapidly and exhibit almost no inactivation when cells are depolarized for 200 ms (Figure 1B, left panel). Longer pulses cause channels to inactivate by a slow ‘C-type' mechanism that results in an ∼20% decay of current amplitude during 1.5 s depolarizations to +70 mV (Figure 1B, right panel). At this potential, inactivation is monoexponential with a time constant of ∼0.8 s. Co-expression of Kv1.5 with Kvβ1.3 subunits introduces an additional, rapid component (‘N-type') of inactivation (Figure 1C). As reported previously (Uebele et al, 1998), Kvβ subunit-induced fast inactivation is eliminated when Kvβ1.3 is truncated by the removal of residues 2–10 (Kvβ1.3Δ2–10; Figure 1C).
To assess the importance of specific residues in the N terminus of Kvβ1.3 for N-type inactivation, we made individual mutations of residues 2–11 of Kvβ1.3 to alanine or cysteine and co-expressed these mutant subunits with Kv1.5 subunits. Alanine residues were substituted with cysteine or valine. Substitution of native residues with alanine or valine introduces or retains hydrophobicity without disturbing helical structure, whereas substitution with cysteine introduces or retains hydrophilicity. In addition, cysteine residues can be subjected to oxidizing conditions to favour crosslinking with another cysteine residue. Representative currents recorded in oocytes co-expressing WT Kv1.5 plus mutant Kvβ1.3 subunits are depicted in Figure 2A and B. Mutations at positions 2 and 3 of Kvβ1.3 (L2A/C and A3V/C) led to a complete loss of N-type inactivation (Figure 2A–D). A similar, but less pronounced, reduction of N-type inactivation was observed for A4C, G7C and A8V mutants. In contrast, mutations of R5, T6 and G10 of Kvβ1.3 increased inactivation of Kv1.5 channels (Figure 2A and B). The effects of all the Kvβ1.3 mutations on inactivation are summarized in Figure 2C and D. In addition, the inactivation of channels with cysteine substitutions was quantified by their fast and slow time constants (τinact) during a 1.5-s pulse to +70 mV in Figure 2E. In the presence of Kvβ1.3, the inactivation of Kv1.5 channels was bi-exponential. With the exceptions of L2C and A3C, cysteine mutant Kvβ1.3 subunits introduced fast inactivation (Figure 2E, lower panel). Acceleration of slow inactivation was especially pronounced for R5C and T6C Kvβ1.3 (Figure 2E, lower panel). The more pronounced steady-state inactivation of R5C and T6C (Figure 2A and B) was not caused by a marked increase of the fast component of inactivation (Figure 2E, upper panel).
Figure 2.

Scanning mutagenesis of the Kvβ1.3 N terminus. Superimposed currents elicited by depolarizations applied in 10-mV increments to test potentials ranging from −70 to +70 mV for Kv1.5 co-expressed with Kvβ1.3 containing either (A) alanine or (B) cysteine mutations as indicated. (C, D) Relative inactivation plotted as a ratio of steady-state current after 1.5 s (Iss) to peak current (Imax) for alanine/valine or cysteine point mutations of the Kvβ1.3 N terminus. A value of 1.0 indicates no inactivation; a value of 0 indicates complete inactivation. (E) Kinetics of inactivation for Kv1.5 and Kv1.5/Kvβ1.3 channel currents determined at +70 mV. Labels indicate cysteine mutations in Kvβ1.3. Upper panel: relative contribution of fast (Af) and slow (As) components of inactivation. Lower panel: time constants of inactivation. For (C–E), *P<0.05; **P<0.005 compared with Kv1.5 plus wild-type Kvβ1.3 (n=4–13).
Kv1.3 mutations change inactivation kinetics independent of intracellular Ca2+
Rapid inactivation of Kv1.1 by Kvβ1.1 is antagonized by intracellular Ca2+. This Ca2+-sensitivity is mediated by the N terminus of Kvβ1.1 (Jow et al, 2004), but the molecular determinants of Ca2+-binding are unknown. The mutation-induced changes in the rate of inactivation could potentially result from an altered Ca2+-sensitivity of the Kvβ1.3 N terminus. Application of the Ca2+ ionophore ionomycine (10 μM) for 3 min removed rapid inactivation of Kv1.1/Kvβ1.1 channels (Figure 3A). However, this effect was not observed when either Kv1.5 (Figure 3B) or Kv1.1 (Figure 3C) was co-expressed with Kvβ1.3 subunits. Thus, alternative splicing of Kvβ1 can alter its Ca2+-sensitivity.
Figure 3.

Ca2+-sensitivity of Kvβ1.1 versus Kvβ1.3. Currents were recorded at +70 mV under control conditions and after the addition of 10 μM ionomycine. (A) Ionomycine prevents N-type inactivation of Kv1.1 by Kvβ1.1. Elevation of intracellular [Ca2+] does not prevent Kvβ1.3-induced N-type inactivation of Kv1.5 (B) or Kv1.1(C).
Mutant Kv1.3 subunits that disrupt inactivation retain ability to alter voltage-dependent gating of Kv1.5 channels
We reported earlier that although mutation of specific residues in the S6 domain of Kv1.5 could disrupt N-type inactivation, these mutations did not alter the ability of Kvβ1.3 to cause shifts in the voltage dependence of channel gating (Decher et al, 2005). This finding suggests that WT Kvβ1.3 can bind to and affect Kv1.5 gating without blocking the pore. Can mutant Kvβ1.3 subunits that no longer induce fast N-type inactivation still cause shifts in the gating of Kv1.5? This question was addressed by comparing the voltage dependence of activation and inactivation of Kv1.5 when co-expressed with WT and mutant Kvβ1.3 subunits. WT subunits shifted the voltage required for half-maximal activation by −15 mV and the voltage dependence of inactivation by −11 mV (Figure 4A and B). Mutant Kvβ1.3 subunits retained their ability to cause negative shifts in the half-points of activation and inactivation, albeit to a variable degree (Figure 4A and B). These findings suggest that point mutations in the N terminus of Kvβ1.3, including those that eliminated N-type inactivation, did not disrupt co-assembly of Kvβ1.3 with the Kv1.5 channel.
Figure 4.

Shifts in voltage dependence of Kv1.5 gating induced by co-expression with wild-type and mutant Kvβ1.3 subunits. (A) Half-point (V1/2) for activation (left panels) and inactivation (right panels) of current determined for oocytes expressing Kv1.5 channels alone or when co-expressed with Kvβ1.3 subunits with alanine/valine substitutions of indicated residues. (B) Same analysis for cysteine substitutions. *P<0.05; **P<0.005 compared with Kv1.5 alone (n=4–11).
Interaction of PIP2 with R5 of Kv1.3
The most pronounced gain of Kvβ1.3-induced inactivation was observed after mutation of R5 or T6 to cysteine or alanine. To further explore the role of charge at position 5 in Kvβ1.3, R5 was substituted with another basic (K), a neutral (Q) or an acidic (E) amino acid. Introduction of neutral or negatively charged residues at position 5 accelerated Kvβ1.3-induced fast inactivation, without increasing the relative contribution of the fast component (Figure 5A and B). In contrast, the conservative exchange R5K did not alter inactivation (Figure 5B, lower panel). All of the R5 mutant Kvβ1.3 subunits retained their ability to cause negative shifts in the half-points of activation and inactivation (Figure 5C).
Figure 5.

Phosphoinositide-binding to R5 of Kvβ1.3 prevents N-type inactivation. (A) Superimposed current traces in response to depolarizations applied in 10-mV increments to test potentials ranging from −70 to +70 for Kv1.5 co-expressed with Kvβ1.3 subunits with indicated mutations of R5. (B) Kinetics of inactivation for Kv1.5 and Kv1.5/Kvβ1.3 channel currents determined at +70 mV (n=4–13). Labels indicate mutations of R5 in Kvβ1.3. Upper panel: relative contribution of fast (Af) and slow (As) components of inactivation. Lower panel: time constants of inactivation. (C) Half-points (V1/2) for activation (left panel) and inactivation (right panel) of current determined for oocytes expressing Kv1.5 channels alone or when co-expressed with WT or R5 mutant Kvβ1.3 subunits (n=4–13). For (B, C), *P<0.05; **P<0.005 compared with Kv1.5 alone. #P<0.05; ##P<0.005 compared with Kv1.5+Kvβ1.3 WT. (D) In vitro PIP2-binding assay of N-terminal Kvβ1.3 fragments. Average-binding data of GST alone and GST-Kvβ1.3 fusion proteins (amino acids 1–33) of WT and mutant subunits. Relative arbitrary units of PIP2-binding are shown for liposomes containing 5 mol% PI(4,5)P2. **P<0.01. (E) Fast inactivation of Kv1.5/Kvβ1.3 channels is removed by application of 10 μM PIP2 to the cytosolic side of an inside-out macropatch from Xenopus oocytes (left panel). Here, 10 μM PIP2 only partially antagonizes inactivation introduced by R5C Kvβ1.3 (right panel). (F) R5C mutation in Kvβ1.3 blunts the slowing of fast inactivation induced by a 15 s application of PIP2 to inside-out macro-patches (n=6–8). (G–J) Effect of PIPs on relative inactivation of current in inside-out macropatches plotted as a ratio of steady-state current after 1.5 s (Iss) to peak current (Imax). For all experiments n=4–7. (G) [PIP2]-dependent reduction of inactivation of Kv1.5/Kvβ1.3 channel current. (H) At 10 μM, PIP, PIP2 and PIP3 have equivalent effects on WT Kv1.5/Kvβ1.3 channel inactivation. (I) At 10 μM, PIP, PIP2 and PIP3 have similar but reduced effects on Kv1.5/R5C Kvβ1.3 channel inactivation. (J) Poly-lysine (polyK, 25 μg/ml) applied to cytosolic side of membrane enhances inactivation of Kv1.5/Kvβ1.3 channels (n=6). For (G–J) *P<0.05 compared with Kv1.5 alone by Student's t-test.
PIP2 antagonizes N-type inactivation of Kv3.4 by binding to its N-terminal residues R13 and K14 and immobilizing the inactivation ball (Oliver et al, 2004). PIP2 also removes fast inactivation of Kv1.1 channels by immobilization of Kvβ1.1 (Oliver et al, 2004). We tested whether the R5C or T6C mutations in Kvβ1.3 altered the binding affinity of PIP2 for the Kvβ1.3 N terminus (residues 1–33). A lipid-binding assay was performed using N-terminal Kvβ1.3 GST fusion proteins and fluorescently labelled liposomes with incorporated PI(4,5)P2, as described earlier (Soom et al, 2001). The PIP2-binding affinity (expressed as relative arbitrary units) of R5C or T6C N-terminal Kvβ1.3 fusion proteins was reduced compared with WT fusion proteins (Figure 5D). Specifically, the fluorescence signal was reduced by 90% for R5C and 41% for T6C Kvβ1.3. The reduction in PIP2-binding affinity for the T6C mutant might be caused by impairment of electrostatic PIP2 interaction with the neighbouring residue R5.
PIP2 (10 μM) eliminated Kvβ1.3-induced N-type fast inactivation of Kv1.5 measured in inside-out macro-patches from Xenopus oocytes (Figure 5E, left panel and 5F). In contrast, PIP2 only partially attenuated the fast channel inactivation mediated by R5C Kvβ1.3 (Figure 5E, right panel and 5F). PIP2 antagonized Kv1.5 inactivation by Kvβ1.3 in a concentration-dependent manner (Figure 5G). Attenuation of Kvβ1.3-induced inactivation was also observed for 10 μM PIP and PIP3 (Figure 5H). In contrast, inactivation was only partially antagonized by these phosphoinositides (PIPs) when Kv1.5 was co-expressed with R5C Kvβ1.3 (Figure 5I). Thus, the sensitivity of Kv1.5 channels to Kvβ1.3 is modulated by PIPs, and R5 is a key residue for this functional modulation.
Endogenous PIP2 modulates Kv1.5/Kv1.3 channels
Next, we tested whether a depletion of endogenous PIP2 can modulate inactivation of the Kv1.5/Kvβ1.3 channel complex. Kv1.5, Kvβ1.3 and the Gq-coupled α1A-adrenoreceptor were co-expressed in Xenopus oocytes. We have shown earlier that activation of α1A-adrenoreceptors by methoxamine triggers Gq-coupled inhibition of the K+ channel TASK-1 in oocytes (Putzke et al, 2007). Stimulation of the α1A-receptor with 1 μM methoxamine accelerated inactivation and led to a small but significant reduction in steady-state current amplitude of the Kv1.5/Kvβ1.3 channel complex. Currents were reduced by 10.5±1.9% (n=8). However, receptor stimulation might not be sufficient to globally deplete PIP2 from the plasma membrane of an Xenopus oocyte, especially if the channel complex and receptors are not adequately colocalized in the cell membrane, an argument used to explain why stimulation of several Gq-coupled receptors (bradykinin BK2, muscarinic M1, TrkA) did not cause the expected shift in the voltage dependence of HCN channel activation (Pian et al, 2007).
The Kv1.5/Kvβ1.3 channel complex expressed in Xenopus oocytes has a more pronounced inactivation when recorded from an inside-out macropatch (Figure 5E, left panel) as compared with two-electrode voltage-clamp recordings (Figure 1C, middle panel). Iss/Imax was significantly decreased from 0.40±0.02 (Figure 2C) to 0.24±0.04 (Figure 5G) in an excised patch. This effect might be partially explained by PIP2 depletion from the patch. Therefore, we performed inside-out macropatches from Xenopus oocytes and applied poly-lysine (25 μg/ml) to the inside of the patch to deplete PIPs from the membrane (Oliver et al, 2004). Poly-lysine enhanced the extent of steady-state inactivation, decreasing the Iss/Imax from 26.0±5.0 to 10.5±4.3% (Figure 5J). Taken together, these findings indicate that endogenous PIPs are important determinants of the inactivation kinetics of the Kv1.5/Kvβ1.3 channel complexes.
Co-expression of mutant Kv1.5 and Kv1.3 subunits
In an attempt to determine the structural basis of Kvβ1.3 interaction with the S6 domain of Kv1.5, single cysteine mutations were introduced into each subunit. Our previous alanine scan of the S6 domain (Decher et al, 2005) identified V505, I508, V512 and V516 in Kv1.5 as important for interaction with Kvβ1.3. Here, these S6 residues (and A501) were individually substituted with cysteine and co-expressed with Kvβ1.3 subunits containing single cysteine substitutions of L2–T6. Potential physical interaction between cysteine residues in the α- and β-subunits was assayed by changes in the extent of current inactivation at +70 mV (Figure 6).
Figure 6.

Co-expression of mutant Kv1.5 and Kvβ1.3 subunits. Upper panels are plots of current inactivation (expressed as % reduction in peak current) induced by a 1.5 s pulse to +70 mV. Lower panels show superimposed currents elicited by shorter (200 ms) pulses to potentials ranging from −70 to +70 mV for channels that altered inactivation the most. Possible interacting residues are indicated by arrows (for example, L2C of Kvβ1.3 with I508C Kv1.5 in (A)). For all experiments n=4–13.
N-type inactivation was eliminated when L2C Kvβ1.3 was co-expressed with WT Kv1.5 or mutant Kv1.5 channels with cysteine residues in pore-facing positions (Figures 2B and 6A). Co-expression of L2C Kvβ1.3 with I508C Kv1.5 slowed C-type inactivation, whereas C-type inactivation was enhanced when L2C Kvβ1.3 was co-expressed with V512C Kv1.5 (Figure 6A). For A3C Kvβ1.3, the strongest changes in inactivation were observed by mutating residues V505, I508 and V512 in Kv1.5 (Figure 6B). For A4C Kvβ1.3, the extent of inactivation was changed by co-expression with Kv1.5 subunits carrying mutations at position A501, V505 or I508 (Figure 6C). The pronounced inactivation observed after co-expression of R5C Kvβ1.3 with WT Kv1.5 was significantly reduced by the mutation A501C (Figure 6D). A501 is located in the S6 segment close to the inner pore helix. The strong inactivation of Kv1.5 channels by T6C Kvβ1.3 was antagonized by cysteine substitution of A501, V505 and I508 of Kv1.5 (Figure 6E). Taken together, these data suggest that R5 and T6 of Kvβ1.3 interact with residues located in the upper S6 segment of Kv1.5, whereas L2 and A3 apparently interact with residues in the middle part of the S6 segment.
Mutations located deep in the pore of Kv1.5 reduce R5C Kv1.3 gain of function
To investigate further whether R5 of Kvβ1.3 interacts with specific residues in the upper region in S6 of Kv1.5, we co-expressed R5C Kvβ1.3 with Kv1.5 subunits harbouring single alanine mutations. In addition, R5C Kvβ1.3 was paired with T480A or A501V Kv1.5 subunits (Figure 7A). These co-expression studies confirmed the putative interaction between R5 and A501, identified with the A501C mutation (Figure 6D) and revealed potential interactions of R5 with V505 and T480 (Figure 7A). We also studied several double alanine mutations in S6 (V505A/I508A, V505A/V512A and I508A/V512A). The double mutation V505A/I508A eliminated N-type inactivation induced by R5C (Figure 7B), whereas the other double mutations had no discernable effect on fast inactivation (Figure 7B and C). Thus, the enhanced N-type inactivation induced by R5C Kvβ1.3 was only attenuated by single-residue mutations when they were located in the upper S6 segment or the pore helix (e.g. V505, A501 and T480; Figure 7A and C). The reduction in inactivation was the more pronounced the deeper the mutations were located in the pore cavity (Figure 7C).
Figure 7.

Arginine 5 of Kvβ1.3 interacts with residues near the selectivity filter of Kv1.5. (A) Traces for mutant channel currents elicited by pulses to potentials ranging from −70 to +70 mV, applied in 10-mV increments. Labels indicate point mutations of pore-facing residues in Kv1.5. All were co-expressed with R5C Kvβ1.3. Mutations A501V and T480A strongly reduce inactivation by R5C Kvβ1.3. (B) Current traces for channels with indicated double mutations of Kv1.5 and co-expressed with R5C Kvβ1.3. Scale bars represent 0.1 μA and 100 ms, respectively. (C) Inactivation (expressed as % reduction in peak current for 200 ms pulse to +70 mV) of WT and indicated mutant Kv1.5/R5C Kvβ1.3 channels. The ability of R5C Kvβ1.3 to induce inactivation is blunted when residues located high in the inner cavity of Kv1.5 are mutated. For all experiments, n=4–13.
Co-expression of WT Kv1.5 with Kv1.3 cysteine mutants under oxidizing conditions
Exposure of oocytes to the membrane-permeable oxidizing agent, t-butyl HO2 (100 μM) had no significant effect on WT Kv1.5/Kvβ1.3 channel inactivation (Figure 8A). However, oxidation of channels formed by co-expression of Kv1.5 with R5C or T6C Kvβ1.3 subunits caused ∼20-fold increase in current magnitude, when analysed at the end of a 1.5s pulse to +70 mV (Figure 8B and C). Oocytes co-expressing Kv1.5 and L2C or A3C Kvβ1.3 were also relatively unaffected by oxidation, whereas A4C Kvβ1.3 had an intermediate sensitivity (Figure 8C). Redox sensitivity was correlated with the extent of inactivation induced by the specific mutation in Kvβ1.3. Unfortunately, these effects prevented crosslinking experiments as an aid to identify specific residue interactions between Kvβ1.3 and Kv1.5. However, the prevention of R5C-mediated channel inactivation by t-butyl HO2 were utilized to gain further evidence that this residue is located deep within the pore. When 100 μM t-butyl HO2 was applied to Kv1.5/R5C Kvβ1.3 channels that were inactivated by a prolonged pulse to +40 mV, only partial relief of inactivation was achieved (Figure 8D). Inactivation was nearly completely removed when the cell was subsequently held at −80 mV for 3 min before recording another voltage step to +40 mV (Figure 8D, right panel). Summary data for multiple cells (n=8) are plotted in Figure 8E. The oxidation-induced alteration of R5C channel gating does not require pulsing. When oocytes were held at −80 mV during perfusion with t-butyl HO2, currents were maximally increased during the first pulse to +40 mV, and this effect was fully reversible upon washout of t-butyl HO2 from the bath solution (Figure 8F). The relatively rapid reversal indicates that the C5 in Kvβ1.3 was probably oxidized to a sulphinic or sulphonic acid (Claiborne et al, 2001; Poole et al, 2004), rather than forming a disulphide bridge with another Cys in the same or another Kvβ1.3 subunit. These findings suggest that when Kvβ1.3 subunit is bound to the channel pore, it is protected from the oxidizing agent.
Figure 8.

Effects of oxidation on WT and cysteine mutant Kv1.5/Kvβ1.3 channels. (A) t-Butyl HO2 (100 μM) has no effect on WT Kv1.5/Kvβ1.3 current elicited by 1 s pulse to +70 mV. Currents were normalized to the peak amplitude before the addition of t-butyl HO2. (B) Normalized currents of Kv1.5/R5C Kvβ1.3 before and after the application of 100 μM t-butyl HO2. (C) Change in Kv1.5/Kvβ1.3 channel current magnitude induced by 100 μM t-butyl HO2. Mutations of Kvβ1.3 are indicated on X-axis. (D, E) State-dependent modification of inactivation by oxidation. Kv1.5/R5C Kvβ1.3 current was first recorded with a voltage step to +40 mV (D, left panel). Subsequently, the oocyte was held at +40 mV while the cell chamber was perfused with 100 μM t-butyl HO2 (D, middle panel). The oocyte was then clamped to −80 mV for 3 min before another voltage step to +40 mV was recorded (D, right panel). (E) Change in current amplitude induced by oxidation when oocytes were held at a potential of +40 mV (*) and for the same cells after holding at −80 mV for 3 min (#). *P<0.05. (F) Voltage steps to +40 mV were repeated every 30 s. After stabilization of the current, the oocyte was held at −80 mV without pulsing while the chamber was perfused with 100 μM t-butyl HO2 for 8 min. The oocyte was then pulsed again to +40 mV. Insets show current traces before the application of 100 μM t-butyl HO2 and with the first voltage step to +40 mV following the 8 min pulse-free period. Scale bars represent 0.5 μA and 50 ms, respectively. Current inactivation was recovered after washout of t-butyl HO2. (G) Double-mutant cycle analysis of channels with indicated mutations in Kv1.5 and Kvβ1.3 subunits.
Double-mutant cycle analysis of Kv1.5–Kv1.3 interactions
The experiments summarized in Figures 6D and E, and 7A–C predict that R5 and T6 of Kvβ1.3 interact with residues in the upper S6 segment, near the selectivity filter of Kv1.5. In contrast, for Kvβ1.1 and Kv1.4 (Zhou et al, 2001), this interaction would not be possible because residue 5 interacts with a valine residue equivalent to V516 that is located in the lower S6 segment (Zhou et al, 2001). To identify residues of Kv1.5 that potentially interact with R5 and T6, we performed a double-mutant cycle analysis. The Kd values for single mutations (α or β subunit) and double mutations (α and β subunits) were calculated to test whether the effects of mutations were coupled. The apparent Kd values were calculated based on the time constant for the onset of inactivation and the steady-state value (% inactivation; see Materials and methods). Figure 8G summarizes the analysis for the co-expressions that resulted in functional channel activity. Surprisingly, no strong deviation from unity for Ω was observed for R5C and T6C in combination with A501C, despite the effects observed on the steady-state current (Figure 6D and E). In addition, only small deviations from unity for Ω were observed for R5C co-expressed with V505A, although the extent of inactivation was altered (Figure 7A). The highest Ω values were for R5C in combination with T480A or A501V. These data, together with the results shown in Figures 6 and 7, suggest that Kvβ1.3 binds to the pore of the channel with R5 near the selectivity filter. In this conformation, the side chain of R5 might be able to reach A501 of the upper S6 segment, which is located in a side pocket close to the pore helix.
Model of the Kv1.3-binding mode in the pore of Kv1.5 channels
Our data suggest that R5 of Kvβ1.3 can reach deep into the inner cavity of Kv1.5. Our observations are difficult to reconcile with a linear Kvβ1.3 structure as proposed for interaction of Kvβ1.1 with Kv1.1 (Zhou et al, 2001). The Kv1.5 residues proposed to interact with Kvβ1.3 are highlighted in Figure 9A. The energy-optimized model of the first 11 residues of the Kvβ1.3 N terminus is shown in Figure 9B. The side chain of R5 points towards A3 leading to a compact hairpin structure that would easily fit into the inner cavity of the Kv1.5 pore. This Kvβ1.3 structure was manually positioned within the confines of the Kv1.5 central cavity before calculating energy-minimized binding poses. Figure 9C illustrates the docking of Kvβ1.3 with a single Kv1.5 subunit. The residues in Kv1.5 described earlier as important for interaction with Kvβ1.3 (Decher et al, 2005) are highlighted with van der Waals surfaces. Figure 9D depicts the docking of Kvβ1.3 with two subunits, displaying important Kv1.5 residues as ball and stick model. A stereo-view of the docking with two Kv1.5 subunits is shown in Figure 9E. In the docking shown, the backbone of the Kvβ1.3 hairpin at position R5 and the residues T6 are in close proximity (2.74 Å) to T480 of the selectivity filter.
Figure 9.

Structural model of Kvβ1.3 bound to the pore of Kv1.5 channels. (A) Amino-acid sequence of the Kv1.5 pore-forming region. Residues that may interact with Kvβ1.3 based on an earlier site-directed mutagenesis study (Decher et al, 2005) are depicted in bold. (B) Structure of the N-terminal region (residues 1–11) of Kvβ1.3. (C) Kvβ1.3 docked into the Kv1.5 pore homology model showing a single subunit. Kvβ1.3 side chains are shown as ball and stick models and residues of the Kvβ1.3-binding site in Kv1.5 are depicted with van der Waals surfaces. The symbol′ indicates the end of long side chains. (D) Kvβ1.3 docked into the Kv1.5 pore homology model showing two subunits. (E) Kvβ1.3 hairpin bound to Kv1.5. Two of the four channel subunits in stereo-view are depicted.
Next, we tested whether bulky side-chains at key residues in the N terminus of Kvβ1.3 affect inactivation. Introducing a tryptophan at positions R5 and T6 (at the tip of the proposed hairpin) enhanced inactivation (Figure 10A) as observed for other substitutions of these residues, consistent with the backbone of R5, and not its bulky side chain interacting with the selectivity filter. Kvβ1.3 has two Gly residues located at positions 7 and 10. Mutation of G10 to Ala or Cys (Figure 2) or Trp (Figure 10B) did not reduce the ability of Kvβ1.3 to induce inactivation. In contrast, although mutation of G7 to Ala had no functional consequence (Figure 2A), substitution with Cys significantly reduced inactivation (Figure 2B). Mutation of G7 to a much bulkier and hydrophobic Trp completely eliminated inactivation (Figure 10B), indicating the requirement for a small residue in this position located near the start of the hairpin loop.
Figure 10.

Tryptophan substitutions of R5, T6, G7 and G10. Currents shown were elicited by 200 ms pulses to test potentials ranging from −70 to +70 mV from a holding potential of −80 mV. Peak current amplitudes were reduced by 78.8±3.1% (n=8) for R5W, by 86.1±2.8% for T6W (n=9), by 12.5±1.8% for G7W (n=10) and by 60.7±2.4 % for G10W (n=9).
Discussion
Occlusion of the central cavity by an inactivation peptide is the mechanism of rapid, N-type inactivation of Kv channels (Hoshi et al, 1990). Depending on the specific Kv channel, the inactivation peptide can either be the N terminus of the Kv α-subunit or a separate, tethered Kvβ subunit. Considering their common function, the N-terminal regions of Kv1.4, Kv3.4 or Shaker B α-subunits and the three Kvβ1 subunit isoforms have a surprisingly low sequence homology. NMR structures of Kv1.4 and Kv3.4 indicated earlier that Kvα inactivation peptides can adopt different tertiary structures. Using systematic site-directed mutagenesis, we studied the mode of binding of Kvβ1.3 subunits to Kv1.5 channels. Comparing earlier work with our new findings suggests that the mode of binding of Kvβ1.x subunits to Kv channels exhibit significant variability. We also found that Kvβ1 isoforms are differentially modulated by Ca2+ and PIP2.
We have identified an arginine residue (R5) located in the proximal N terminus of Kvβ1.3 subunits as a likely binding site for intracellular PIP2. Binding of PIPs to R5 prevents N-type inactivation mediated by Kvβ1.3. Although Kvβ1.1 is also sensitive to PIP2, the first 10 amino acids of this subunit do not include an arginine residue. Thus, the PIP2 sensor of Kvβ1.1 remains to be discovered. In our lipid-binding assay, the N terminus of Kvβ1.3 binds PIP2 with high affinity. For the N terminus of Kvβ1.3, we observed a strong PIP2-binding signal with 5 mol% of PIP2. With the same assay, addition of 10 and 35 mol% PIP2 was required for significant binding to the Kv3.4 and Kv1.4 N termini (Oliver et al, 2004). In addition, we were able to show that a single residue substitution in the Kvβ1.3 N terminus can almost completely abolish PIP2-binding. When bound to PIP2, Kvβ1.3 may be positioned near the channel pore, but incapable of blocking the channel. This putative resting state might correlate with the pre-bound or pre-blocking state (O′), as was proposed earlier for Kvβ1 subunits (Zhou et al, 2001). Binding of Kvβ1.3 to the O′ state might induce shifts in the voltage dependence of steady-state activation and C-type inactivation, even for mutant forms of Kvβ1.3 that are no longer capable of inducing N-type inactivation. The modulation of N-type inactivation in native Kv1.x–Kvβ1.3 complexes by PIP2 might be important for the fine-tuning of neuronal excitability. As a result, fluctuations in intracellular PIP2 levels due to Gq-coupled receptor stimulation might be relevant for the inactivation of K+ channels and thus, for electrical signalling in the brain.
The variation in the amino-acid sequence of the proximal N termini also determines the different redox sensitivities of Kvβ1.1 and Kvβ1.3. Normally, Kvβ1.3 subunits are redox insensitive. However, we found that a single cysteine residue introduced at any position between amino acids 3–11 is sufficient to confer redox sensitivity to Kvβ1.3. Also in contrast to Kvβ1.1, we found that Kvβ1.3 was not sensitive to increased intracellular Ca2+ concentrations. Thus, an important physiological consequence of N-terminal splicing of the Kvβ1 gene might be the generation of rapidly inactivating channel complexes with different sensitivities to redox potential and intracellular Ca2+.
We propose that Kvβ1.3 binds to the pore of Kv1.5 channels as a hairpin-like structure, similar to the N-terminal inactivation particles of Kv1.4 and Kv3.4 α-subunits (Antz et al, 1997). This is in contrast to Kvβ1.1, which was reported to bind to the central cavity of the Kv1 channel as a linear peptide (Zhou et al, 2001). For Kvβ1.1, interactions of residue 5 (Ile) were observed with sites in the distal S6 segment of Kv1.4, three helix turns distal to the PVP motif (Zhou et al, 2001). The interaction of R5 and T6 from Kvβ1.3 with the S6 segment residues high in the inner cavity and residues near the selectivity filter of Kv1.5 is only plausible if Kvβ1.3 blocks the channel as a small hairpin, as in the energy-minimized conformation illustrated in our model. The narrowing of the pore by the four S6 segments near the PVP motif with a diameter of 0.9–1.0 nm suggests that Kvβ1.3 can enter the inner cavity configured as a small hairpin. In addition, this hairpin structure is smaller than the N-terminal ball domains that were proposed earlier for the Kv1.4 and Kv3.4 N termini (Antz et al, 1997). On the basis of the crystal structures available, these inactivation balls are too large to pass the PVP barrier and enter the inner cavity. Accordingly, these N-terminal ball domains might bind more distally in the S6 segments and block the pore as ‘shallow plugs' (Antz et al, 1997).
Mutation of R5 in Kvβ1.3 to E, C, A, Q and W accelerated the Kv1.5 channel inactivation. Thus, the acceleration of inactivation by R5 mutations is independent of the size and charge of the residue introduced. Together with our PIP2-binding assay, these findings suggest that PIP2 immobilizes Kvβ1.3 and prevents it from entering the central cavity to induce N-type inactivation. Our model predicts that the backbone of the hairpin, near R5, interacts with the selectivity filter. This is in good agreement with our observation that the nature of the side chain introduced at position 5 was not relevant for the blocking efficiency of the hairpin.
N-terminal splicing of Kvβ1 produces the Ca2+-insensitive Kvβ1.3 isoform that retains the ability to induce Kv1 channel inactivation. We propose that the N terminus of Kvβ1.3 exists in a pre-blocking state when PIPs located in the lipid membrane bind to R5. We further propose that when Kvβ1.3 dissociates from PIPs, it assumes a hairpin structure that can enter the central cavity of an open Kv1.5 channel to induce N-type inactivation.
Materials and methods
Molecular biology
Kv1.5 cDNA in the pSGEM oocyte expression vector and the methods of site-directed mutagenesis were described earlier (Decher et al, 2004). The Kv1.5 sequence (NM_002234) has an N terminus with two additional residues compared with an earlier database entry (M60451). This results in a shift of the amino acid numbering of +2 when compared with older literature. Restriction mapping and DNA sequencing were used to confirm the presence of the desired mutation and the lack of extra mutations in the PCR-amplified segment. Complementary RNA (cRNA) for injection into oocytes was prepared with T7 Capscribe (Roche) after linearization with NheI. The Kvβ1.3 construct in a modified pSP64T vector was described previously (England et al, 1995) and cRNA was made with SP6 Capscribe (Roche) after linearization with EcoRI. The quality and quantity of cRNA were determined by gel electrophoresis and UV spectroscopy.
Lipid-binding assays
For the lipid-binding assay, the nucleotide sequence encoding amino acids 1–33 of WT Kvβ1.3 and mutants R5C and T6C were subcloned with EcoRI–SalI into the pGEX4T-1 vector (Amersham Pharmacia Biotech) to produce an in-frame GST fusion protein. Proteins and liposomes were prepared and assayed as described (Soom et al, 2001). Briefly, GST, GST-fused Kvβ1.3 (residues 1–33), Kvβ1.3 (residues 1–33) R5C and Kvβ1.3 (residues 1–33) T6C were overexpressed in Escherichia coli strain BL-21 Codon Plus and immobilized on GSH Sepharose according to the manufacturer's instructions (Amersham Pharmacia Biotech). Mixed liposomes were prepared from PI(4,5)P2, phosphatidylcholine (PC), phosphatidylethanolamine (PE), cholesterol (ChS) and rhodamine-PE (Rh-PE) to obtain a lipid composition of 5 mol% PI(4,5)P2. The PE, ChS and Rh-PE contents were always 50, 32 and 1 mol%, respectively. Immobilized GST proteins (0.01 mM) were incubated with liposomes with subsequent washing. Binding of liposomes to immobilized proteins was quantified by fluorescence measurement using excitation/emission wavelengths of 390/590 nm (cutoff at 570 nm). The data were corrected by subtracting the fluorescence of control liposomes without PI(4,5)P2 from the values obtained in assays with liposomes containing PI(4,5)P2 and normalized to the binding of GST-fused Kvβ1.3 WT peptide. Results are presented as mean±s.e.m. of three parallel experiments.
Two-electrode voltage-clamp
Stage IV and V Xenopus laevis oocytes were isolated and injected with cRNA encoding WT or mutant Kv1.5 and Kvβ1.3 subunits as described earlier (Decher et al, 2004). Oocytes were cultured in Barth's solution supplemented with 50 μg/ml gentamycin and 1 mM pyruvate at 18°C for 1–3 days before use. Barth's solution contained (in mM): 88 NaCl, 1 KCl, 0.4 CaCl2, 0.33 Ca(NO3)2, 1 MgSO4, 2.4 NaHCO3, 10 HEPES (pH 7.4 with NaOH). For voltage-clamp experiments, oocytes were bathed in a modified ND96 solution containing (in mM): 96 NaCl, 4 KCl, 1 MgC12, 1 CaC12, 5 HEPES (pH 7.6 with NaOH). Currents were recorded at room temperature (23–25°C) with standard two-microelectrode voltage-clamp techniques (Stuhmer, 1992). The holding potential was −80 mV. The interpulse interval for all voltage-clamp protocols was 10 s or longer to allow for full recovery from inactivation between pulses. The standard protocol to obtain current–voltage (I–V) relationships and activation curves consisted of 200 ms or 1.5 s pulses that were applied in 10-mV increments between −70 and +70 mV, followed by a repolarizing step to −40 mV.
The voltage dependence of the Kv1.5 channel activation (with or without co-expression with Kvβ1.3) was determined from tail current analyses at −40 mV. The resulting relationship was fit to a Boltzmann equation (equation (1)) to obtain the half-point (V1/2act) and slope factor (kact). 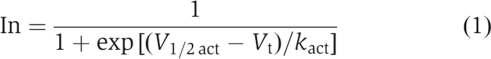
The voltage dependence of Kv1.5 inactivation was determined by using a two-pulse protocol. A prepulse of 1 s was applied to potentials ranging from −90 to +70 mV and was immediately followed by a 200 ms test pulse to +70 mV. The relative amplitude of peak current during the test pulse was plotted as a function of the prepulse voltage and the relationship fit to a Boltzmann function to obtain the V1/2inact for inactivation. Other voltage pulse protocols are described in the Results and figure legends. Data are expressed as mean±s.e.m. (n=number of oocytes).
Excised macropatches from Xenopus oocytes
Recordings from inside-out macropatches were performed as described previously (Oliver et al, 2004). Pipettes (0.2–0.4 MΩ) were filled with extracellular solution (mM): 115 NaCl, 5 KCl, 10 HEPES and 1 CaCl2 (pH 7.2 with NaOH). Intracellular solution contained (mM): 100 KCl, 10 EGTA and 10 HEPES (pH 7.2 with KOH). A hypertonic solution used to shrink oocytes and facilitate removal of the vitelline membrane contained (mM): 200 K-aspartate, 20 KCl, 1 MgCl2, 10 EGTA and 10 HEPES (pH 7.4 with KOH).
Double-mutant cycle analysis
The double-mutant cycle parameter Ω (equation (2)) was calculated to quantify the degree of coupling between two mutations, as described previously (Hidalgo and MacKinnon, 1995; Gulbis et al, 2000). 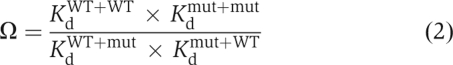
A value of Ω greater than unity indicates that the effects of two mutations are coupled. For Ω values smaller than 1, the reciprocal was taken to facilitate the display of changes from unity, as described previously (Hidalgo and MacKinnon, 1995). The Kd values were obtained from the apparent rate constants for binding (α) and unbinding (β), according to: 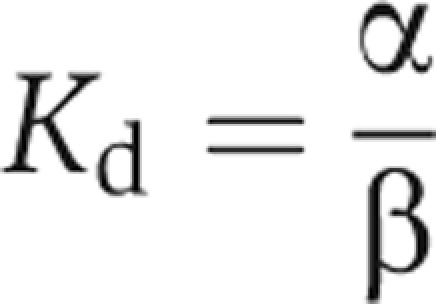
The α and β values were obtained from the inactivation time constant (τin) and the steady-state probability (ss) at the end of 1.5-s pulses. For the double-cycle analysis, only the predominant (fast) component of the inactivation was analysed: 
Molecular modelling and docking
Molecular modelling was based on the crystal structure of the mammalian Shaker Kv1.2 potassium channel subunit complex (PDB 2A79) and the sequence of potassium channel subunit Kv1.5 (NP_037104). A hairpin-shaped structural model of the first 11 residues of the Kvβ1.3 N terminus (AAD37855) was built and energy-optimized (Figure 9B). Interactions of the Kvβ1.3 N terminus and Kv1.5 were determined by molecular docking. The docking was initiated from random configurations (n=200) of the Kvβ1.3 N terminus in the cytoplasmic cavity of the Kv1.5 tetramer and performed by using simulated annealing techniques with an initial temperature of 500 K and a final temperature of 300 K. Residues of Kv1.5 facing the pore region and the Kvβ1.3 N terminus were flexible during the optimization procedure. Modelling and docking were performed with the Insight II modules Homology, Builder and Docking (version 8.2; Accelrys, San Diego, CA).
Acknowledgments
We thank Pradeep Kumar, Paul David Sonntag, Krista Kinard and Meng San Pun for technical assistance. This study was supported by NHLBI, National Institutes of Health Grant R01 HL065299 to MCS, SFB 604 A4 to SHH, SFB593 TPA4 to JD and by the PE Kempkes Stiftung 09/05 and 11/06 to ND.
References
- Antz C, Fakler B (1998) Fast inactivation of voltage-gated K+ channels: from cartoon to structure. News Physiol Sci 13: 177–182 [DOI] [PubMed] [Google Scholar]
- Antz C, Geyer M, Fakler B, Schott MK, Guy HR, Frank R, Ruppersberg JP, Kalbitzer HR (1997) NMR structure of inactivation gates from mammalian voltage-dependent potassium channels. Nature 385: 272–275 [DOI] [PubMed] [Google Scholar]
- Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D (2001) Structural, redox, and mechanistic parameters for cysteine–sulfenic acid function in catalysis and regulation. Adv Protein Chem 58: 215–276 [DOI] [PubMed] [Google Scholar]
- Decher N, Kumar P, Gonzalez T, Renigunta V, Sanguinetti MC (2005) Structural basis for competition between drug binding and Kvbeta 1.3 accessory subunit-induced N-type inactivation of Kv1.5 channels. Mol Pharmacol 68: 995–1005 [DOI] [PubMed] [Google Scholar]
- Decher N, Pirard B, Bundis F, Peukert S, Baringhaus KH, Busch AE, Steinmeyer K, Sanguinetti MC (2004) Molecular basis for Kv1.5 channel block: conservation of drug binding sites among voltage-gated K+ channels. J Biol Chem 279: 394–400 [DOI] [PubMed] [Google Scholar]
- Encinar JA, Fernandez AM, Molina ML, Molina A, Poveda JA, Albar JP, Lopez-Barneo J, Gavilanes F, Ferrer-Montiel AV, Gonzalez-Ros JM (2002) Tyrosine phosphorylation of the inactivating peptide of the shaker B potassium channel: a structural–functional correlate. Biochemistry 41: 12263–12269 [DOI] [PubMed] [Google Scholar]
- England SK, Uebele VN, Kodali J, Bennett PB, Tamkun MM (1995) A novel K+ channel beta-subunit (hKv beta 1.3) is produced via alternative mRNA splicing. J Biol Chem 270: 28531–28534 [DOI] [PubMed] [Google Scholar]
- Fernandez-Ballester G, Gavilanes F, Albar JP, Criado M, Ferragut JA, Gonzalez-Ros JM (1995) Adoption of beta structure by the inactivating ‘ball' peptide of the Shaker B potassium channel. Biophys J 68: 858–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez T, Navarro-Polanco R, Arias C, Caballero R, Moreno I, Delpon E, Tamargo J, Tamkun MM, Valenzuela C (2002) Assembly with the Kvbeta1.3 subunit modulates drug block of hKv1.5 channels. Mol Pharmacol 62: 1456–1463 [DOI] [PubMed] [Google Scholar]
- Gulbis JM, Zhou M, Mann S, MacKinnon R (2000) Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science 289: 123–127 [DOI] [PubMed] [Google Scholar]
- Heinemann SH, Rettig J, Graack HR, Pongs O (1996) Functional characterization of Kv channel beta-subunits from rat brain. J Physiol 493 (Part 3): 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo P, MacKinnon R (1995) Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science 268: 307–310 [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW (1990) Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250: 533–538 [DOI] [PubMed] [Google Scholar]
- Jow F, Zhang ZH, Kopsco DC, Carroll KC, Wang K (2004) Functional coupling of intracellular calcium and inactivation of voltage-gated Kv1.1/Kvbeta1.1 A-type K+ channels. Proc Natl Acad Sci USA 101: 15535–15540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak YG, Navarro-Polanco RA, Grobaski T, Gallagher DJ, Tamkun MM (1999) Phosphorylation is required for alteration of kv1.5 K+ channel function by the Kvbeta1.3 subunit. J Biol Chem 274: 25355–25361 [DOI] [PubMed] [Google Scholar]
- Lee CW, Aldrich RW, Gierasch LM (1993) Conformational studies of the N-terminal domain of the shaker B potassium channel by CD and NMR. Biophys J 61: 379a (Abstract) [Google Scholar]
- Leicher T, Bahring R, Isbrandt D, Pongs O (1998) Coexpression of the KCNA3B gene product with Kv1.5 leads to a novel A-type potassium channel. J Biol Chem 273: 35095–35101 [DOI] [PubMed] [Google Scholar]
- Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B (2004) Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science 304: 265–270 [DOI] [PubMed] [Google Scholar]
- Pian P, Bucchi A, Decostanzo A, Robinson RB, Siegelbaum SA (2007) Modulation of cyclic nucleotide-regulated HCN channels by PIP2 and receptors coupled to phospholipase C. Pflugers Arch 455: 125–145 [DOI] [PubMed] [Google Scholar]
- Poole LB, Karplus PA, Claiborne A (2004) Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol 44: 325–347 [DOI] [PubMed] [Google Scholar]
- Putzke C, Wemhoner K, Sachse FB, Rinne S, Schlichthorl G, Li XT, Jae L, Eckhardt I, Wischmeyer E, Wulf H, Preisig-Muller R, Daut J, Decher N (2007) The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res 75: 59–68 [DOI] [PubMed] [Google Scholar]
- Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O (1994) Inactivation properties of voltage-gated K+ channels altered by presence of beta-subunit. Nature 369: 289–294 [DOI] [PubMed] [Google Scholar]
- Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M (1991) Regulation of fast inactivation of cloned mammalian IK(A) channels by cysteine oxidation. Nature 352: 711–714 [DOI] [PubMed] [Google Scholar]
- Soom M, Schonherr R, Kubo Y, Kirsch C, Klinger R, Heinemann SH (2001) Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett 490: 49–53 [DOI] [PubMed] [Google Scholar]
- Stephens GJ, Robertson B (1995) Inactivation of the cloned potassium channel mouse Kv1.1 by the human Kv3.4 ‘ball' peptide and its chemical modification. J Physiol 484 (Part 1): 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer W (1992) Electrophysiological recording from Xenopus oocytes. Methods Enzymol 207: 319–339 [DOI] [PubMed] [Google Scholar]
- Uebele VN, England SK, Gallagher DJ, Snyders DJ, Bennett PB, Tamkun MM (1998) Distinct domains of the voltage-gated K+ channel Kv beta 1.3 beta-subunit affect voltage-dependent gating. Am J Physiol 274: C1485–C1495 [DOI] [PubMed] [Google Scholar]
- Zhou M, Morais-Cabral JH, Mann S, MacKinnon R (2001) Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature 411: 657–661 [DOI] [PubMed] [Google Scholar]