

SUMMARY
Cyclic AMP-induced phosphorylation of the transcription factor CREB elicits expression of genes mediating diverse biological functions. In lymphoid organs, the neurotransmitter norepinephrine stimulates β2-adrenergic receptors on B lymphocytes to promote CREB-dependent expression of genes like the B-cell Oct 2 coactivator (OCA-B). Although CREB phosphorylation recruits co-factors such as CBP/p300 to stimulate transcription, bona fide endogenous inhibitors of CREB-coactivator or CREB-DNA interactions have not emerged. Here, we identified RGS13, a member of the Regulator of G protein Signaling (RGS) protein family, as a nuclear factor that suppresses CREB-mediated gene expression. cAMP or Ca2+ signaling promoted RGS13 accumulation in the nucleus, where it formed a complex with phosphorylated CREB and CBP/p300. RGS13 reduced the apparent affinity of pCREB for both the CRE and CBP. B lymphocytes from Rgs13−/− mice had more β2-agonist-induced OCA-B expression. Thus, RGS13 inhibits CREB-dependent transcription of target genes through disruption of complexes formed at the promoter.
INTRODUCTION
The ubiquitously expressed transcription factor CREB functions in many biological processes including metabolism, neuronal development, and cancer progression (Conkright and Montminy, 2005; Dragunow, 2004). Over 10,000 CRE-containing promoter sequences and more than 4,000 potential CREB target genes exist in the human genome, suggesting broad involvement of CREB in gene expression (Zhang et al., 2005). Creb−/− mice die at birth due to respiratory failure caused by insufficient lung maturation and defects in T cell development (Rudolph et al., 1998). Transgenic expression of dominant negative forms of CREB showed that CREB also mediates cell survival and growth in mature mice (Zhang et al., 2002).
Over 300 extracellular signals activate CREB including hormones, growth factors, and neurotransmitters (Johannessen et al., 2004; Mayr and Montminy, 2001). G-protein-coupled receptors (GPCR) such as the β2-adrenergic receptor induce CREB transactivation through the Gαs-cAMP-PKA pathway. Phosphorylation of CREB on S133 by PKA recruits the co-activator CREB binding protein (CBP) or its paralogue p300 to induce gene transcription. In B lymphocytes, β2-adrenergic receptor-induced expression of OCA-B promotes expression of genes involved in physiological B cell functions (Podojil and Sanders, 2005; Wolf et al., 1998). OCA-B is essential for proper immune function as mice deficient in OCA-B do not form adequate germinal centers in lymphoid organs after immunization (Qin et al., 1998).
The detailed mechanism of transcriptional activation by the CREB-CBP complex remains incompletely understood. Although CREB phosphorylation is required for CBP recruitment to promoters, this does not strictly correlate with induction of cAMP-responsive genes (Zhang et al., 2005). Chromatin immunoprecipitation (ChIP) analysis of mammalian gene promoters suggested that only a fraction of promoters occupied by phosphoCREB (pCREB) also contains CBP (Zhang et al., 2005). Thus, additional factors may control CBP recruitment and transcriptional activation (Geiger et al., 2007). It is also unclear whether CREB-CBP promoter occupancy is subject to regulation (Mayr and Montminy, 2001). CREB binding to identical target gene CRE sites in vivo may differ in distinct cell types (Cha-Molstad et al., 2004). Thus, other nuclear factors may regulate CREB-CRE and CREB-coactivator interactions to specify the subset of genes turned on by CREB phosphorylation.
Proteins of the RGS family (~30 members in mammalian cells) regulate GPCR signaling at the plasma membrane through their GTPase activating protein (GAP) activity on Gα subunits, which may inhibit signal output by shortening the lifetime of activated G proteins (Willars, 2006). RGS13, one of the smallest RGS family members, regulates B cell chemotaxis in vitro, presumably through its GAP activity on proteins of the Gαi family, which are coupled to chemokine receptors (Shi et al. 2002). We found that RGS13 expression also reduced cAMP production in human embryonic kidney (HEK)293T cells in response to β2-adrenergic receptor stimulation (Johnson and Druey, 2002). Since RGS13 lacks GAP activity toward Gαs (Johnson and Druey, 2002), it might affect cAMP signaling independently of the G protein. We investigated how RGS13 regulates the β2-adrenergic receptor-evoked signaling route and found that PKA activation lead to RGS13 accumulation in the nucleus, where it interacted with the pCREB-CBP/p300 complex. Further, RGS13 reduced both pCREB binding to the CRE and its association with CBP/p300. The physiological relevance of these findings was supported by the observation that B lymphocytes from Rgs13−/− mice had more OCA-B expression. RGS13 provides an uncommon example of a cell surface receptor’s regulatory protein that is also capable of influencing nuclear processes directly.
RESULTS
RGS13 inhibits Gαs-mediated signaling at the level of CREB
To investigate whether RGS13 regulated the β2-adrenergic receptor signaling pathway downstream of the G protein, we tested the effect of RGS13 expression on the activity of a CRE reporter gene (CRE-Luciferase) in HEK293T cells stimulated with forskolin, a direct activator of adenylyl cyclase, or with a membrane-permeable cAMP analogue [8-(4-chlorophenylthio)-adenosine 3’ 5’-cyclic monophosphate, 8-pCPT cAMP]. Expression of RGS13 fused to green fluorescent protein (GFP), but not GFP-RGS4, attenuated both forskolin- and cAMP-evoked CRE-Luc activity compared to GFP alone (Figure 1A, 1B) although the proteins exhibited comparable expression (Figure S1). Thus, RGS13 regulates CRE-Luc activation independently of Gαs. Since RGS13, but not RGS4, also impaired CRE-Luc activation by constitutively active Gαq (GαqQ209→L209), which stimulates the reporter in a Ca2+-dependent manner that is independent of PKA (Shaywitz and Greenberg, 1999; Johnson and Druey, 2002), it may inhibit CREB-mediated transcription downstream of kinase activation. Consistent with this hypothesis, RGS13 reduced CRE-Luc activity induced by constitutively active VP16-CREB, a chimeric transcription factor consisting of the herpes virus VP16 activation domain fused to full length CREB(aa1-341) (Figure 1C). From these studies, we concluded that RGS13 inhibits cAMP signaling at the level of CREB. The fact that RGS13 did not block activation of a serum response element (SRE)-dependent reporter gene induced by constitutively active forms of Gα12 and Gα13 indicated that this RGS protein is not a general transcriptional repressor (Johnson and Druey, 2002).
Figure 1. RGS13 attenuates Gαs-coupled signaling at the level of CREB.
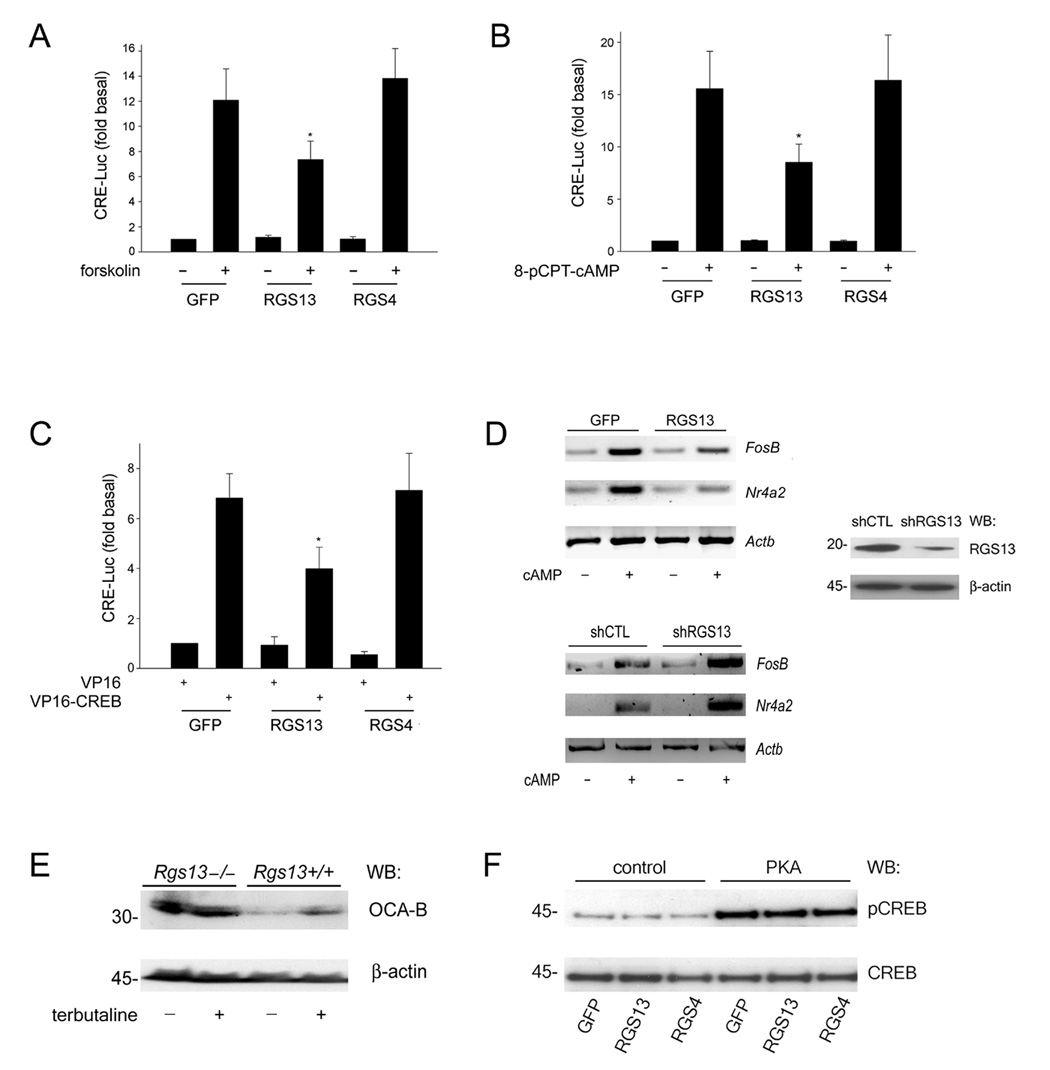
(A–C) GFP, GFP-RGS13 or GFP-RGS4 was expressed with a CRE-luciferase reporter and β-galactosidase in HEK293T cells. Cells were stimulated with 10 µM forskolin (A) or 2 mM 8-pCPT-cAMP (B) for 6 hrs. prior to measurement of luciferase activity. Luciferase values were normalized to β-galactosidase activity. In (C), cells were co-transfected with a plasmid encoding VP16 or VP16-CREB(aa1-341). Values are the ‘fold’ stimulation relative to control (GFP), set as 1 (mean ± S.E.M. of at least 3 experiments; *P=0.02 (A–B); *P=0.002 (C), one way ANOVA). (D) RGS13 abundance affects CREB target gene expression. GFP or GFP-RGS13 plasmids (upper panel) or a control or RGS13-specific shRNA plasmid (lower panel) was expressed in HEK293T cells. Cells were stimulated with 2 mM 8-bromo-cAMP for 1 hr. followed measurement of FosB, Nr4a2, or Actb by RT-PCR. RGS13 knockdown was assessed by immunoblotting (right panel). Images show a single experiment representative of 3–4 experiments with similar results. (E) Increased CREB target gene expression in Rgs13−/− B cells. Splenic B lymphocytes from wild-type or Rgs13−/− mice were stimulated with 10 µM terbutaline for 72 hrs. OCA-B expression was analyzed by immunoblotting. Images are representative of 3 independent experiments. (F) RGS13 does not affect PKA-mediated phosphorylation of CREB. HEK293T cells were co-transfected with GFP, GFP-RGS13, or GFP-RGS4 plasmids together with empty vector or the catalytic subunit of PKA. Nuclear extracts (NE) were prepared and immunoblotted as indicated. Blot represents 3 experiments with nearly identical results.
RGS13 modulates expression of endogenous CREB target genes
We evaluated expression of widely studied CREB target genes (FosB and Nr4a2) (Holmes and Zachary, 2004; Tullai et al., 2007) in HEK293T cells treated with a cAMP analog (8-bromo-cAMP) by RT-PCR. Compared to GFP alone, RGS13 overexpression attenuated the increase in Nr4a2 and FosB mRNAs induced by cAMP treatment by ~50% (Figure 1D, upper left panel). HEK293T cells express endogenous RGS13 (Figure 1D, right panel; Figure S2). An RGS13-specific short, hairpin RNA (shRNA) substantially reduced RGS13 protein content compared to a control shRNA (Figure 1D, right panel). RGS13 knockdown resulted in an almost 2-fold increase in FosB and Nr4a2 mRNAs following cAMP exposure relative to control (Figure 1D, lower left panel).
RGS13 is expressed primarily in mast cells and B lymphocytes (Bansal et al., 2007). To determine whether RGS13 also regulated CREB-mediated gene expression in untransformed primary cells or tissues, we compared OCA-B expression in splenic B lymphocytes from wild-type and Rgs13−/− mice. The β2-adrenergic receptor agonist terbutaline induced a modest increase in OCA-B content in B cells from wild-type mice. In contrast, B cells from Rgs13−/− mice had more OCA-B than wild-type at baseline and after terbutaline exposure (Figure 1E).
CREB phosphorylation at S133 by kinases such as PKA, Akt, p90Rsk, or CaMKIV is required for transcription at CRE sites (Johannessen et al., 2004; Mayr and Montminy, 2001). To determine whether RGS13 affected CREB phosphorylation, we co-expressed RGS13 or RGS4 together with the active catalytic subunit of PKA in HEK293T cells. Neither RGS13 nor RGS4 expression attenuated CREB phosphorylation at S133 by PKA compared to GFP alone (Figure 1F). These data suggest that RGS13 inhibits the CREB transcriptional apparatus directly.
RGS13 binds phosphorylated CREB
Since RGS13 reduced cAMP-induced transcription but not CREB phosphorylation, we investigated whether it regulates gene expression by interacting with CREB. Immunoprecipitation studies revealed that RGS13 associated with CREB in the presence of PKA while GFP did not interact with CREB in the presence or absence of PKA (Figure 2A). Thus, CREB phosphorylation by PKA could be required for RGS13-CREB complex formation. Accordingly, CREB containing a substitution at the PKA phosphorylation site (S133→A133) did not co-immunoprecipitate RGS13 (Figure 2B). Recombinant protein ‘pulldown’ experiments demonstrated a direct interaction between RGS13 and CREB, which required both p300 (Figure 2C) and CREB phosphorylation by PKA (Figure 2D). RGS13 did not bind p300 or PKA alone (Figure S3 and data not shown). Thus, RGS13, pCREB, and p300 may form a trimolecular complex.
Figure 2. RGS13 interacts with phosphorylated CREB.
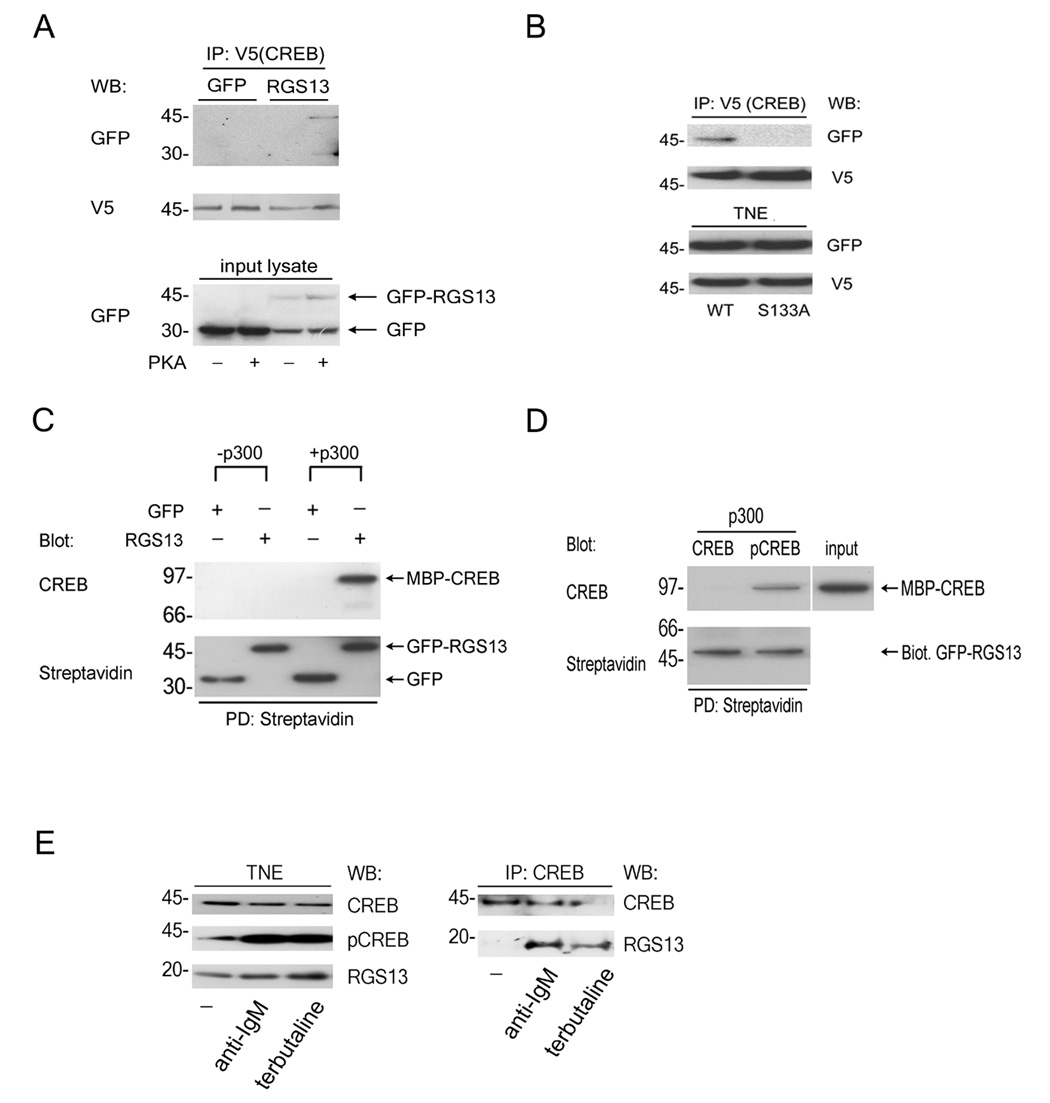
(A–B) Co-immunoprecipitation of RGS13 with wild type (WT) CREB in the presence of PKA. (A) HEK293T cells were co-transfected with plasmids for CREB-V5 and GFP or GFP-RGS13 with or without the catalytic subunit of PKA. Cell lysates were immunoprecipitated with anti-V5 antibody followed by immunodetection as indicated. Protein expression in total lysates was assessed by immunoblotting (bottom panel). (B) Co-immunoprecipitation was done as in (A) except that V5-tagged WT and S133A CREB were analyzed in parallel. Expression of proteins in total NE was determined by immunoblotting (bottom panel). (C) p300 is required for pCREB-RGS13 interaction. Recombinant H6MBP-CREB (Mr~97 kD) was phosphorylated by PKA prior to incubation with streptadivin-coupled agarose beads containing biotinylated GFP or GFP-RGS13 in the presence or absence of p300. Beads were washed and analyzed by immunoblotting. (D) CREB phosphorylation is required for interaction with RGS13. Biotinylated GFP-RGS13 immobilized on streptavidin beads was incubated with unmodified CREB or PKA-phosphorylated CREB in the presence of p300. Beads were washed and analyzed by immunoblotting as in (C). 10% of the input CREB was run as an immunoblot control. (E) Co-immunoprecipitation of RGS13 and CREB in B lymphocytes. Ramos B cells were left untreated or stimulated with anti-IgM (10 µg/ml) or terbutaline (10 µM) for 15 min. NE were immunoprecipitated with anti-CREB followed by detection of protein complexes by immunoblotting (right panel). Expression of proteins in NE was determined by immunoblotting (left panel). Images represent at least 3 experiments with similar results.
Interaction of native RGS13 and CREB was also observed in Ramos B lymphocytes, a Burkitt lymphoma cell line with endogenous RGS13 (Figure 2E). CREB co-immunoprecipitated RGS13 from nuclear lysates of cells treated with anti-immunoglobulin M (IgM) or terbutaline, which elicit CREB phosphorylation by protein kinase C (PKC) or PKA activation, respectively (Parker et al., 1996; Zhang et al., 2002), but not in unstimulated cell lysates (Figure 2E). These results demonstrate agonist-induced interaction of RGS13 with pCREB and support the hypothesis that RGS13 regulates CREB-mediated gene expression in B cells.
PKA-induced RGS13 nuclear translocation and co-localization with pCREB
We detected more RGS13 in the nuclear fraction of B cells stimulated with anti-IgM or terbutaline than in unactivated cells (Figure 2E). Prior studies demonstrated translocation of cytosolic RGS13-GFP to the nucleus of HeLa cells in the presence of Gαs(Q213→L213), a constitutively active G protein that activates the PKA/CREB pathway (Shi et al., 2002). Therefore, RGS13 might accumulate in the nucleus after PKA activation due to its interaction with pCREB. We co-expressed GFP-RGS13 and PKA and examined GFP localization in living HEK293T cells. RGS13 was located diffusely throughout the cell in the absence of PKA (Figure 3A). In contrast, RGS13 co-expressed with PKA localized predominantly in the nucleus (Figure 3A, 3B). We also evaluated GFP-RGS13 localization by DNA staining of fixed NIH3T3 cells with 4’-6-diamidino-2-phenylindole (DAPI) (Figure 3B). We observed two patterns of GFP-RGS13 localization in cells expressing PKA: diffuse but incomplete nuclear accumulation (Figure 3B, second row from top), or condensed/nearly complete nuclear localization (Figure 3B, third row from top). The compact nuclear GFP-RGS13 in the latter cells did not overlay with DAPI, and these cells had irregular or rounded morphology. Since PKA did not affect the shape of cells expressing GFP alone (Figure 3A), these morphological changes may be related to accumulation of GFP-RGS13 in the nucleus. Because RGS13 affects the function of such a fundamental transcription factor, further studies will be needed to determine the magnitude of downstream effects induced by RGS13 overexpression.
Figure 3. Nuclear accumulation of RGS13 and co-localization with pCREB.
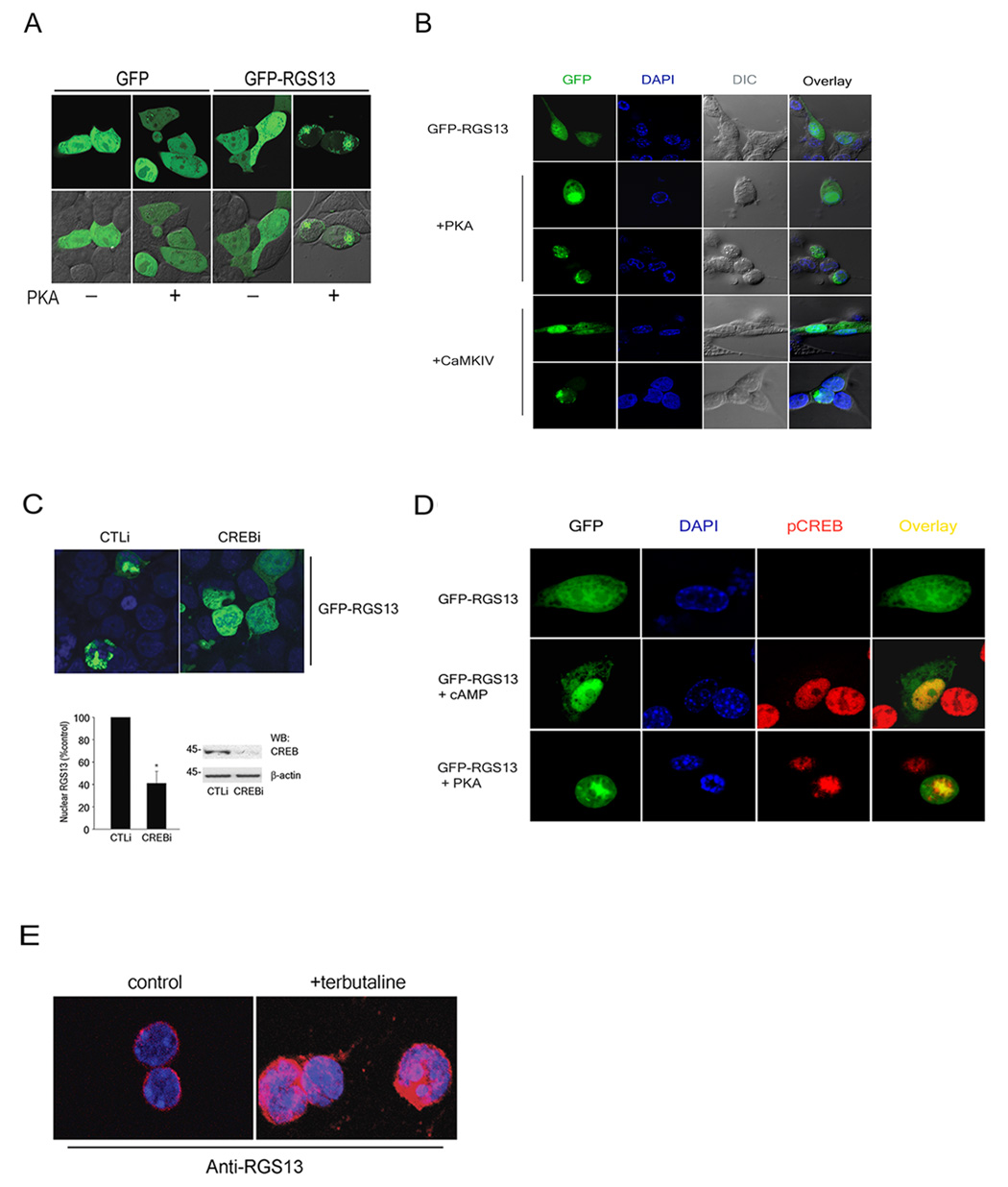
(A–B) HEK293T (A) or NIH3T3 cells (B) were transfected on chamber slides with plasmids encoding the indicated proteins. Fluorescence images of live cells (A) or DAPI-stained fixed cells (B) were analyzed by confocal microscopy. (C) Control (CTLi) or CREB-specific (CREBi) siRNAs (100 nM) were transfected into HEK293T cells with plasmids encoding GFP-RGS13 and PKA. Fixed cells were analyzed by fluorescence microscopy (upper panel). The frequency of RGS13 nuclear accumulation was determined by examining 200 or more cells in random fields for each experiment. Bar graph represents the mean ± S.E.M. of 3 independent experiments (*, P=0.03, paired t-test). Representative immunoblots of lysates from cells expressing either control or CREB siRNAs are shown on the right. (D) Co-localization of pCREB and RGS13. NIH3T3 cells were transfected with GFP-RGS13 and stimulated with vehicle or cAMP for 30 min. (upper and middle panels as indicated) or co-transfected with PKA (lower panel) followed by immunofluorescent staining with anti-pCREB antibody. (E) Terbutaline-induced nuclear translocation of endogenous RGS13. Splenic B lymphocytes were stimulated with terbutaline (10 µM) for 20 min., followed by plating on glass coverslips. Cells were stained with anti-RGS13 and DAPI. All images represent at least 3 experiments with similar results.
We also evaluated whether a kinase other than PKA promoted RGS13 nuclear localization. CaMKIV phosphorylates CREB independently of PKA in a Ca2+-dependent manner (Soderling, 1999). RGS13 expressed with CaMKIV in NIH3T3 cells accumulated in the nucleus in a pattern similar to that induced by PKA (Figure 3B, bottom two panels). To determine how cellular CREB content affected PKA-induced GFP-RGS13 nuclear concentration, we used CREB-specific siRNA, which substantially reduced endogenous CREB compared to a control siRNA (Figure 3C, lower panel). Cells expressing CREB-specific siRNA had less GFP-RGS13 in the nucleus in the presence of PKA than cells containing a control siRNA (Figure 3C). Thus, CREB phosphorylation may promote RGS13 accumulation in the nucleus through the interaction of CREB and RGS13. Consistent with this hypothesis, GFP-RGS13 co-localized with pCREB in NIH3T3 cells co-expressing PKA or stimulated with cAMP (Figure 3D). Agonist-induced nuclear localization of endogenous RGS13 was also observed in B lymphocytes stimulated with terbutaline (Figure 3E).
RGS13 interacts with CREB through diverged residues at its amino terminus
To elucidate the molecular mechanism whereby RGS13 inhibited CREB activity, we determined the region in RGS13 necessary for CREB binding (Figure S4). Immunoprecipitation of CREB revealed that two discontinuous regions of RGS13, aa1-33 and 93-117, were responsible for its interaction with pCREB (Figure 4A) and its capacity to inhibit PKA-evoked CRE-Luc activation (Figure 4B). In contrast, an RGS13 mutant containing the carboxyl terminus (aa118-159) did not bind CREB (Figure S5) or suppress CRE-Luc activity (Figure 4B).
Figure 4. Distinct regions of RGS13 attenuate CREB-mediated transcription independently of GAP activity.
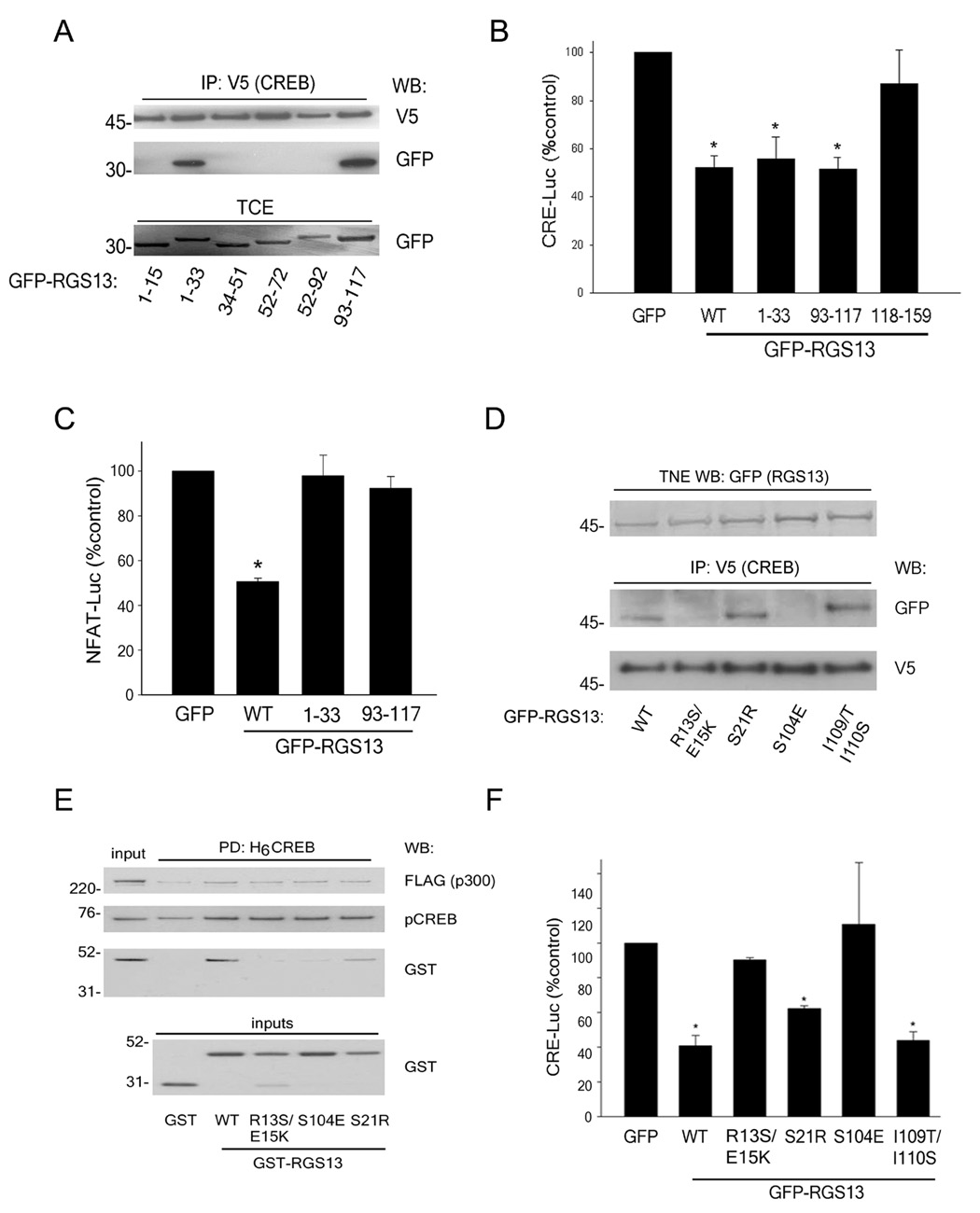
(A) Plasmids encoding GFP-RGS13 truncation mutants were co-expressed with CREB-V5 and PKA in HEK293T cells. Total cell extracts (TCE) were immunoprecipitated with anti-V5 and blotted as indicated. Expression of the mutants in TCE was evaluated by immunoblotting. Similar results were obtained in 3 independent experiments. (B–C) Plasmids encoding PKA and CRE-Luc (B) or Gαq(R183→C183) and NFAT-Luc (C) were transfected into 293T cells together with β-galactosidase and either GFP, full length GFP-RGS13 or GFP-RGS13 truncation mutants. Luciferase activity was measured as in Fig. 1 (B–C values are mean ± S.E.M of 5–6 experiments; *, P<0.006, one way ANOVA. (D) Plasmids encoding CREB-V5, PKA, and GFP-RGS13 (wild-type or various point mutants) were transfected into 293T cells. NE were immunoprecipitated with anti-V5 followed by immunoblotting with anti-V5 and anti-GFP. Amounts of GFP-RGS13 in TNE were analyzed by immunoblotting (top panel). Blot is representative of 3 experiments with similar results. (E) Same experiment as (B) except that the activity of WT GFP-RGS13 was compared to RGS13 containing the indicated point mutations (values are mean ± S.E.M of 6 experiments; *, P<0.005, one way ANOVA). (F) H6MBP-CREB (25 nM) was phosphorylated by PKA followed by incubation with GST or GST-RGS13 (WT or mutants, 30 nM) and p300 (2 nM). Protein complexes were recovered with Ni2+/NTA beads followed by immunoblot analysis. 10% of the input GST proteins were run separately as immunoblot controls (bottom panel).
These results also suggested that RGS13 GAP function was not necessary for its regulation of CREB-mediated transcription as an intact RGS domain (aa31-150 in RGS13) is required for GAP activity (Popov et al., 1997). To test this hypothesis, we utilized a nuclear factor of activated T cells-luciferase (NFAT-Luc) reporter system evoked by constitutively active Gαq(R183→C183)[GαqRC]. This G protein mutant, which induces NFAT activity through a phospholipase Cβ-mediated increase in cytosolic Ca2+, has been utilized to assess GAP activity of RGS proteins (Carman et al., 1999; Chidiac and Ross, 1999). Wild-type RGS13 inhibited GαqRC-evoked NFAT-Luc activation whereas neither RGS13(aa1-33) nor RGS13(aa93-117) had a significant effect on reporter activity (Figure 4C). Thus, RGS13 does not utilize GAP activity to inhibit CREB-mediated transcription.
The RGS13 amino terminus, which diverges considerably from other RGS proteins of the R4/B subfamily such as RGS4 and RGS16, lacks a core amphipathic α-helix contained in the other R4 family members (Gu et al., 2007) (Figure S6). We mutated individual residues in each of the CREB-interacting regions of RGS13 that differed substantially from other R4 RGS proteins. CREB did not immunoprecipitate GFP-RGS13 proteins containing R13E15→S13K15 or S104→E104 substitutions (Figure 4D), and neither mutant effectively reduced PKA-evoked CRE-Luc activation (Figure 4E). Recombinant GST-RGS13 proteins containing either of these substitutions also failed to bind pCREB in a ‘pulldown’ assay (Figure 4F). By contrast, mutation of RGS13 residues in close proximity to these amino acids (S21→R21 or I109I110→T109S110) did not affect RGS13-pCREB binding or reduce its ability to inhibit reporter activity (Figures 4D–F). Thus, several residues in two separate regions in RGS13 (aa1-33 and 93-117) are critical for its impairment of CREB-dependent transcription.
RGS13 binds the kinase-inducible domain (KID) of CREB
We determined the region(s) in CREB necessary for interaction with RGS13. CREB contains several domains with distinct functions (see Figure S7). Various CREB domains fused to the DNA binding domain of yeast GAL4(aa1-147) were used for co-immunoprecipitation studies. While no RGS13 was present in anti-GAL4 immunoprecipitates of polypeptides containing GAL4 alone, GAL4-CREB(aa1-119, Q1), (aa165-280, Q2), or (aa281-341, B-ZIP) domains, RGS13 was readily detected in GAL4-CREB(aa1-165) immunoprecipitations (Figure 5A). Thus, RGS13 may contact CREB through the KID domain, encompassing residues 119-165. Since CREB phosphorylation promotes complex formation between CREB and RGS13 (Figure 2), RGS13 probably binds phosphorylated KID (pKID).
Figure 5. RGS13 interacts with the kinase-inducible domain (KID) of CREB.
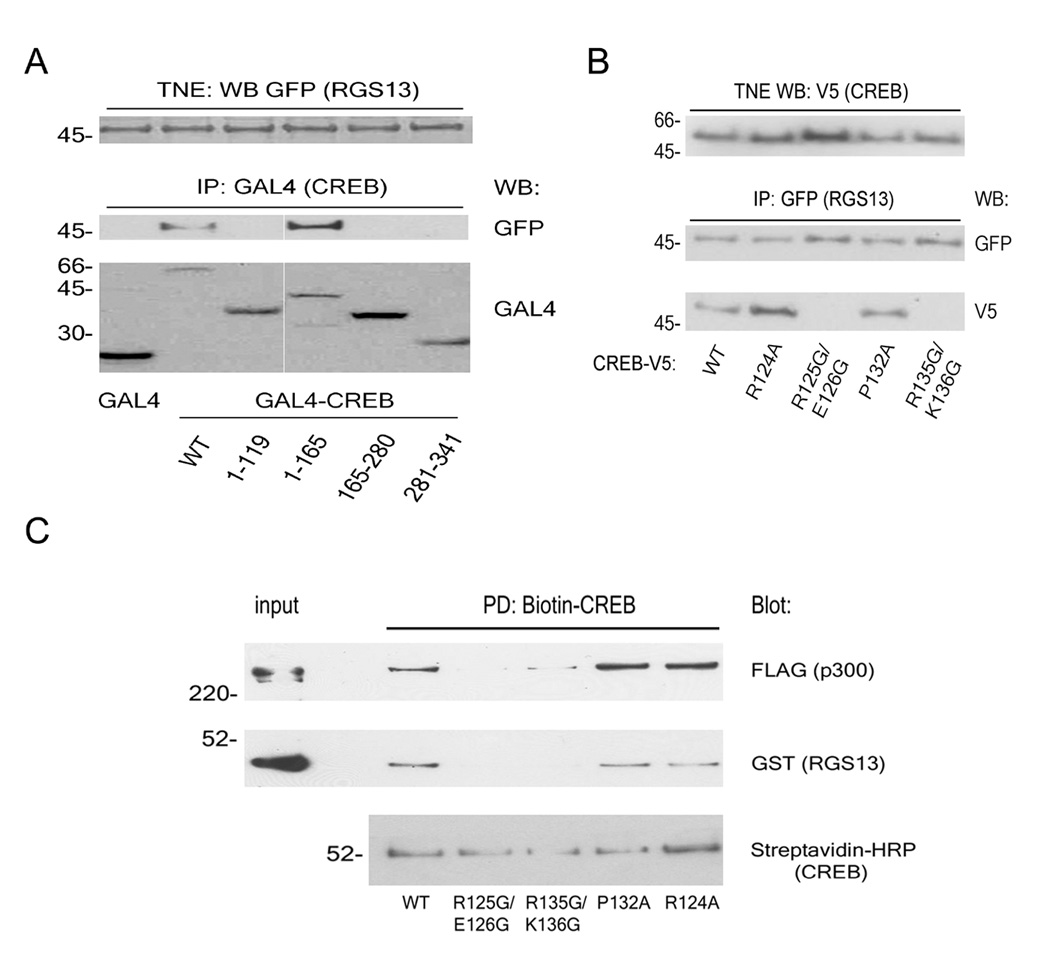
(A) Plasmids encoding GFP-RGS13, PKA, and various CREB domains fused to the DNA binding domain of GAL4(aa1-147) were expressed in HEK293T cells. NE were immunoprecipitated with anti-GAL4 followed by immunoblotting with anti-GFP and anti-GAL4. (B) Plasmids encoding CREB-V5 (wild-type or point mutants) were transfected into HEK293T cells together with PKA and wild-type GFP-RGS13 as in (A). RGS13 was immunoprecipitated from TNE with anti-GFP followed by immunodetection of CREB proteins with anti-V5. Expression of the various CREB-V5 proteins in TNE was assessed by immunoblotting (upper panel). (C) Biotinylated CREB proteins (WT or various point mutants) bound to streptavidin beads were incubated with p300 (2 nM) and GST-RGS13 (30 nM). Beads were washed extensively and protein complexes evaluated by immunoblotting. Blots in (A–C) represent at least 3 experiments with similar results.
Free KID is largely unordered in solution, and binding of pKID to a specific domain in CBP/p300 termed KIX induces formation of two α-helices [αA (aa119-129) and αB(aa134-144)] in KID (Parker et al., 1998; Radhakrishnan et al., 1997; Turjanski et al., 2008; Zor et al., 2002). Because p300 was required for RGS13-pCREB complex formation (Figure 2C), we hypothesized that the ‘active’ helical conformation of CREB facilitates RGS13 binding. To evaluate the importance of KID secondary structure and CBP/p300 interaction for RGS13 binding, we mutated individual residues in KID and expressed the CREB mutants together with GFP-RGS13 and PKA in HEK293T cells. RGS13 co-immunoprecipitated V5-tagged CREB proteins containing substitutions of two KIX contact residues (e.g. R124→A124 or P132→A132) that are not critical for helix formation or CBP binding (Parker et al., 1998; Zor et al., 2002) (Figure 5B). In contrast, RGS13 did not associate with CREB proteins containing helix-destabilizing glycine mutations in either of the αA or αB helices (R125→G125/E126→G126 or R135→G135/K136→G136). Such mutations in KID have been shown previously to reduce affinity for KIX (Zor et al., 2002). Similarly, recombinant CREB proteins containing these mutations did not bind p300 or GST-RGS13 effectively (Figure 5C). These studies suggest that α-helix formation or other secondary structures in pKID induced by p300 binding play key roles in complex formation between pCREB and RGS13.
RGS13 reduces pCREB DNA and CBP binding
Although we could not detect endogenous CBP or p300 in CREB immunoprecipitations, recombinant p300 bound pCREB equally well in the presence or absence of GST-RGS13 (Figure 4F). These results suggest that RGS13 does not impair pCREB-p300 interactions in the absence of DNA. We hypothesized that RGS13 could attenuate transcription by reducing pCREB binding to CRE sites. By using an enzyme-linked immunosorbent assay (ELISA), we observed a 2.5-fold increase in the amount of pCREB bound to CRE olignonucleotides induced by PKA (Figure 6A). This result is similar to the increase in pCREB at CRE-containing promoters in vivo after cAMP analog treatment determined by ChIP (Zhang et al., 2005). Expression of GFP-RGS13 significantly attenuated PKA-induced pCREB-CRE interactions compared to GFP alone (Figure 6A). To evaluate the impact of RGS13 deficiency on CREB-DNA interactions, we analyzed pCREB binding to CRE oligonucleotides in nuclear extracts of splenic B cells from wild-type or Rgs13−/− mice. The amount of CRE-bound pCREB was significantly higher in control and terbutaline-treated B lymphocytes from Rgs13−/− mice compared to wild-type (Figure 6B).
Figure 6. RGS13 reduces binding of pCREB to DNA and CBP.
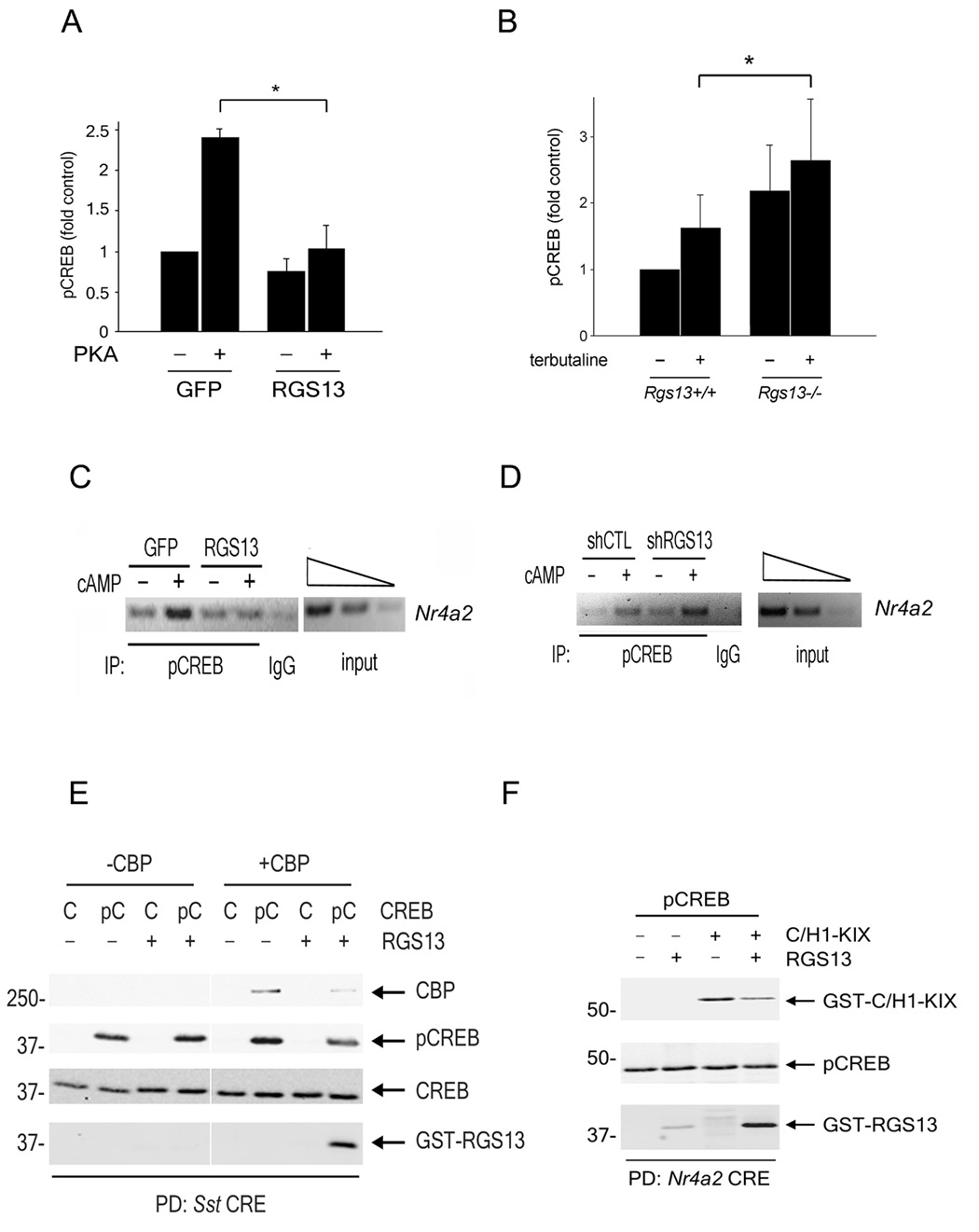
(A) HEK293T cells were transfected with plasmids encoding CREB with or without the catalytic subunit of PKA and either GFP or GFP-RGS13. NE were prepared and pCREB-DNA binding measured by quantitative ELISA. Values are the mean ± S.E.M. (‘fold’ increase over control [GFP], set as 1) of at least 3 independent experiments (*, P<0.05, one way ANOVA). (B) Augmented pCREB-CRE binding in RGS13-deficient B lymphocytes. Splenic B cells from WT or Rgs13−/− mice were incubated with IL-4 and anti-CD40 followed by stimulation with 10 µM terbutaline for 15 min. Nuclei were extracted and pCREB-DNA binding was measured as in (A) (mean ± S.E.M. from 6 mice in each group; *P=0.04, one-way ANOVA. (C–D) ChIP using an anti-pCREB antibody and amplification of Nr4a2 was done on chromatin from HEK293T cells expressing GFP or GFP-RGS13 (C) or control or RGS13-specific shRNAs (D). Images represent 3 independent experiments with similar results (E–F). Recombinant untagged pCREB (100 nM) and either full-length CBP (50 nM) (E) or C/H1-KIX (200 nM) (F) were incubated in the presence or absence of GST-RGS13 (100 nM) followed by incubation with biotinylated CRE oligonucleotides [either consensus CRE from the Sst gene (E) or Nr4a2 CRE (F)] bound to streptavidin-coupled beads. DNA-bound proteins were analyzed by immunoblotting as indicated. In (E), CREB amounts are not accurately represented in this immunoblot because prior probing for pCREB contributed to overall subsequent detection of CREB.
To determine whether these results correlated with pCREB promoter occupancy in vivo, we performed ChIP using an anti-pCREB antibody on chromatin preparations from HEK293T cells expressing GFP or GFP-RGS13. 8-bromo-cAMP treatment increased the amount of pCREB bound to the Nr4a2 promoter (Figure 6C). Expression of GFP-RGS13 reduced pCREB association with Nr4a2 in cAMP-treated cells compared to GFP alone, which paralleled the reduced amounts of Nr4a2 mRNA in cells overexpressing RGS13 (Figure 6C, Figure 1D). In contrast, knockdown of endogenous RGS13 in HEK293T cells by a specific shRNA increased Nr4a2 promoter occupancy by pCREB compared to a control shRNA (Figure 6D).
To visualize pCREB-DNA interactions directly, we used a DNA ‘pulldown’ assay with souble-stranded CRE-containing oligonucleotides to precipitate recombinant proteins. As expected, we observed RGS13 binding only to the pCREB/DNA complex (not the CREB/DNA complex) and only in the presence of CBP (Figure 6E). However, equimolar amounts of RGS13 and pCREB reduced pCREB binding to the CRE 26.6 ± 5.5% (n=7, P<0.001) (Figure 6E). Surprisingly, the amount of CBP associated with pCREB was decreased nearly five-fold by RGS13 (Figure 6E), indicating that recruitment of RGS13 to the pCREB/CBP/CRE complex not only reduces pCREB DNA binding affinity but also impairs its interaction with CBP. Relative to a positive control (Tax and pCREB, data not shown), pCREB recruitment of recombinant full length CBP to the CRE was quite weak, consistent with previously published data (Geiger et al., 2008). Therefore, we did ‘pulldown’ assays using truncations of CBP that strongly bind pCREB in the absence of co-factors. Although the KIX domain of CBP was not sufficient to recruit RGS13 to the pCREB-CRE complex (not shown), a protein containing an extended domain encompassing C/H1 and KIX (C/H1-KIX, aa302-683) (Livengood et al., 2002), behaved like full-length CBP to promote RGS13 binding to pCREB (Figure 6F). Interestingly, although RGS13 reduced the amount of C/H1-KIX retained by pCREB/CRE more than 4-fold, it did not significantly affect pCREB binding to the CRE in the presence of C/H1-KIX (90.2 ± 7.7% of control). Collectively, these studies support two possible mechanisms by which RGS13 inhibits CREB function. RGS13 reduces the apparent affinity of pCREB for the CRE in the presence of full-length CBP (Figures 6A–E). pCREB undergoes a conformational change upon binding the CRE (Sharma et al., 2007). Once pCREB is CRE-bound, it may exist in a conformation unable to accommodate CBP and RGS13 simultaneously, resulting in reduced CBP recruitment (Figure 7).
Figure 7. Proposed model for inhibition of CREB-dependent gene transcription by RGS13.
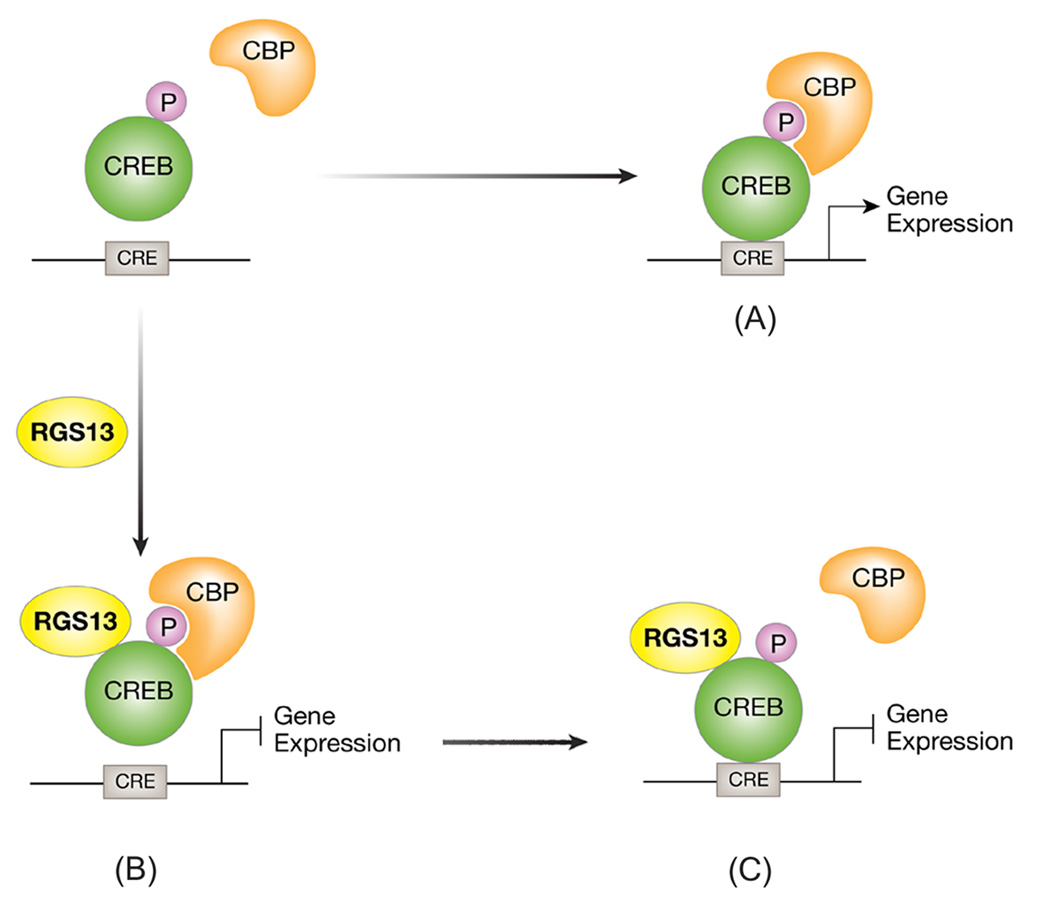
PKA phosphorylates CREB on S133, which recruits the co-activator CREB binding protein (CBP, or its paralogue p300) and promotes CRE-dependent transcription (A). RGS13 binds pCREB in the presence of CBP in solution, reducing the affinity of pCREB for promoter CRE sites (B). pCREB bound to DNA in the presence of RGS13 may be unable to bind CBP effectively (C). Either possibility would result in attenuated transcription of CREB target genes in the presence of RGS13.
DISCUSSION
This study illuminates an unexpected function of an RGS protein, RGS13, as a direct repressor of CREB-mediated gene expression. Although a large body of evidence supports a biological role for RGS proteins as regulators of GPCR signaling through their GAP function, RGS13 inhibited CREB independently of its classical GAP activity. Rather, RGS13 impaired cAMP-evoked gene transcription by accumulating in the nucleus, binding pCREB in a complex with CBP/p300 in the absence of DNA, and reducing the subsequent interaction of pCREB with CRE promoter sites. In addition, RGS13, in complex with CRE-bound pCREB, attenuated CBP recruitment. The biological relevance of these findings was supported by the enhanced CREB-dependent gene expression in HEK293T cells or primary B lymphocytes deficient in RGS13.
An unresolved issue in gene regulation is how CREB binding to DNA is regulated. Although CREB interaction with consensus CREs may be sensitive to Mg2+ concentration (Craig et al., 2001) and methylation of a central CpG within the CRE (Martinowich et al., 2003), the effect of CREB phosphorylation at S133 on its binding to naked DNA templates in vitro or CRE-containing promoters in vivo is controversial. Although it has been assumed that CREB binding to gene promoters was constitutive, ChIP and footprinting studies have shown signal-dependent recruitment of CREB to the CRE and cell-type-specific CREB promoter binding to identical CREs (Hiroi et al., 2004; Cha-Molstad et al., 2004). Our results suggest that additional nuclear factors such as RGS13 could contribute to the selective binding of CREB to target CREs.
CREB-coactivator interactions may also be subject to acute regulation. Immunofluorescence experiments with antiserum specific for the KID-KIX complex demonstrated that stimulation of cells with cAMP and non-cAMP signals induced formation of the complex to differing degrees (Wagner et al., 2000). Potentiation of CREB-CBP complex formation is mediated by the mixed lineage leukemia protein (MLL), which binds to KIX (Ernst et al., 2001). Recently, a small molecule antagonist of CREB-CBP binding was identified that also interacts with KIX (Best et al., 2004). By contrast, no endogenous cellular inhibitors of CREB-CBP interactions have been isolated. Our results suggest that RGS13 reduces the affinity of DNA-bound pCREB for CBP. As CBP ChIP would not discriminate between reduced CBP promoter binding due to diminished pCREB association with the CRE or impaired pCREB-CBP interaction, immunostaining of RGS13hi cells with the KID-KIX antiserum may illuminate how RGS13 affects CREB-coactivator interactions in vivo.
The precise mechanism whereby RGS13 impairs pCREB-DNA binding requires further study. RGS13 did not co-immunoprecipitate with the DNA binding domain of CREB (B-ZIP) when B-ZIP was expressed as a GAL4 fusion protein. It seems unlikely that RGS13’s association with KID blocks CREB-DNA binding competitively as it was recovered in pCREB-CBP-CRE complexes by DNA ‘pulldown’. Rather, since we detected less pCREB at the Nr4a2 promoter in chromatin from RGS13hi cells by ChIP, RGS13 may act allosterically to transmit a conformational change to pCREB that a) reduces its apparent affinity for the CRE and/or b) decreases its association with CBP/p300 at the CRE. Our in vitro analyses suggest that both occur. Further biochemical studies such as crystallization of the pKID-CH/1-KIX-RGS13 complex may resolve the specific details of how RGS13 interferes with CREB induction of the transcriptional machinery.
Most likely because unmodified CREB contains a largely disordered KID, RGS13 did not interact with CREB in the absence of agonist or PKA-induced phosphorylation. Accordingly, we found that KID residues that contribute to the CBP/p300-induced α-helical conformation of pCREB are crucial for RGS13 binding. Regulation of CREB-mediated transcription by RGS13 may also depend on the duration and intensity of agonist stimulation since RGS protein expression is often low in unstimulated cells but is highly transcriptionally regulated (Bansal et al., 2007). Further control of this process may be achieved by recruitment of RGS13 to the nucleus by certain stimuli.
The physiological impact of increased CREB target gene transcription in B lymphocytes deficient in RGS13 remains to be determined. RGS13 is also concentrated in neuroendocrine cells in the gastrointestinal and respiratory tracts (data not shown), suggesting possible regulation of CREB-dependent hormone secretion (Gevrey et al., 2004). Given the ubiquity and importance of CREB as a transcription factor, direct repression of CREB-mediated gene transcription by RGS13 could have an impact on numerous cellular processes for which CREB has an essential function.
EXPERIMENTAL PROCEDURES
Cells and reagents
HEK293T and NIH3T3 cells were purchased from ATCC, and Ramos cells were obtained from Dr. John Kehrl (NIAID/NIH). H6MBP-CREB and FLAG-p300 were from Invitrogen and Active Motif, respectively. PKA (catalytic subunit) was from EMD Biosciences. pEGFP-RGS13 and pEGFP-RGS4 were constructed as described (Johnson and Druey, 2002; Sullivan et al., 2000). RGS13 and CREB truncation mutants were generated by PCR and subcloned into pEGFP-C3 (BD Clontech) or pCMV-DBD (Stratagene) vectors, respectively. Point mutagenesis of GST- and GFP-RGS13 or CREB-V5 plasmids was done using the QuikChange mutagenesis kit (Stratagene). VP16-CREB(aa1-341) was from Carter Bancroft (Mt. Sinai School of Medicine) and Gαq(R183→C183) was the gift of Ken Blumer (Washington University St. Louis). pcDNA3.1-CaMKIV was the gift of J. Silvio Gutkind (NIDCR/NIH). pFc-PKA, NFAT-Luc, and CRE-Luc plasmids were purchased from Stratagene. Antibodies sources: OCA-B, GFP, GAL4 (Santa Cruz); CREB and pCREB (Cell Signaling Technology); V5 (Invitrogen). Polyclonal anti-RGS13 has been characterized previously (Shi et al., 2002). Terbutaline, forskolin, 8-bromo cAMP, and 8-pCPT cAMP were purchased from Sigma, and anti-IgM F(ab’)2 was from Jackson Immunoresearch.
Protein pulldown
Biotinylated GFP or GFP-RGS13, or CREB was synthesized from pCDNA3.1 TOPO plasmids using Transcend biotinylated lysine tRNA and TNT T7 quick-coupled in vitro transcription/translation system (Promega) and purified on streptavidin-agarose (EMD Biosciences). Recombinant CREB (25 nM) was phosphorylated by PKA in labeling buffer (25 mM Tris pH 7.4, 2 mM EDTA, 10 mM MgCl2, 100 µM ATP, 0.1 mg/ml BSA, 2 mM β-mercaptoethanol, 10 mM NaF) followed by incubation with p300 (2 nM) and biotinylated proteins (~10 nM) in buffer containing 200 mM Tris pH 7.5, 200 mM NaCl, 5% glycerol, and 1% Triton X-100.
DNA pulldown
Recombinant GST-RGS13 (100 nM), unphosphorylated CREB (100 nM) (Lopez et al., 2007) or S133-phosphorylated CREB (Geiger et al., 2007) and CBP (50 nM) (Kraus et al., 1999), GST-KIX (Giebler et al., 1997) or GST-C/H1-KIX (200 nM) (Livengood et al., 2002) were incubated in HM buffer (20 mM HEPES pH 7.9, 100 mM KCl, 6.25 mM MgCl2, 10% glycerol, 0.1% NP-40, 2 mM DTT) plus 50 µM ATP, 40 ng/µl poly dA:dT and 80 µM acetyl CoA for 15 min. at 30°C followed by addition of biotinylated CRE-containing oligonucleotides (50 nM) coupled to streptavidin magnetic beads (Dynal) for 15 min. at 30°C. The top strand, 5'-biotinylated sequences of the complementary oligonucleotides were (CRE sequences underlined): Nr4a2, 5'-Bio/TCCCACCTTAAAATCGGCCCTGCTCGTGACGTCAGGTCGGAAATATACCAAAGCG; somatostatin (Sst), 5'-Bio/ATCAGGCTTCCTCCTCCTAGCCTGACGTCAGAGAGAGAGGTCGCC. Following binding, beads were washed and analyzed by immunoblotting. Quantitation of pCREB levels was done using Quantity One software (Bio-Rad).
RNA interference
CREB-specific (SMARTpool) or scrambled control siRNA (Millipore) was transfected into HEK293T cells using Lipofectamine 2000 (Invitrogen). For RGS13 knockdown, a double-stranded oligonucleotide predicted to form a short, hairpin siRNA (shRNA) (AAGGCGATTGGTCAGTCGATTGATATCCGTCGACTGACCAATCGCCTT) was subcloned into pSIREN-RetroQ Zs-Green (Clontech). This plasmid or a similar control plasmid containing a scrambled shRNA sequence was transfected into 293T cells using Lipofectamine.
Reporter assays
We analyzed activity of CRE-Luc and NFAT-Luc reporters as described (Johnson and Druey, 2002).
RT-PCR
Total RNA was isolated and reverse transcribed into cDNA using the Superscript III RT kit (Invitrogen). Primers for FosB and Nr4a2 have been described (Holmes and Zachary, 2004; Tullai et al., 2007).
Immunoprecipitation and immunofluorescence
Inmunoprecipitation was performed essentially as described (Bansal et al., 2007). For immunofluorescence, cells were seeded in chamber wells and transfected as indicated. We obtained live cell images using a Leica SP2 laser scanning confocal microscope. For co-localization studies, the slides were stained with rabbit anti-pCREB (87G3, Cell Signaling Technology, 1:30) overnight followed by Alexa594-conjugated anti-rabbit IgG.
pCREB DNA binding assay
We quantitated pCREB binding to consensus CRE oligonucleotide sequences using TransAM ELISA (ActiveMotif) per the manufacturer’s protocol.
Chromatin immunoprecipitation (ChIP)
HEK293T were fixed with 1% formaldehyde after stimulation. Enzymatically sheared chromatin (ChIP Express kit, Active Motif) was immunoprecipitated with polyclonal rabbit anti-pCREB (the gift of M. Montminy) or rabbit IgG using the EZ ChIP kit (Millipore). Purified DNA was used for amplification of an Nr4a2 CRE-containing sequence previously described (Conkright et al., 2003).
Splenic B cell isolation and stimulation
We isolated naïve B lymphocytes from minced spleens by negative selection using anti-CD43 magnetic beads (Miltenyi Biotech). B cells (routinely 85–90% pure) were stimulated with 10 µM terbutaline for 3 days in the presence of IL-4 (1 ng/ml, Peprotech) and anti-CD40 antibody (10 µg/ml, BD Biosciences) as described (Podojil et al., 2004) before lysis and assessment of OCA-B expression by immunoblotting. For measurement of pCREB DNA binding, cells were incubated with IL-4 and anti-CD40 for 24 hrs. followed by 15 min. stimulation with 10 µM terbutaline.
Rgs13−/− mice
These mice were generated as described (Bansal et al., 2008). All animal experiments were approved by and conducted in accordance with the Animal Care and Use Committee, NIAID, NIH.
Statistical analysis
Sigma Plot software was used to determine statistical significance by Students t-test for two groups or analysis of variance (ANOVA) for multiple groups. P values <0.05 were considered to be significant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank K. Ravnskjaer and M. Montminy (Salk Institute, La Jolla, California, H. Rosenberg (NIAID/NIH) for reagents and helpful advice, J. Kehrl (NIAID, NIH) for the RGS13 antibody, and D. Metcalfe for his support. This research was supported by the Intramural Research Program of the NIH, NIAID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bansal G, Druey KM, Xie Z. R4 RGS proteins: Regulation of G-protein signaling and beyond. Pharmacol Ther. 2007;116:473–495. doi: 10.1016/j.pharmthera.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal G, Xie Z, Rao S, Nocka KH, Druey KM. Suppression of immunoglobulin E-mediated allergic responses by regulator of G protein signaling 13. Nat Immunol. 2008;9:73–80. doi: 10.1038/ni1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best JL, Amezcua CA, Mayr B, Flechner L, Murawsky CM, Emerson B, Zor T, Gardner KH, Montminy M. Identification of small-molecule antagonists that inhibit an activator: coactivator interaction. Proc Natl Acad Sci U S A. 2004;101:17622–17627. doi: 10.1073/pnas.0406374101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T. Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem. 1999;274:34483–34492. doi: 10.1074/jbc.274.48.34483. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Keller DM, Yochum GS, Impey S, Goodman RH. Cell-type-specific binding of the transcription factor CREB to the cAMP-response element. Proc Natl Acad Sci U S A. 2004;101:13572–13577. doi: 10.1073/pnas.0405587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chidiac P, Ross EM. Phospholipase C-beta1 directly accelerates GTP hydrolysis by Galphaq and acceleration is inhibited by Gbeta gamma subunits. J Biol Chem. 1999;274:19639–19643. doi: 10.1074/jbc.274.28.19639. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Guzman E, Flechner L, Su AI, Hogenesch JB, Montminy M. Genome-wide analysis of CREB target genes reveals a core promoter requirement for cAMP responsiveness. Mol Cell. 2003;11:1101–1108. doi: 10.1016/s1097-2765(03)00134-5. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Montminy M. CREB: the unindicted cancer co-conspirator. Trends Cell Biol. 2005;15:457–459. doi: 10.1016/j.tcb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Craig JC, Schumacher MA, Mansoor SE, Farrens DL, Brennan RG, Goodman RH. Consensus and variant cAMP-regulated enhancers have distinct CREB-binding properties. J Biol Chem. 2001;276:11719–11728. doi: 10.1074/jbc.M010263200. [DOI] [PubMed] [Google Scholar]
- Dragunow M. CREB and neurodegeneration. Front Biosci. 2004;9:100–103. doi: 10.2741/1197. [DOI] [PubMed] [Google Scholar]
- Ernst P, Wang J, Huang M, Goodman RH, Korsmeyer SJ. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol Cell Biol. 2001;21:2249–2258. doi: 10.1128/MCB.21.7.2249-2258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger TR, Sharma N, Kim YM, Nyborg JK. The HTLV-1 Tax Protein Confers CBP/p300 Recruitment and Transcriptional Activation Properties to Phosphorylated CREB. Mol Cell Biol. 2007 doi: 10.1128/MCB.01657-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger TR, Sharma N, Kim YM, Nyborg JK. The human T-cell leukemia virus type 1 tax protein confers CBP/p300 recruitment and transcriptional activation properties to phosphorylated CREB. Mol Cell Biol. 2008;28:1383–1392. doi: 10.1128/MCB.01657-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gevrey JC, Malapel M, Philippe J, Mithieux G, Chayvialle JA, Abello J, Cordier-Bussat M. Protein hydrolysates stimulate proglucagon gene transcription in intestinal endocrine cells via two elements related to cyclic AMP response element. Diabetologia. 2004;47:926–936. doi: 10.1007/s00125-004-1380-0. [DOI] [PubMed] [Google Scholar]
- Giebler HA, Loring JE, van Orden K, Colgin MA, Garrus JE, Escudero KW, Brauweiler A, Nyborg JK. Anchoring of CREB binding protein to the human T-cell leukemia virus type 1 promoter: a molecular mechanism of Tax transactivation. Mol Cell Biol. 1997;17:5156–5164. doi: 10.1128/mcb.17.9.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S, He J, Ho WT, Ramineni S, Thal DM, Natesh R, Tesmer JJ, Hepler JR, Heximer SP. Unique hydrophobic extension of the RGS2 amphipathic helix domain imparts increased plasma membrane binding and function relative to other RGS R4/B subfamily members. J Biol Chem. 2007 doi: 10.1074/jbc.M702685200. [DOI] [PubMed] [Google Scholar]
- Hiroi H, Christenson LK, Chang L, Sammel MD, Berger SL, Strauss JF., 3rd Temporal and spatial changes in transcription factor binding and histone modifications at the steroidogenic acute regulatory protein (stAR) locus associated with stAR transcription. Mol Endocrinol. 2004;18:791–806. doi: 10.1210/me.2003-0305. [DOI] [PubMed] [Google Scholar]
- Holmes DI, Zachary I. Placental growth factor induces FosB and c-Fos gene expression via Flt-1 receptors. FEBS Lett. 2004;557:93–98. doi: 10.1016/s0014-5793(03)01452-2. [DOI] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16:1211–1227. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Johnson EN, Druey KM. Functional characterization of the G protein regulator RGS13. J Biol Chem. 2002;277:16768–16774. doi: 10.1074/jbc.M200751200. [DOI] [PubMed] [Google Scholar]
- Kraus WL, Manning ET, Kadonaga JT. Biochemical analysis of distinct activation functions in p300 that enhance transcription initiation with chromatin templates. Mol Cell Biol. 1999;19:8123–8135. doi: 10.1128/mcb.19.12.8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livengood JA, Scoggin KE, Van Orden K, McBryant SJ, Edayathumangalam RS, Laybourn PJ, Nyborg JK. p53 Transcriptional activity is mediated through the SRC1-interacting domain of CBP/p300. J Biol Chem. 2002;277:9054–9061. doi: 10.1074/jbc.M108870200. [DOI] [PubMed] [Google Scholar]
- Lopez DI, Mick JE, Nyborg JK. Purification of CREB to apparent homogeneity: removal of truncation products and contaminating nucleic acid. Protein Expr Purif. 2007;55:406–418. doi: 10.1016/j.pep.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Parker D, Ferreri K, Nakajima T, LaMorte VJ, Evans R, Koerber SC, Hoeger C, Montminy MR. Phosphorylation of CREB at Ser-133 induces complex formation with CREB-binding protein via a direct mechanism. Mol Cell Biol. 1996;16:694–703. doi: 10.1128/mcb.16.2.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker D, Jhala US, Radhakrishnan I, Yaffe MB, Reyes C, Shulman AI, Cantley LC, Wright PE, Montminy M. Analysis of an activator:coactivator complex reveals an essential role for secondary structure in transcriptional activation. Mol Cell. 1998;2:353–359. doi: 10.1016/s1097-2765(00)80279-8. [DOI] [PubMed] [Google Scholar]
- Podojil JR, Kin NW, Sanders VM. CD86 and beta2-adrenergic receptor signaling pathways, respectively, increase Oct-2 and OCA-B Expression and binding to the 3'-IgH enhancer in B cells. J Biol Chem. 2004;279:23394–23404. doi: 10.1074/jbc.M313096200. [DOI] [PubMed] [Google Scholar]
- Podojil JR, Sanders VM. CD86 and beta2-adrenergic receptor stimulation regulate B-cell activity cooperatively. Trends Immunol. 2005;26:180–185. doi: 10.1016/j.it.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Popov S, Yu K, Kozasa T, Wilkie TM. The regulators of G protein signaling (RGS) domains of RGS4, RGS10, and GAIP retain GTPase activating protein activity in vitro. Proc Natl Acad Sci U S A. 1997;94:7216–7220. doi: 10.1073/pnas.94.14.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin XF, Reichlin A, Luo Y, Roeder RG, Nussenzweig MC. OCA-B integrates B cell antigen receptor-, CD40L- and IL 4-mediated signals for the germinal center pathway of B cell development. Embo J. 1998;17:5066–5075. doi: 10.1093/emboj/17.17.5066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan I, Perez-Alvarado GC, Parker D, Dyson HJ, Montminy MR, Wright PE. Solution structure of the KIX domain of CBP bound to the transactivation domain of CREB: a model for activator:coactivator interactions. Cell. 1997;91:741–752. doi: 10.1016/s0092-8674(00)80463-8. [DOI] [PubMed] [Google Scholar]
- Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B, Schutz G. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci U S A. 1998;95:4481–4486. doi: 10.1073/pnas.95.8.4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Lopez DI, Nyborg JK. DNA binding and phosphorylation induce conformational alterations in the kinase-inducible domain of CREB. Implications for the mechanism of transcription function. J Biol Chem. 2007;282:19872–19883. doi: 10.1074/jbc.M701435200. [DOI] [PubMed] [Google Scholar]
- Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–861. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- Shi GX, Harrison K, Wilson GL, Moratz C, Kehrl JH. RGS13 regulates germinal center B lymphocytes responsiveness to CXC chemokine ligand (CXCL)12 and CXCL13. J Immunol. 2002;169:2507–2515. doi: 10.4049/jimmunol.169.5.2507. [DOI] [PubMed] [Google Scholar]
- Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- Sullivan BM, Harrison-Lavoie KJ, Marshansky V, Lin HY, Kehrl JH, Ausiello DA, Brown D, Druey KM. RGS4 and RGS2 bind coatomer and inhibit COPI association with Golgi membranes and intracellular transport. Mol Biol Cell. 2000;11:3155–3168. doi: 10.1091/mbc.11.9.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullai JW, Chen J, Schaffer ME, Kamenetsky E, Kasif S, Cooper GM. Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J Biol Chem. 2007;282:9482–9491. doi: 10.1074/jbc.M700067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turjanski AG, Gutkind JS, Best RB, Hummer G. Binding-induced folding of a natively unstructured transcription factor. PLoS Comput Biol. 2008;4:e1000060. doi: 10.1371/journal.pcbi.1000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BL, Bauer A, Schutz G, Montminy M. Stimulus-specific interaction between activator-coactivator cognates revealed with a novel complex-specific antiserum. J Biol Chem. 2000;275:8263–8266. doi: 10.1074/jbc.275.12.8263. [DOI] [PubMed] [Google Scholar]
- Willars GB. Mammalian RGS proteins: multifunctional regulators of cellular signalling. Semin Cell Dev Biol. 2006;17:363–376. doi: 10.1016/j.semcdb.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Wolf I, Pevzner V, Kaiser E, Bernhardt G, Claudio E, Siebenlist U, Forster R, Lipp M. Downstream activation of a TATA-less promoter by Oct-2, Bob1, and NF-kappaB directs expression of the homing receptor BLR1 to mature B cells. J Biol Chem. 1998;273:28831–28836. doi: 10.1074/jbc.273.44.28831. [DOI] [PubMed] [Google Scholar]
- Yan G, Chen X, Bancroft C. A constitutively active form of CREB can activate expression of the rat prolactin promoter in non-pituitary cells. Mol Cell Endocrinol. 1994;101:R25–R30. doi: 10.1016/0303-7207(94)90255-0. [DOI] [PubMed] [Google Scholar]
- Zhang CY, Wu YL, Boxer LM. Impaired proliferation and survival of activated B cells in transgenic mice that express a dominant-negative cAMP-response element-binding protein transcription factor in B cells. J Biol Chem. 2002;277:48359–48365. doi: 10.1074/jbc.M209329200. [DOI] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zor T, Mayr BM, Dyson HJ, Montminy MR, Wright PE. Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J Biol Chem. 2002;277:42241–42248. doi: 10.1074/jbc.M207361200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.