

Abstract
Apoptosis, or programmed cell death (PCD), is an important physiological mechanism, through which the human immune system regulates homeostasis and responds to diverse forms of cellular damage. PCD may also be involved in immune counteraction to microbial infection. Over the past decade, the amount of research on bacteria-induced PCD has grown tremendously, and the implications of this mechanism on immunity are being elucidated. Some pathogenic bacteria actively trigger the suicide response in critical lineages of leukocytes that orchestrate both the innate and adaptive immune responses; other bacteria proactively prevent PCD to benefit their own survival and persistence. Currently, the microbial virulence factors, which represent the keys to unlocking the suicide response in host cells, are a primary focus of this field. In this review, we discuss these bacterial “apoptosis regulatory molecules” and the apoptotic events they either trigger or prevent, the host target cells of this regulatory activity, and the possible ramifications for immunity to infection. Gram-positive pathogens including Staphylococcus, Streptococcus, Bacillus, Listeria, and Clostridia species are discussed as important agents of human infection that modulate PCD pathways in eukaryotic cells.
Keywords: Bacteria, apoptosis, programmed cell death, pathogenesis, immunity
INTRODUCTION
A number of excellent reviews have been published in recent years on the topic of bacteria-induced apoptosis, or programmed cell death (PCD) [1-7]. These comprehensive reports have brought together the plethora of research studies conducted over the past decade in this field, beginning with the first demonstration that enteropathogenic Shigella induces PCD in macrophages under certain conditions [8]. Following that seminal study by Zychlinsky et al. (1992), other investigators illustrated convincingly that many other species of pathogenic bacteria such as Streptococcus [9] and Mycobacterium [10] and certain viruses, including human immunodeficiency virus (HIV) [11], directly manipulate the apoptotic machinery within human cells to benefit their own survival and persistence. This explosion of new information on bacteria-induced apoptosis and the diverse molecular mechanisms triggered by bacteria to bring about PCD argues that apoptosis plays a key role in microbial pathogenesis and antibacterial immunity.
Gram-negative and gram-positive bacteria can trigger the suicide response in selected lineages of eukaryotic host cells [12, 13]. This suggests that some common bacterial structural component may function meaningfully or coincidentally with infection to bring about PCD in the harboring host cell. To some degree, this concept is supported by available data on genera such as Mycobacterium, in which purified lipoarabinomannan or its precursors from different species influences apoptotic pathways (albeit in contrasting ways) [14-16]. In larger divisions of bacteria such as gram-negative Gracilicutes [17] the lipopolysaccharide component of the outer cell membrane exerts a proapoptotic effect in several dissimilar situations [13, 18, 19], although it can also prevent PCD in certain cell types under different conditions [20, 21]. In the case of gram-positive pathogens, there have been few reports of shared molecules that are able to modulate PCD pathways. Moreover, it appears that the molecular mechanisms of PCD induced by gram-positive bacteria differ from those triggered by their gram-negative counterparts [22]. One conserved gram-positive factor studied to date is lipotechoic acid, though investigations into its role as a PCD regulator in eukaryotic host cells have indicated disparate effects on different cell types [23-25]. Other apoptogenic molecules identified in gram-positive pathogens so far include exotoxins (e.g., lethal factor of Bacillus anthracis) [25, 26], cytolysins (e.g., listeriolysin O of Listeria monocytogenes) [27], and bacterial hemolysins (e.g., β-hemolysin of Streptococcus agalactiae) [28, 29].
At the most basic level, apoptosis regulatory molecules (ARMs) in gram-positive bacteria can be classified as secreted exoproducts (e.g., exotoxins such as pneumolysin) [30] and nonsecreted endoproducts (e.g., lipotechoic acid) [31]. Functionally, the molecular mechanisms resulting in PCD that are brought about by gram-positive bacteria are heterogeneous. PCD pathways involving both the intrinsic mechanism (i.e., mitochondrial function disruption) [32, 33] and extrinsic signaling through death receptors [34, 35] are triggered by various gram-positive bacteria (Table 1). Thus, similar to PCD triggered by parasitic infections [36], there appears to be limited conservation of the mechanisms underlying the PCD response of eukaryotic host cells to gram-positive bacteria. This review summarizes the current knowledge about ARMs produced by gram-positive bacteria, their known target cell repertoire, the PCD events that these ARMs trigger, and the potential consequences for immunity to infection.
Table 1. Apoptosis Regulatory Molecules of Gram-Positive Bacteria and their Cellular Targets.
| Bacteria & Disease Presentations | ARMs* | Identified Target Cell Type* | Reference |
|---|---|---|---|
|
Staphylococcus aureus (skin and soft tissue, infections of bones and joints, respiratory tract infections, endovascular, gastrointestinal systems, sepsis) |
SEB TSST-1 α-toxin SpA SEA, D, E ?† |
CD4+, T, B, Epi, Endo, Eosin B, Mono T, Mono, Endo B T keratinocytes, Osteo, Chondr |
[37-75] [76, 77] [49, 78-81] [82, 83] [84-86] [87-90] |
|
Streptococcus agalactiae (pneumonia, meningitis, septic arthritis, osteomyelitis, sepsis) |
β-hemolysin GRF ? |
MØ, Hep MØ neurons, Mono |
[28, 29, 91] [92] [93-96] |
|
Streptococcus pneumoniae (otitis media, pneumonia, sinusitis, meningitis, sepsis) |
PLY H2O2 cell wall ? |
MØ, neurons, microglia neurons, microglia Mono DCs, PMNLs, Epi, T |
[97-99] [97] [100] [101-109] |
|
Streptococcus pyogenes (pharyngitis-tonsillitis, STTS, necrotizing fasciitis, impetigo, erysipelas, scarlet fever, post infection autoimmune disease) |
NADase SLO SpeB glycoprotein ? |
Keratinocytes Keratinocytes MØ, Epi fibrosarcoma cells PMNLs |
[110] [110] [111-113] [114] [115] |
|
Bacillus anthracis (anthrax, edema, sepsis) |
LeTx PA |
MØ, Endo, PMNLs Mono |
[25, 26, 116-119] [116, 120-122] |
|
Listeria monocytogenes (granulomatosis infantiseptica, gastroenteritis, meningitis) |
LLO ? |
T, B, DCs Hep, Epi, γδ, CD4+, CD8+, B, Neurons, NK cells |
[27, 123-126] [127-131] [132-134] |
|
Clostridium difficile (diarrhea, colitis, megacolon) |
exotoxin A exotoxin B |
Col, Mast, T, Epi, Eosin, Hep Epi, Hep, fibroblasts, Endo |
[135-143] [143-148] |
|
Clostridium perfringens (diarrhea, enteritis, gangrene) |
CPE | Epi | [149, 150] |
PCD induced in most cell types; those cell types in which PCD is inhibited are underlined.
Question mark denotes unidentified ARMs.
Abbreviations: ARMs, apoptosis regulatory molecules; SEB, staphylococcal enterotoxin B; CD4+, CD4+ T cells; T, T cells; B, B cells; Epi, epithelial cells; Endo, endothelial cells; Eosin, eosinophils; TSST-1, toxic shock syndrome toxin-1; Mono, monocytes; SpA, superantigenic protein A; SEA, staphylococcal enterotoxin A; SEE, staphylococcal enterotoxin E; Osteo, osteoblasts; Chondr, chondrocytes; MØ, macrophages; Hep, hepatocytes; GRF, glucose-regulated factor; PLY, pneumolysin; DCs, dendritic cells; PMNLs, polymorphonuclear leukocytes; NADase, nicotinamide adenine dinucleotide-glucohydrolase; SLO, streptolysin O; SpeB, streptococcal pyrogenic exotoxin B; LeTx, lethal toxin; PA, protective antigen; LLO, listeriolysin O; γδ, γδ T cells; CD8+, CD8+ T cells; NK, natural killer cells; Col, colonocytes; Mast, mast cells; CPE, Clostridium perfringens enterotoxin.
STEADY-STATE PCD MACHINERY AND FUNCTION
Apoptosis plays an important role in immune system function; PCD that occurs normally in development and homeostasis is balanced with that stimulated after cytotoxic insult, metabolic imbalance, or infectious attack [151-156]. Removal of useless or autoreactive cells by apoptotic clonal deletion (negative selection) is central to the development of B- and T-cell repertoires. In immune responses to infection, PCD maintains appropriately sized T-cell memory pools after disease resolution by PCD-driven removal of antigen-expanded T-cell clones [157-160]. PCD also regulates the balance between T-cell proliferation and T-cell death in some infections [161-163]. Human cells commit to PCD via one of two generalized activation pathways: First, death receptor-independent deregulation of mitochondrial function, during which cytosolic cytochrome c binds with apoptotic protease activating factor-1 (APAf-1) and cleaved procaspase-9 to form the apoptosome (intrinsic PCD) [32, 33, 164-168]. Second, activation of the death receptor pathway through ligation of CD95 (FAS/APO-1) [169], tumor necrosis factor (TNF)-α receptor 1, or other death receptors at the eukaryotic cell surface may lead to the activation of caspase-8, cleavage of procaspase-3 (or effector caspases-6 or -7), and terminal extrinsic PCD events [34, 35, 170] (Fig. 1). Toxic proteins released from cytotoxic lymphocytes and natural killer (NK) cells such as perforin and granzyme B may also activate extrinsic PCD [171]. Both pathways are tightly regulated so that under normal conditions unnecessarily high levels of energy-dependent apoptosis are circumvented [172], yet a state of readiness is maintained. The capacity for rapid PCD responses, which can be completed within 30 minutes of initial signaling, is tied to the presence of large pools of enzymatically inactive forms of cysteine-dependent aspartate-directed specific proteases (caspases) within the cytoplasm of eukaryotic cells under resting conditions [34, 173, 174]. In human cells, a family of at least 13 caspases act in concert, functioning as initiators (e.g., caspase-8, -9, -10) and effectors (e.g., caspase-3, -6, -7) of PCD [175-179]. Caspases can also cleave proforms of cytokines, such as interleukin-1, that are involved in inflammation [176]. Procaspase zymogens undergo rapid proteolytic cleavage at specific aspartate residues, which can be self-induced or triggered by other caspases after appropriate proapoptotic stimulation [173, 177]. Upon reaching a critical threshold, complete execution of PCD is achieved, resulting in cleavage of protein substrates required for cellular integrity, e.g., poly(ADP-ribose)polymerase (PARP), inhibitor of caspase-activated deoxyribonuclease (ICAD), and DNA degradation [180-182]. Caspases target as many as 280 proteins [183]. Functional redundancy and compensation in the caspase system ensures PCD can be achieved when necessary [184]. Ultimately, both PCD pathways converge at the point of effector caspase-3 or -6 activation [160]. This leads to chromatin condensation, externalization of cell membrane lipid phosphatidylserine [185], membrane blebbing, cell shrinkage, and cell disassembly into apoptotic bodies. Phagocytosis of suicidal cells and apoptotic bodies prevents the spillage of intracellular contents from dying cells, thereby limiting inflammation [173]. Macrophages and other phagocytic cells recognize apoptotic cells by phosphatidylserine receptors [186], and can actually lead to the release of anti-inflammatory cytokines [187] and desensitization [188]. Complex regulatory checkpoints for PCD exist at all levels of transcription, translation and posttranslational modification [177]. In particular, the network of nuclear factor κB (NF-κB) transcription factors lies at the crossroads of many PCD signaling pathways and typically exerts prosurvival influence [189]. An additional transcription factor that is central to the regulation of PCD is p53, which influences cell cycle arrest, DNA repair, cell proliferation, and can induce PCD in cells harboring genetic mutations [190].
Fig. (1).
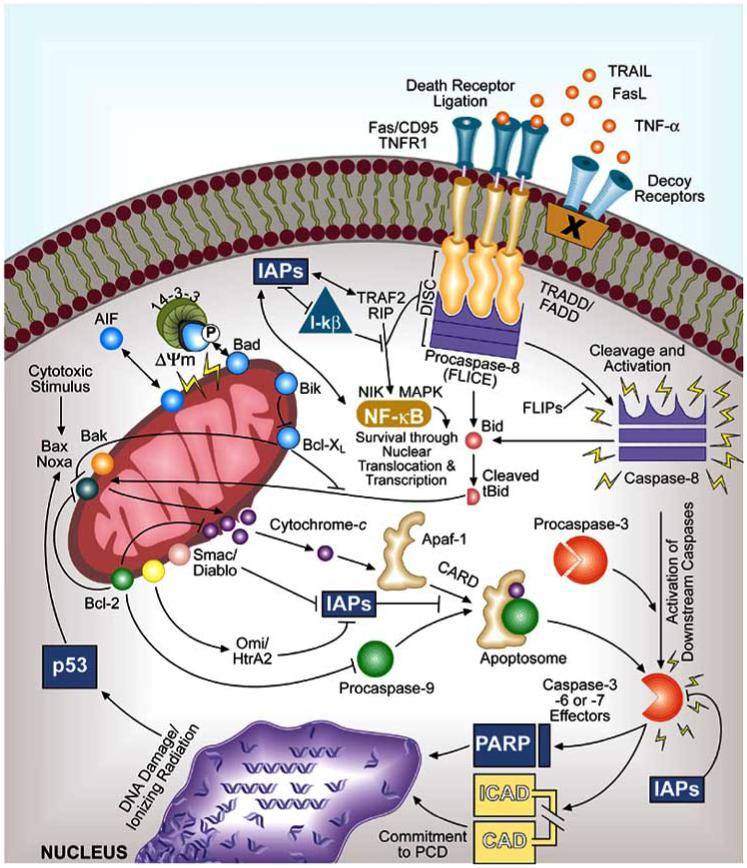
Mechanisms of intrinsic and extrinsic programmed cell death (PCD) in eukaryotic cells. Shown on the left is the current paradigm for intrinsic PCD, a pathway characterized by mitochondrial dysfunction, loss of mitochondrial membrane integrity (potential), and integrated signal transduction through various BCL-2 family members. Numerous BCL-2 proteins reside in the inner mitochondrial membrane space and contribute to cytochrome c release, subsequent caspase-9 activation, apoptosome formation, procaspase-3 cleavage, and terminal PCD events. As shown on the upper right, membrane-displayed death receptors relay initial receptor ligation by apoptotic ligands that provoke extrinsic PCD through death-effector domain-containing intracellular adaptor molecules TRADD and FADD with their corresponding procaspase protein-docking regions; these regions act as signal transducers for initiator caspase-8 or -10 activation. Subsequent downstream effector caspase function results from cleavage of procaspase-3, -6, or -7 and then acts on suicide effectors such as PARP, which causes DNA breakdown and PCD. Various activators, inhibitors, and decoys regulate both intrinsic and extrinsic pathways; these regulators keep PCD pathways in check at various levels of early-, intermediate-, and late-stage events. Induction of the nuclear transcription factor NF-κB by its inducing kinase NIK (or MAPK) activates antiapoptotic gene transcription and regulates death pathways through factors such as IAPs and FLIPs. In this manner, NF-κB exerts prosurvival effects [189]. Normally, NF-κB activity is suppressed by I-κB, which is also regulated through interactions with IAPs.
Intrinsic Mitochondria-Mediated PCD
Once the intrinsic pathway of PCD is triggered, the ensuing cellular changes are characterized by loss of mitochondrial transmembrane potential (ΔΨm) and cellular reduction-oxidation steady state, disruption of the electron transport chain, oxidative phosphorylation, and adenosine triphosphate (ATP) production, and release of proteins that activate caspases [32, 33]. Environmental cues that can induce intrinsic PCD include stressors such as cytotoxic treatments (e.g., UV light, irradiation), depletion of essential nutrients or growth factors (e.g., cytokines), chemical imbalances (e.g., Ca2+ overload) and chemotherapeutic insults [160, 191]. Several bacterial toxins also stimulant intrinsic PCD [163]. Cytotoxic signals open permeability transition pores in the inner mitochondrial membrane, which induces loss of membrane potential. Subsequent release of mitochondrial cytochrome c into the cytoplasm may occur as a result of swelling of the mitochondria and outer membrane rupture. Alternatively, direct pore formation in the outer mitochondrial membrane due to the actions of BAK and/or BAX may facilitate the passage of cytochrome c. Free cytochrome c then binds to APAf-1 in the cytoplasm [177, 192, 193]. Subsequent binding with procaspase-9 forms the apoptosome, which leads to cleavage and activation of caspase-3 [177] and PCD (Fig. 1). Mitochondria-induced PCD can also occur independently of caspase activation [194]. Intrinsic PCD is regulated by an intricate system of agonistic and antagonistic mitochondrial proteins that display sequence and structural similarity to the B-cell leukemia (BCL)-2 homology domain [160, 195]. Proteins containing BCL-2 homology domains (BH1 through BH4) regulate the balance of PCD events occurring at the mitochondria by homodimerizing, heterodimerizing, and neutralizing the activities of functionally competing partner proteins through complementary binding domains [191, 196]. A key member of the BCL-2 family of proteins is BCL-XL, which resides in the inner mitochondrial membrane space and promotes cell survival by binding to and suppressing the activities of apoptogenic BCL-2 family member proteins such as BAX [191, 196]. Twenty BCL-2 family member proteins have been identified: A1, BCL-W, BCL-XL, BOO, MCL-1, BAX, BAK, BOK, BCL-XS, BCL-GL, BAD, BCL-GS, BID, BIK, BIM, BLK, BMF, HRK, NOXA and PUMA (reviewed in [160]). Several gram-positive bacteria and their ARMs manipulate the levels, phosphorylation status, and functional availability of BCL-2 homology proteins, thereby influencing intrinsic PCD.
Extrinsic Death Receptor-Mediated PCD
Death receptors on the surface of host cells include CD95, TNF receptors, and death receptors 3 through 5 [34, 35, 170, 197]. Ligation of death receptors induces cleavage of procaspase-8 (also called FLICE), which, in turn, activates procaspase-3 and leads to DNA fragmentation and cell death (Fig. 1) [34]. Signal transduction via recruitment of the FAS-associated death domain (FADD) adaptor protein and formation of the death-inducing signaling complex is essential for caspase-8 activation by both CD95-FAS ligand (FASL) and the TNF-TNF receptor 1-TNF receptor-associated death domain (TRADD) adaptor protein complex [34]. Transmembrane death receptors initiate PCD by binding to death domain regions of cytoplasmic adaptor proteins, causing structural rearrangements and recruitment of procaspase-8 or procaspase-10 to the complex, where the caspases are activated [160]. BCL-2 homology proteins, with the notable exception of BID [198, 199], play a minimal role in regulating death receptor-mediated PCD [160]. Instead, FLICE inhibitory proteins (FLIPS), which mimic procaspase-8 structurally but have no active site for death effector domain-induced activation, are important regulators of extrinsic PCD [200]. FLIPS compete with death receptor signal transduction at the adaptor protein level by binding to the activating regions in the cytoplasmic death domain sites of FADD/TRADD and competing with signal transduction for activation of caspase-8 [160]. Additional regulators of extrinsic PCD include components that exert effects on the signaling complex, such as receptor-interacting protein (RIP) and TNF receptor-associated factor-2 (TRAF2); RIP and TRAF2 interact with the ubiquitous cellular inhibitor of apoptosis proteins (IAPs) [34, 201]. IAPs can also prevent PCD induced by external stimuli by directly binding to and inactivating caspases [177]. Decoy receptors, which compete with death receptors for binding of apoptotic ligands such as FASL and TNF-related apoptosis-inducing ligand (TRAIL), also regulate extrinsic PCD [34]. As with intrinsic PCD, various pathogenic bacteria and their products manipulate extrinsic PCD pathways in eukaryotic cells.
APOPTOSIS-REGULATORY MOLECULES OF GRAM-POSITIVE BACTERIA
Pathogenic bacteria use several strategies to circumvent normal immune responses, including seizing control of apoptosis machinery [202]. Indeed, the number of bacterial pathogens shown to exploit PCD pathways in human cells for their own advantage is growing at a rapid pace [203]. Gram-positive pathogens included in this review include species of Staphylococcus, Streptococcus, Bacillus, Listeria, and Clostridium (Table 1). These organisms cause serious infections in humans, and all produce toxins or other structural moieties that regulate PCD pathways in eukaryotic cells. The PCD regulatory activities of selected gram-negative bacteria and protozoa have recently been reviewed elsewhere [163, 204-206].
An initial comparison of the PCD regulatory activities of gram-positive bacteria with Shigella flexneri [2] noted that these organisms induce different forms of PCD in different host cell types. In a similar comparison, intracellular and extracellular pathogens trigger PCD via distinct mechanisms [163]. Since these reports, our understanding of the complex interactions between human cells and gram-positive pathogens and their toxins that culminate in or prevent PCD has advanced. It is now clear that different pathways of PCD can be activated by different bacterial species and strains [204]. Thus, PCD is an unexpectedly complex process. Indeed, some forms of cell death that resemble necrosis may involve enzymatic cascades more typical of classical apoptotic cell death [206b]. Here, we discuss microbial triggers of cell death involving hallmarks of apoptotic PCD; activation of initiator and effector caspase cascades, DNA fragmentation, nuclear/cytoplasmic condensation, disassembly to apoptotic bodies, and non-inflammatory outcomes. It should be noted that many of the models of infection discussed here have not described all of these features of PCD.
Gram-positive bacteria are equipped with an array of virulence determinants (Table 1). In many of the diseases caused by these organisms, the bacterial toxins produced during infection are largely or wholly responsible for the clinical presentations [207]. Most bacterial toxins act within the cytoplasm of eukaryotic cells, causing host cell dysfunction by affecting ion channels, forming pores in host cell membranes, through protease or nuclease activity, and altering intracellular signal transduction pathways [208]. Other bacterial toxins act at the host cell surface [209]. Bacterial toxins that directly affect PCD [13] can be broadly differentiated into four groups: (1) superantigens (e.g., S. aureus enterotoxins, TSST-1 and S. pyogenes pyrogenic exotoxins), (2) pore-forming toxins (e.g., S. aureus α-toxin, S. pneumoniae pneumolysin, S. pyogenes streptolysin O, and L. monocytogenes listeriolysin O), (3) nonpore-forming toxins with enzymatic activity (e.g., B. anthracis lethal toxin and C. difficile exotoxins), and (4) non-enzymatic structural components (e.g., lipotechoic acid) [2, 23, 24, 207].
Staphylococcus aureus
Staphylococcus aureus, a commensal organism that normally colonizes as many as half of healthy adults, is a versatile pathogen that causes community-acquired and nosocomial infections [210]. Exotoxigenic virulence factors produced by S. aureus include the pyrogenic superantigen enterotoxins A through E, G, H, and I (not superantigenic), toxic shock syndrome toxin-1, the hemolysins α-toxin, β-toxin, δ -toxin, and γ-hemolysin, the exfoliative toxins epidermolytic toxins A and B, and Panton-Valentine leukocidin [207, 210, 211]. These exoproducts are robust and stable to chemical inactivation, proteolysis, and heat [211, 212]. Other secreted products include protease, lipase, and hyaluronidase. Membrane-spanning endoproducts on the S. aureus surface include fibronectin-binding protein, collagen-binding protein, and elastin-binding protein, the B-lymphocyte superantigenic toxin protein A [213], coagulase, and clumping factor. Finally, the peptidoglycan cell wall and capsule of S. aureus also contribute to human infection [210].
Whole live S. aureus induces PCD in monocytes [214, 215], chondrocytes [87], keratinocytes [88, 89], endothelial cells [81, 216], epithelial cells [217, 218], and osteoblasts [90]. The ability of an S. aureus exotoxin to promote apoptosis and thereby influence immune responses [219] was first reported in enterotoxin B-stimulated murine T cells [37] and human CD4+ T lymphocytes obtained from asymptomatic HIV-infected individuals [38, 39]. Superantigens, unlike conventional antigens, interact directly with MHC class II molecules on accessory cells and with particular variable regions of the T-cell receptor β chain (TCRVβ) without the need for prior processing. Direct cross-linking of the TCRVβ to the MHC class II molecule results in polyclonal T-cell activation [220-229] and may ultimately lead to PCD. In this manner, staphylococcal superantigens like enterotoxin B circumvent the normal rules for antigen processing and presentation and trigger the death of specific human T-cell subsets, potentially interfering with the host’s ability to respond to conventional antigens. Enterotoxin B has been used extensively as an model to study aspects of tolerance (i.e., deletion and anergy), clonal expansion, and maintenance of peripheral T-cell pools; much is known about its structure and synthesis [211]. Interestingly, there is significant structural homology between enterotoxin B and toxic shock syndrome toxin-1 [211], but only enterotoxin B induces T-cell apoptosis. Toxic shock syndrome toxin-1, in fact, prevents apoptosis in monocytes [76] but induces dose-dependent PCD in B cells [77]. Neither toxin has direct cytotoxic affects on neutrophils, but inhibition of PCD in neutrophils can be brought about by both indirectly as a result of their pro-T helper 2 cell (Th2) stimulatory activity [230]. Many studies have confirmed that enterotoxin B causes activation-induced cell death (AICD) of T lymphocytes, a process involving sequential T cell receptor (TCR) binding, activation, clonal expansion, FAS expression, and eventual PCD [46, 50-55, 68]. AICD, which typically involves extrinsic PCD [163], is now recognized as a key process through which effector CD8+ T cells are removed from the lymphocyte pool after disease resolution [231]. AICD caused by enterotoxin B is restricted to activated [44], post-proliferative T cells [43, 67], depends on the number of cell divisions [56], and is prevented by costimulation with CD28 [57]. Processes of T-lymphocyte sensitization to PCD after antigen-triggered proliferation are reviewed elsewhere [231] and may differ from those involved in maintenance of lymphocyte homeostasis after microbial infection [130]. Enterotoxin B-triggered fratricidal PCD (i.e., killing of neighboring cells) has also been demonstrated in T lymphocytes [58, 232]. Thus, not only does enterotoxin B induce PCD directly in target T cells in which it engages the TCR, but also it causes bystander PCD in proximal lymphocytes in a paracrine fashion. In monocytes, S. aureus similarly stimulates the release of soluble FASL from host cells, thereby inducing PCD in infected and bystander host cells [214, 233]. Enterotoxin B also induces PCD in human immunoglobulin (Ig)-secreting B cells, a process that occurs in a CD4+ T-cell-contact-restricted, CD95-dependent manner [47]. CD4+ T-lymphocyte-dependent PCD in endothelial cells is also triggered by enterotoxin B [48, 49]. Finally, enterotoxin B inhibits PCD in eosinophils [69] and is cleared rapidly after induction of PCD in vivo [234].
At the molecular level, enterotoxin B induces TCRVβ-specific PCD in T cells in a manner that mimics normal host-driven, FAS-mediated PCD for peripheral deletion of antigen-expanded T-cell clones [59, 60, 162, 232]. However, whereas ion channels are involved in SEB-triggered PCD in thymic T cells, this is not characteristic of SEB-driven peripheral deletion [235]. The precise series of intracellular signaling events leading to AICD of enterotoxin B-reactive Vβ8+ T lymphocytes is not yet defined, and it should be noted that the role of FAS signaling is controversial [13]. However, mounting evidence of CD95/FASL dependence [59-64, 70-72, 161, 162] suggests an essential role for extrinsic PCD. Regulation of staphylococcal enterotoxin-triggered PCD might occur at several levels: autonomous FASL-CD95 engagement involving expansion of T lymphocytes and deletion by clonal suicide [59, 60, 73], alternative regulation by activated CD8+ T cells [74, 75], and expression of FASL on nonlymphoid cells as opposed to signal derivation in the activated T cells themselves [232]. FASL-CD95 engagement also appears essential for enterotoxin B-induced PCD in epithelial cells [48]. Other experiments have yielded conflicting results indicating that FAS/FASL signaling has a minimal role in enterotoxin B-stimulated PCD [65]. However, FAS-deficient mice are more susceptible to SEB-triggered lethal shock [236], suggesting that FAS-mediated extrinsic PCD helps to contain overzealous lymphocyte responses during staphylococcal superantigen exposure [13]. FAS-independent mechanisms might compensate for enterotoxin B-triggered, CD95-initiated PCD in experimental systems, where CD95 signaling has been absolutely removed. In mice, FAS-independent PCD in enterotoxin B-stimulated T cells involves decreased expression of BCL-2 and depends on the presence of BIM; this is consistent with the involvement of intrinsic rather than extrinsic signaling [65]. Nitric oxide (NO) appears to exert regulatory influence on enterotoxin B-mediated PCD on CD4+Vβ8+ T cells in vivo [66]. Structure-function relationship studies, similar to those performed to define the structural basis for superantigenicity [211], will help to identify the structural components necessary for the ARM functions of enterotoxin B and toxic shock syndrome toxin-1. A second enterotoxin of S. aureus with PCD regulatory activity is enterotoxin A, which induces apoptosis in Vβ3+ T cells and Vβ11+ T cells (but not in Vβ6+ T cells), akin to enterotoxin B-stimulated PCD in Vβ8+ T cells (and Vβ3+ T cells) [50]. The implications of this T cell-subtype specificity and mechanisms are unknown, but interleukin 2 (IL-2) deprivation and not CD95-FASL interactions appears to contribute to enterotoxin A-induced PCD in these cells [84]. Interestingly, enterotoxin A-induced apoptosis of T cells does not appear to be MHC Class II-restricted, as is AICD triggered by enterotoxin E, despite the fact that these exotoxins share more than 80% homology at the amino acid sequence level [85].
S. aureus hemolysin α-toxin, another ARM [79, 211], is secreted in monomeric form, penetrates and integrates into the target cell membrane, and forms cylindrical heptamers [211]. Nanomolar concentrations of α-toxin cause pores to form in the membrane of T lymphocytes, and the ensuing fluxes of monovalent ions such as Na+ (influx) and K+ (efflux) appear to trigger PCD [13, 78]. Alpha-toxin also causes pore formation and PCD in endothelial cells [81, 237] and pore formation in neutrophils [238] and neuroendocrine cells [239]; however, hallmarks of apoptotic PCD such as DNA fragmentation and caspase activation have not been demonstrated in these cells. In monocytes, α-toxin induces caspase-dependent PCD [79] that occurs independently of death receptors and involves mitochondrial dysfunction [80]. In keratinocytes, however, α-toxin selectively induces necrosis rather than PCD [240]. This dichotomy reflects the pore size produced by oligomerization of α-toxin monomers, which is concentration-dependent. Smaller pores restrict the passage of larger molecules (e.g., ATP) and divalent ions (e.g., Ca2+) [78], favoring PCD. Toxin concentration dependency has also been reported in other cell types [241]. In contrast to enterotoxin B-induced PCD (i.e., TCR engagement and FASL-CD95 signal transduction), the PCD cascade induced by α-toxin involves caspase activation via the intrinsic pathway with mitochondrial cytochrome c release and BCL-2 regulation [49, 80]. This presents an interesting scenario where two distinct ARMs, namely enterotoxin and hemolysin, from a single organism exert different effects on host cells and trigger distinct forms of PCD. A more recent study suggested that α-toxin can also induce an unusual form of necrosis in T cells that involves coincidental caspase activation [242].
Finally, the B-cell superantigenic protein A of S. aureus induces dose-dependent PCD in murine lymphocytes by altering the concentration of BAX [82]. Ligation of the antigen receptor in B cells induces dissipation of mitochondrial membrane potential and activation of caspases, a process regulated by BCL-2, but independent of CD95/TNF receptor [83]. Thus, similar to PCD induced by α-toxin, that induced by protein A appears to trigger intrinsic PCD. Earlier studies in murine cells demonstrated that protein A-induced B-cell proliferation involves the overexpression of BCL-2 [243], a process associated with increased cell survival in some experimental systems [244] and concomitant Th1 stimulatory activity [245]. It is not known if the effects of protein A on human cells are similar to those observed on murine cells.
Streptococcus spp
Streptococcus agalactiae
Streptococcus agalactiae, or Group B Streptococcus (GBS), is a common cause of pneumonia, sepsis, and meningitis in newborns, pregnant women, and adults with underlying medical conditions [246]. Neonates acquire GBS at birth from their asymptomatically colonized mothers (35%-40% of pregnant women have lower gastrointestinal or genitourinary GBS colonization) [247, 248]. The neonatal lung may be infected with a substantial inoculum of GBS at birth, because the bacteria can grow to high numbers in human amniotic fluid [246]. Therefore, the proficiency of the antibacterial function of newborns’ alveolar macrophages [249-251] is considered a crucial component of early immune responses against GBS [252]. Pulmonary epithelial and endothelial cells, and neutrophils also respond to GBS [246, 253]. GBS persist inside macrophages for an extended period of time after nonopsonic phagocytosis and eventually trigger PCD in macrophages and monocytes [91, 92, 254, 255]. Intracellular persistence and manipulation of PCD pathways and antibacterial activity in macrophages may represent an important virulence mechanism whereby GBS contributes to the characteristically poor inflammatory response in the neonatal lung. GBS-induced PCD is also a prominent feature of hepatocytes in a rabbit model of GBS sepsis [256] and in neurons of the dentate gyrus in GBS meningitis [93]. PCD in the latter is mediated, in part, by reactive oxygen intermediates [95].
Exotoxigenic virulence factors produced by GBS include hyaluronidase (hyaluronate lyase), CAMP factor, superoxide dismutase, protease, nuclease, platelet-activating factor, and collagenase/oligopeptidase, protein c, RIB, R protein, and C5a peptidase [246, 257, 258]. The structure of the antiphagocytic capsule also influences pathogenicity (reviewed in [259]). GBS lipotechoic acid is cytotoxic to a variety of human monocytic cell lineages, but the precise form of cell death has not yet been defined. This cytotoxicity may contribute to disease by promoting adhesion and invasion [246]. Among the several toxins produced by GBS, investigations of PCD have focused on β-hemolysin [246, 258].
GBS β-hemolysin is bound to the bacterial cell wall and is responsible for the β-hemolysis characteristic of most clinical GBS isolates [246, 260]. Under experimental conditions, the β-hemolysin is relatively unstable and difficult to purify, which has hindered its study. PCD induced by GBS infection of macrophages was initially attributed to β-hemolysin, because GBS grown in conditions minimizing β-hemolysin expression, namely high glucose concentrations, were not able to induce PCD in macrophages [91]. The ability of acellular β-hemolysin preparations to induce PCD in macrophages has also been demonstrated [29]. In another study, however, the ability of a β-hemolysin-deficient GBS mutant to induce PCD in macrophages did not differ from that of its hemolytic parental strain, suggesting that PCD can be triggered by additional GBS factors [92].
GBS-induced PCD in macrophages is inhibited by the caspase-3 inhibitor DEVD-CHO [91, 261], and is associated with alterations in BAD, BAX, 14-3-3, and OmiHtr/A2 expression. Alterations in mitochondrial transmembrane potential leading to loss of mitochondrial membrane integrity further indicate involvement of the intrinsic pathway of PCD [261]. Similarly, death of GBS-infected human monocytes is caspase-dependent [94]. NO secretion triggered by GBS infection contributes to PCD in murine macrophages, but similar findings have not been shown in human macrophages, suggesting an alternative NO-independent mechanism in human cells [262]. Interestingly, TNF-α is required for GBS-induced PCD in neurons [93], but appears dispensable for GBS-induced PCD in macrophages [262]. Oxygen intermediates also contribute to GBS-induced PCD in neurons in vivo [263]. GBS triggers increases in intracellular Ca2+, and activates selected mitogen-activated protein kinases (MAPKs) including c-Jun NH2-terminal kinase and p38 [264]. In monocytes, GBS causes up-regulation and secretion of TRAIL, which is associated with PCD [94], although functional studies using TRAIL depletion or deletion have not been reported. GBS also induces phosphatidylserine exposure, which probably contributes to the clearance of the dying apoptotic cells [265]. Investigation of GBS ARMs is in its infancy and further study of the effects of GBS products on eukaryotic cells will be important to define how GBS-triggered PCD influences disease pathogenesis and immune responses.
Streptococcus pneumoniae
Streptococcus pneumoniae is part of the normal microflora of the human respiratory tract and a common cause of respiratory infections and meningitis [101, 266]. Pneumococcal virulence, like that of GBS, is in large part determined by the chemical composition and synthesis of its capsular polysaccharide [101]. S. pneumoniae expresses several additional virulence factors, as reviewed elsewhere [267], that may initiate PCD in phagocytes after microbial uptake. Pneumolysin [268, 269] has direct proapoptotic activity on several cell types including microglia, neurons [97, 99], and macrophages [98] but not on epithelial cells [105]. Other factors produced by S. pneumoniae, i.e., super oxide dismutase, NADH oxidase, zinc metalloproteinases, and H2O2, also contribute to virulence [101], but among these, only H2O2 has proapoptotic activity toward human cells [97]. Surface endoproducts of S. pneumoniae include the capsule, hyaluronate lysase, pneumolysin (classified here as a surface molecule that is also secreted), neuraminidases, autolysin, choline-binding protein A, pneumococcal surface antigen A, lipotechoic acid, LPXTG-anchored proteins (e.g., surface protein A and surface protein C), and other lipoproteins [270]. In one study, pneumococcal cell wall extracts induced PCD in monocytes [100]; however, with the exception of pneumolysin, there are no reports linking apoptosis to individual highly purified S. pneumoniae endoproducts.
S. pneumoniae stimulates PCD in a variety of human cell types. Whole live S. pneumoniae initiates PCD in dendritic cells by two distinct mechanisms [101] and NO-dependent PCD in monocytes, macrophages [98] and neutrophils [102-104]. Pneumococcus also induces PCD in human alveolar and bronchial epithelial cells [105, 107] and in neurons [97]. Increased numbers of apoptotic T cells have been reported in patients infected with S. pneumoniae. [106]. In an animal model of S. pneumoniae infection, inhibition of PCD in lymphocytes in pneumococcal septicemia improved host survival, a finding that suggests that T-cell PCD triggered by the bacterium is detrimental to effective immunity [108, 109]. The majority of studies on pneumococcus-induced PCD, however, have focused on macrophages because of their crucial role in early immune responses and ability to bridge innate and adaptive immunity through pathogen-associated molecular pattern recognition [271, 272].
PCD in S. pneumoniae-infected macrophages requires internalization of bacteria and continued macrophage protein synthesis [9]. This differs from GBS-induced PCD of macrophages, where external signal transduction resulting from bacterial attachment to the macrophage surface can result in PCD without bacterial internalization [91]. The concept that a particular ARM of S. pneumoniae instigates PCD in macrophages to facilitate immune escape is not entirely supported by the ultimate intracellular fate of the bacterium, i.e., death, as discussed elsewhere [9]. Indeed, inhibition of pneumococcus-induced PCD in vivo promotes bacterial dissemination and implicates PCD as a crucial component of an immune response that helps to contain and eliminate S. pneumoniae [273]. Indeed, opsonization enhances pneumococcus-induced PCD in macrophages by increasing the number of intracellular bacteria [100]. In contrast, recent data on GBS have indicated that, unlike S. pneumoniae, a proportion of intracellular GBS can avoid intracellular killing and persist inside apoptotic macrophages, which may facilitate bacterial escape from infected cells [92, 262]. Therefore, host-driven PCD in macrophages appears to contribute to immune control of pneumococcus, but PCD may play a distinct role in infections caused by GBS.
Pneumolysin, like β-hemolysin of GBS, is a pore-forming exotoxin [268, 269]. Similar to S. aureus α-toxin, pneumolysin inserts into the host cell membrane, where it forms pores [274]. The role of pneumolysin in PCD was initially unclear, because early observations indicated it inhibited bacterial phagocytosis and prevented PCD in macrophages [9]. More recent genetic studies have confirmed that pneumolysin triggers PCD in macrophages [98] and other cell types [97]. S. pneumoniae-induced PCD occurs by FAS-independent mechanisms in infected monocytes and by FAS-dependent mechanisms in noninfected bystander monocytes [9]. In macrophages, the PCD response is associated with mitochondrial membrane permeabilization, translocation of cytochrome c to the cytoplasm, and downregulation of the antiapoptotic BCL-2 family membrane protein myeloid cell leukemia sequence (MCL-1) [98]. BAX and BID may also contribute to S. pneumoniae-induced PCD in human macrophages [98]. Interestingly, encapsulated S. pneumoniae does not induce PCD in macrophages as well as nonencapsulated bacteria, suggesting that the capsule may cloak pneumococcal ARMs and reduce their effect. Alternatively, reduced PCD in response to encapsulated pneumococcus may reflect impaired bacterial uptake, since internalization is required for S. pneumoniae-induced PCD [9, 100]. Caspase-3 is required for S. pneumoniae-induced PCD in macrophages, similar to PCD triggered by GBS [9].
Pneumococcus-induced PCD in neurons involves different mechanisms than those responsible for PCD of macrophages or monocytes [204]. Neuronal PCD results from rapid H2O2-mediated mitochondrial damage, release of apoptosis-inducing factor (AIF) [275], and intracellular Ca2+ fluxes with [276] or without caspase activation [97, 277, 278]. Pneumolysin contributes directly to these processes [97], whereas the capsule does not [97, 275]. S. pneumoniae-infected pulmonary epithelial cells undergo PCD through mechanisms that depend on caspase-6, -8, and -9 but not caspase-3, and result in cleavage of BID and BAX and downregulation of BCL-2 [105]. As with neurons, PCD in parenchymal cells of the lung is independent of the pneumococcal capsule [105].
Streptococcus pyogenes
Streptococcus pyogenes, or Group A Streptococcus, is the etiologic agent of scarlet fever, necrotizing fasciitis, impetigo, and various postinfectious autoimmune sequelae [279-281]. S. pyogenes colonization of the skin or nasopharynx precedes local and invasive infection, and the site of infection is related to the expression of specific S. pyogenes virulence factors [281]. Secreted exotoxigenic virulence factors produced by S. pyogenes include the pore-forming β-hemolysin toxins streptolysins O and S, DNases A through D, hyaluronidase, NADase, streptococcal inhibitor of complement, various streptococcal superantigenic pyrogenic exotoxins (A-C, F-H, J-L, SmeZ, SmeZ-2), streptococcal pyrogenic exotoxins I and M, exotoxin F (mitogenic factor), the cAMP cohemolysin, and streptokinase [281-283]. Streptococcal pyrogenic exotoxin A shares homology with S. aureus toxic shock syndrome toxin-1 and a similar mechanism of T-cell activation [284]. Streptococcal pyrogenic exotoxin B, on the other hand, shares significant homology with S. aureus enterotoxins A and C and has cysteine protease activity that cleaves immunoglobulins, fibronectin, vitronectin, extracellular matrix proteins, histamine, and IL-1β [282, 283]. Surface moieties and endoproducts of S. pyogenes include lipotechoic acid, the outer hyaluronic acid capsule, M protein, protein F, multiple fibronectin-binding proteins, serum opacity factor, C5a peptidase, GAPDH, and vitronectin-binding, galactose-binding and collagen-binding proteins [281-283]. Several S. pyogenes exotoxins have been characterized in detail; however, the knowledge on PCD induced by S. pyogenes is limited.
In contrast to other pathogenic Streptococcus spp., S. pyogenes induces large-scale necrosis of a range of host cells. Widespread necrosis, inflammation, and vascular permeability is characteristic of severe S. pyogenes infections [112, 285] and is linked to the exotoxins and endoproducts expressed by the organism. Several studies have suggested a role for S. pyogenes-induced PCD in the infectious process. In experimental models of S. pyogenes infection, whole live S. pyogenes induces PCD in human neutrophils [115], keratinocytes [110], and epithelial cells [22, 111, 286, 287]. With the exception of one study in patients with severe S. pyogenes infection in which S. pyogenes-induced PCD was associated with depletion of specific T-lymphocyte subsets expressing particular Vβ regions [288], the occurrence of PCD in patients infected with S. pyogenes has not been reported. In keratinocytes, S. pyogenes-induced PCD requires delivery of secreted bacterial NADase into the host cell cytoplasm, where it acts as an intracellular cytotoxin. This process appears to depend on prior streptolysin O-mediated pore formation in the host cell membrane, which facilitates NADase transfer [110]. Thus, it appears that NADase and streptolysin O act as cooperative ARMs of S. pyogenes for PCD in some cells.
Streptococcal pyrogenic exotoxin B enhances PCD in epithelial cells [111] and macrophages [113] by a mechanism that may involve cleavage and activation of an IL-1β precursor [289]. Recently, Tamura et al. (2004) demonstrated that the proapoptotic activity of streptococcal pyrogenic exotoxin B is associated with the ability of the toxin to activate eukaryotic matrix metalloproteinases that cause the release of TNF-α and FASL and activate PCD [112]. Nakagawa et al. (2004) demonstrated that S. pyogenes triggers caspase-9 activation in epithelial cells resulting in dysfunctional permeability transition pores and mitochondrial cytochrome c release [286]. Finally, BAX translocates to the mitochondria, and the prosurvival proteins BCL-2 and BCL-XL are depleted [22]. These observations implicate intrinsic PCD in S. pyogenes-infected epithelial cells. The fibronectin-binding F1 surface protein of S. pyogenes appears to be essential for the organism to invade and induce PCD in epithelial cells [22]. One study investigating the contribution of M protein to host cell PCD responses indicated that PCD after S. pyogenes infection might be influenced by the specific M protein serotype [22]. Finally, purified glycoprotein of S. pyogenes induces PCD in fibrosarcoma cells via mechanisms that appear to involve inhibition of tyrosine kinases and dephosphorylation of host proteins [114].
The marked necrosis and severe inflammation characteristic of S. pyogenes infections suggest that PCD may play a minimal role in disease pathogenesis. These characteristics of infection, however, do not preclude involvement of PCD in the host cell response to S. pyogenes. It appears plausible, for example, that selected pyrogenic exotoxins of S. pyogenes might expand particular T-cell subsets and cause AICD in a manner that parallels lymphocyte-pathogen interactions described for S. aureus exotoxins. Current studies demonstrate that S. pyogenes can directly manipulate particular apoptotic programs in several human cell types, and future studies will elucidate what role these interactions play in the infectious process.
Bacillus anthracis
The host responses involved in Bacillus anthracis infection are reviewed elsewhere [290, 291]. In inhalational anthrax, alveolar macrophages phagocytose inhaled bacterial spores but are incapable of efficiently killing the organism. Indeed, macrophages may facilitate spread of the bacteria by carrying it to regional lymph nodes [291, 292]. Spores germinate in regional lymph nodes, producing vegetative bacteria that cross endothelial cells to the bloodstream [293, 294]. B. anthracis produces a number of exotoxins, collectively known as the anthrax “toxin complex” [293]. The complex is comprised of three polypeptide subunits; lethal factor, edema factor, and protective antigen [291, 295-297]. Separately, none of these protein subunits are toxic [293]. Instead, the lethal factor and edema factor form A-B binary units, and when paired with the nontoxic protective antigen, they produce lethal toxin and edema toxin, respectively [290, 291, 293]. Protective antigen mediates entry of the dimeric toxin into the host cell cytosol [290] by forming protective antigen self-heptamers at the host cell membrane; these self-heptamers bind specifically to the anthrax toxin receptor, which is ubiquitous on host cell surfaces [298]. This receptor-heptamer complex permits the subsequent docking of lethal factor or edema factor and the transfer of the cytotoxic components into the cytosol via receptor-mediated endocytosis [291,293,296]. Preassembled toxin complexes may also bind directly to the receptor [295]. A second receptor for anthrax toxin was recently identified [299]. Additional B. anthracis exoproducts and endoproducts include the surface-layer protein SAP (secreted and cell-bound) [291], the antiphagocytic poly-γ-D-glutamic acid capsule, the surface-layer protein EA1, and cell wall structural polysaccharides and peptidoglycan [291, 300, 301]. Lipotechoic acid and membrane lipoproteins of B. anthracis have not yet been described, although these products are present in other Bacillus spp. [301].
Necropsy of anthrax victims demonstrates apoptotic cells in tissues of the lower respiratory tract [302]. Virtually all types of eukaryotic cells studied to date are sensitive to edema toxin, which is an adenylate cyclase [290, 291, 295]. Edema toxin causes cytokine deregulation and impaired phagocytic cell function by increasing intracellular cAMP [290,293]. Lethal toxin, on the other hand, is a metalloproteinase that can replicate the hypotension, shock, and death caused by systemic infection and appears selectively toxic to macrophages [2, 26, 290, 291, 294]. Macrophage specificity is thought to reflect the presence of a unique intracellular target within these cells or a unique form of toxin processing that activates the lethal toxin after internalization [293]. Compared with rodent macrophages, human macrophages are relatively resistant to the actions of lethal toxin; therefore, PCD in these cells probably has a more limited role in the pathology of infection in humans [303]. However, under certain conditions, human macrophages do undergo lethal toxin-induced PCD [25, 116, 117, 304, 305], which may contribute to escape and dissemination of the organism from these cells [303].
Reports about the effect of interferon-γ (IFN-γ) on lethal toxin-induced PCD in human macrophages have been conflicting. Popov et al. (2002) reported a sensitizing effect leading to increased cell death [116]; however, Gold et al. (2004) described a prosurvival effect [304]. TNF-α may also sensitize macrophages to the cytotoxic effects of lethal toxin [117]. Lethal toxin also induces PCD in human neutrophils [116] and immunologically incapacitates, rather than kills, dendritic cells, thereby rendering them inefficient as antigen presenters and T-cell activators [306]. Two recent studies have demonstrated that the proapoptotic activity of lethal toxin toward human endothelial cells involves decreased BCL-2 [118] and caspase activation [119]. Although generally reported as nontoxic, protective antigen can induce PCD in human monocytes under some conditions [116].
The mechanisms responsible for the proapoptotic activity of lethal toxin have been studied in murine macrophages and in vivo models of inhalation anthrax. Lethal toxin-induced PCD is associated with FASL binding and activation of caspases-1, -3, -4, and -8 [26, 116]. In mice challenged with B. anthracis, administration of a pan-caspase inhibitor improved survival; thus, PCD appears to contribute to severe anthrax [307]. At high concentrations, lethal toxin triggers substantial production of reactive oxidants, causing macrophage necrosis [293]. Necrosis and PCD can also occur simultaneously in cells treated with low doses of lethal toxin [120, 121, 308]. Macrophage PCD induced by low concentrations of lethal toxin dampens local immune responses and impairs innate immunity [116]. Lethal toxin inhibits MAPK kinases (MKKs) and inactivates MAPKs [25, 290, 309-312]. The later is essential to PCD triggered by lethal toxin [25, 309, 313], as these signaling molecules promote survival by inducing NFκB and the transcription of related genes, such as those encoding IAPs [314, 315]. Signal transduction through Toll-like receptor 4 and repression of antiapoptotic p38 MAPK, in a process that requires the protein kinase PKR, are particularly important to the ability of lethal toxin to trigger PCD in macrophages [122]. The rapidly expanding body of knowledge on anthrax and the mechanisms of B. anthracis toxicity will further define how these molecules direct human cells through death pathways. It will be particularly important to define the PCD responses of host cells to B. anthracis in the context of combined treatment with lethal toxin, edema toxin, and other toxins, because these combinations are likely to occur in systemic anthrax infection.
Listeria monocytogenes
The incidence of human listeriosis is relatively low compared to the large number of patients afflicted each year with staphylococcal or streptococcal infections. The overall mortality rate, however, is much higher for listeriosis [316-318]. Listeriosis is a foodborne illness that is most often characterized by gastroenteritis, meningitis, encephalitis, or sepsis [317, 318]. L. monocytogenes is capable of persistent intracellular survival and replication in epithelial cells, endothelial cells, dendritic cells, fibroblasts, and macrophages [317]. Hepatocytes represent the principle site for intracellular replication [319], although macrophages may also be an important reservoir [320]. In fact, L. monocytogenes can completely avoid the extracellular environment within the host by moving from cell to cell via polarized actin polymerization-dependent mechanisms [316, 317, 321]. Thus, L. monocytogenes is often regarded as the prototypical organism for study of intracellular pathogenesis [316, 321]. A detailed picture of the molecular events involved in the organism’s intracellular lifestyle has been compiled and has provided insight into how L. monocytogenes modulates eukaryotic host cell function. The PCD regulatory activities of L. monocytogenes are only now beginning to be elucidated.
L. monocytogenes attachment, phagocytosis/invasion, intracellular survival, replication, and intercellular trafficking are mediated by distinct virulence factors. Virulence determinants [316, 322] and their role in immunity [323, 324] are reviewed elsewhere. Despite intracellular replication of the bacteria, most types of eukaryotic cells are not rapidly killed by L. monocytogenes [325]. This suggests that PCD has a role in immunity. Indeed, the organism itself uses multiple mechanisms to avoid host cell cytotoxicity [325], perhaps as a means of sustaining its intracellular habitat. Genetically engineered L. monocytogenes that kill host cells prematurely, for example, are avirulent compared to their wild-type parent strains [325]. The Listeriae, in effect, are shielded from extracellular immune factors whilst they remain intracellular [325]. Bacilli released from dead host cells into the extracellular environment may be targeted for destruction by neutrophils and humoral components [326]. Host-driven PCD, therefore, might benefit immunity [327]. This premise, however, is based on the assumption that all host cell types play similar roles in immunity, which is not the case. Antigen-presenting dendritic cells, for example, are probably not a significant reservoir for L. monocytogenes [320] but are critical for induction of T-cell responses. Moreover, Listeriae may preferentially take advantage of selected cell types such as hepatocytes for survival and intracellular growth. These facets lend support to the theory that the requirements for successful immunity against a pathogen residing preferentially in macrophages are likely to differ from those immune-effector mechanisms required to combat a pathogen residing in parenchymal cells [326]. Thus, it is not surprising that L. monocytogenes infection is associated with PCD in several types of host cells, depending on the stage of infection.
Apoptosis in response to infection with whole, viable L. monocytogenes has been described in hepatocytes [328-330], epithelial cells [128], CD4+ T cells, CD8+ T cells, and B lymphocytes [125, 130, 134], neurons, Purkinje cells [127], dendritic cells [124, 331], and natural killer (NK) cells [129] but not in macrophages [8, 124, 332]. Whether intracellular Listeriae actively impede PCD in macrophages to preserve their intracellular niche is an interesting and unresolved question [333]. Macrophages die by delayed necrosis after extensive intracellular bacterial replication [334].
Activation of PCD in infected hepatocytes could contribute to resolution of disease in two ways: First, this eliminates infected cells and reduces the available pool of target host cells for intercellular replication and spread of infection [316, 334]. Second, PCD would expose the bacteria to the extracellular milieu, where they are susceptible to extracellular effectors of immunity [319, 335]. Neutrophils may mediate hepatocyte death and abort the cycle of cell-to-cell spread [13, 326, 336], though PCD may also occur in hepatocytes in the absence of neutrophils [328, 337]. Caspase-3 appears to contribute to PCD in hepatocytes, but without cleavage of the typical caspase-3 substrate PARP [319]. Selective host-mediated depletion of activated non-pathogen-specific T lymphocytes during the early phase of listerial infection [338] might allow for maximal expansion of the antigen-specific T-cell compartment [318], which could bolster the rapidity and intensity of the adaptive response. PCD in post-proliferative T lymphocytes, after resolution of active listeriosis, may maintain only enough memory T cells required for an efficient response to reinfection, thereby contributing to the regulation of homeostatic lymphocyte pools [130].
In contrast to these potentially immune-beneficial scenarios, Listeriae-induced PCD in antigen-presenting cells such as dendritic cells may reduce numbers of professional phagocytes, diminishing antigen presentation and hindering efficient adaptive immune responses [124, 316, 334]. Listeriae-induced PCD in dendritic cells can occur independently of bacterial internalization [13, 124]. Similarly, Listeriae-driven PCD in specific subsets of lymphocytes, such as CD8+ T cells, would eliminate important adaptive mediators before the resolution of infection [27, 123]. Direct evidence of a detrimental role of PCD in immunity to listeriosis was provided by Kagi et al. (1994) who demonstrated decreased innate and CD8+ T lymphocyte-driven cell-mediated immune responses to L. monocytogenes in perforin knock-out mice [339] (perforin is integral to host cell-driven PCD pathways in listeriosis). Merrick et al. (1997) demonstrated that more severe infection triggers PCD in a nonspecific manner in T and B lymphocytes; this nonspecific response could also hinder adaptive responses [134]. These observations of PCD in B cells are important in light of new data [340, 341] that challenge the long-standing belief that B cells do not contribute to immunity against listeriosis [316, 325, 342].
Caspases-1 and -11 do not appear to contribute to Listeria-induced PCD in the cell types studied to date [319, 343]. In contrast, perforin and IFN-γ have been implicated [344]. Several studies have demonstrated that cytotoxic CD8+ T lymphocytes kill Listeria-infected hepatocytes by perforin-granzyme-mediated PCD [132, 319, 339, 345] (early), FAS-FASL-mediated PCD [346-348] (later), or both. This is believed to be an important acquired immunologic effector mechanism [349]. Cytotoxic NK cells might also kill infected hepatocytes by similar mechanisms [319]. TNF-α appears to be crucial in directing a separate, CD8+ T-lymphocyte-mediated PCD pathway in hepatocytes in the absence of perforin and FAS [350]. Miura et al. (2000), however, reported that TNF-α and IL-6 play a protective role in mediating survival of hepatocytes during listeriosis [329]. In support of this finding, pancreatic cells in mice lacking TRp55, a key receptor for TNF-α, are prone to PCD triggered by L. monocytogenes [351].
The most widely studied exotoxigenic virulence factor of L. monocytogenes is the pore-forming hemolysin listeriolysin O (Hly in earlier studies) [316]. Other secreted virulence factors include the phospholipases A and B, which are involved in escape from the phagosome [316]. There are approximately 140 surface proteins of L. monocytogenes [316, 352]. Only listeriolysin O, which is similar in structure and function to Group A streptococcal streptolysin O [316], and possibly phospholipase C [353, 354], modulate PCD in host cells. Listeriolysin O promotes PCD in T cells [27, 123] and dendritic cells [124], thereby negating the need for bacterial internalization for PCD of the host cell. A recent study by Menon et al. (2003) implicated listeriolysin O as the major virulence factor directing PCD in B cells [125]. The PCD activity of listeriolysin O, like that of S. aureus α-toxin [78] and S. pyogenes streptolysin O, is dose dependent. This characteristic may affect the disease process in distinct ways. For example, severe disease associated with higher bacterial burdens (more exotoxin) might cause inflammatory necrotic pathology, whereas lower bacterial burdens (less toxin) cause PCD [124].
Functional aspects of listeriolysin O activity are discussed elsewhere [316]. Unlike other bacterial poreforming toxins, listeriolysin O is not cytotoxic under normal conditions. This is believed to be related to the limited pH range in which the toxin is active (pH 4.5-6.5); thus, listeriolysin O activity is compartmentalized to acidified vacuoles [316, 325]. Listeriolysin O-induced PCD in T lymphocytes is associated with the activation of caspases-9 and -3 and loss of mitochondrial membrane potential [27], pointing to intrinsic PCD. In addition, listeriolysin O stimulates caspase-independent PCD mechanisms [27], which have not yet been identified. Most T lymphocytes undergoing apoptosis in response to L. monocytogenes and listeriolysin O are activated/postproliferative [27, 56, 67, 130]. Removal of γδ T cells, which link innate and acquired effector mechanisms [355, 356], early during low-grade infection has been associated with FAS-FASL death receptor signaling [131]. However, FAS-dependent PCD appears to play a more prominent role in the elimination of T cells after resolution of infection [132, 133, 206]. Ironically, γδ T cells are also thought to be important in the induction of PCD in activated macrophages to aid in resolution of inflammation [357]. In murine listeriosis, TRAIL contributes to PCD in lymphocytes and myeloid cells during the acute phase of infection, and TRAIL-deficient mice are more resistant to infection than their wild-type counterparts [358]. These observations suggest that TRAIL-mediated PCD represents an important Listeriae-driven mechanism for depletion of cells that contribute to immune responses. Type I IFNs contribute to PCD in splenocytes [126], macrophages [359], and T cells [123] in response to listeriolysin O demonstrating the importance of cytokines induced by the organism [360-362]. Finally, high levels of expression of CD94 limit PCD in CD8+ T lymphocytes, which may maintain T-cell pools during infection [129]. Whether these described apoptotic processes contribute to disease pathogenesis or host defense remains unknown [333]. Clearly, PCD in listeriosis is emerging as a complex, multifaceted process that will require careful investigation.
Clostridium spp
The genus Clostridium comprises approximately 20 species known to be pathogenic to humans including, most notably, C. difficile, C. perfringens, C. botulinum, and C. tetani. As a group, the Clostridia produce a vast number of toxigenic products ranging from mild cytolysins to the most toxic biological substance known, the botulinum paralytic neurotoxin [297, 363]. PCD regulatory activities and ARMs have been studied in detail in C. difficile, the most important cause of nosocomial diarrhea in adults [364-368], and C. perfringens, a common agent of gas gangrene and food poisoning [363].
C. difficile produces several secreted and surface-bound virulence factors including flagellae, adhesins, hydrolytic enzymes, an ADP-ribosylating toxin [369], and an anti-phagocytic polysaccharide capsule. After ingestion, acid-resistant spores facilitate survival and allow the organism to colonize the lower intestinal tract [364, 367]. The best-characterized virulence determinants of C. difficile are the proteinaceous exotoxins A and B [366]. These toxins, which are similar in function and mechanism of action, are largely responsible for the clinical syndromes of diarrhea and colitis [367], and both exert PCD regulatory activity. Immunity to C. difficile infection is conferred by specific anti-exotoxin immunoglobulin [364, 367]. Colonic epithelium appears to be the main target of exotoxins A and B [365].
Typically, C. difficile antibiotic-associated diarrhea is characterized by extensive colonic inflammation and widespread necrosis [364, 365, 368]. Exotoxins A and B stimulate necrosis in certain cell types (e.g., enterocytes and monocytes) [365, 370-373] and are thought to contribute to this pathology. The toxins also cause degranulation in mast cells and induce reactive oxygen metabolites [374-377]. Initial studies on exotoxin A by several groups [378-382] and on toxin B by two groups [383, 384] described toxin-induced cytopathology in various cell types; however, those actions have since been studied mostly in enterocytes. After binding to receptors on enterocytes and subsequent endocytosis, the toxins cause disaggregation of actin in colonocytes, and glucosylate or inactivate Rho family signaling proteins. These GTPases, which regulate the formation of actin cytoskeleton stress fibers and act as molecular switches, control various signal transduction pathways, including NF-κB activation and the expression of apoptosis regulators such as BCL-2 [209, 385, 386]. This leads to epithelial cell rounding and opening of epithelial cell tight junctions. Second, the toxins induce the release of cytokines by activating MAPK [365-367, 374]. Loss of tight junction integrity further leads to detachment and rounding of host cells, a known stimulus of PCD [365, 367, 374, 387, 388].
Exotoxins A and B trigger PCD in several epithelial cell types. Mahida et al. (1996) provided the first direct evidence of toxin A-triggered PCD in intestinal enterocytes [135]. Other reports of PCD-stimulating activity of toxin A towards mast cells, hepatocytes [138], and colonic lamina propria cells (T lymphocytes and eosinophils) [135-137] and endothelial cells [148] followed. Toxin B-induced PCD in intestinal epithelial cells was subsequently demonstrated [144]. C. difficile toxin-induced PCD regulatory activities have since been studied by several groups [143, 389-391]. Within minutes of endocytosis, the toxins cause a decrease in ATP [139, 140], a loss of mitochondrial membrane potential [141], the release of mitochondrial cytochrome c, and the generation of toxic reactive radicals [139, 142]. Activation of NF-κB is believed to be involved in these responses [374, 376]. Mitochondrial dysfunction occurs before a decrease in Rho protein activity [374], which appears necessary for PCD [139]. This is consistent with Rho protein prosurvival signaling in other systems [392-394]. Activation of caspases-1, -3, -6, -8, and -9 [141] indicates extensive procaspase cleavage culminating in activation of both the intrinsic and extrinsic PCD pathways. In endothelial cells, C. difficile toxins activate intrinsic PCD via alterations in BCL-2, MCL-1 and BID [148]. Toxin B-induced PCD in epithelial cells is also associated with Rho protein dysfunction and inhibition of cell adhesion due to actin depolymerization [144, 366]. Toxin A also activates BID [139], which may link the intrinsic and extrinsic PCD pathways. TNF-α and FASL do not appear to be involved in the early pathway of toxin A-triggered PCD [137]. Toxin B has been used to stimulate apoptosis in various systems [389, 391]. Like toxin A, toxin B activates caspase-3, causes PARP cleavage [145], and reduces BCL-2 levels [395], in a pathway that may be p53-dependent [146]. The proapoptotic effects of toxin B can be reversed by host cell MAPK activity, under certain conditions [143, 145]. In addition, caspase-independent pathways triggered by toxin B exist, and these also appear to involve inactivation of Rho proteins [147].
C. perfringens, unlike C. difficile, produces an array of toxins implicated in eukaryotic cell cytotoxicity including enterotoxin, collagenase, hyaluronidase, sialidase, pore-forming θ-toxin (perfringolysin), α-toxin (phospholipase), β-toxin, ε-toxin, and τ-toxin [208, 363, 396, 397]. Enterotoxin induces PCD by binding to as yet unidentified enterocyte surface receptors and activating caspases-3 and -7, causing mitochondrial cytochrome c release, extracellular Ca2+ influx [150], and eventual host cell death [149], which is dose-dependent [150, 398]. Although β-toxin appears to be an oligomerizing pore-forming toxin that permits movement of cations through susceptible membranes, no studies have addressed its role in PCD [399]. Similarly, although the α-toxin has cytotoxic activity [400], no PCD regulatory activity has been reported.
Collectively, studies on different Clostridia spp. suggest that the intrinsic pathway of PCD involving mitochondrial dysfunction and inactivation of Rho protein function is a common mechanism by which distinct exotoxins from these organisms trigger PCD in host cells [209]. Exoenzyme C3, the major exotoxin of C. botulinum, also exerts inhibitory activity on Rho GTPases and causes PCD in host cells such as lymphocytes [388, 392]. Whether C. perfringens enterotoxin targets Rho proteins in the host cell is an important unresolved question. It is also important to note that additional non-Rho protein-related pathways of PCD are stimulated by clostridial toxins, as exemplified by C. sordellii lethal toxin [401]. The contribution of actin cytoskeleton disruption brought about by toxin-mediated inactivation of Rho GTPases to these apoptotic processes is at present unclear [209]. What role the PCD plays in immunity to the different infections caused by Clostridium spp. remains to be determined; however, several possible influences on immunity can be predicted.
During C. difficile infection, intestinal cells undergoing PCD as a result of toxin A exposure could alter the cytokine environment in unique ways. This could sway the balance of helper T cell responses occurring in the lamina propria microenvironment and thereby, affect humoral and cell-mediated immunity. Toxin-induced disruption of colonic epithelium would expose lamina propria T lymphocytes, which activate the suicide response after toxin exposure, and thereby compromise adaptive immunity. Inflammation is a key aspect of the pathologic changes that occur in colitis, and this may be related to toxin dose, i.e., low doses of toxin trigger PCD that may reduce inflammation, and higher doses trigger inflammatory enterocyte death and infiltration. Finally, deletion of mast cells in the lamina propria by toxin-induced PCD could hinder the immune response to infection by affecting the release of histamine, cytokines, and NO [136]. Understanding the role that PCD plays in these infections will be an important step in elucidating the significance of clostridial toxin-induced PCD in human disease. Future use of appropriate animal models will help to address these questions [135, 144].
IMPLICATIONS OF BACTERIA-MODULATED PCD FOR IMMUNITY TO INFECTION
The clinical spectrum of disease caused by the organisms listed in Table 1 is broad; therefore, effective immune responses to these pathogens involve different elements of the immune system. There are a number of crucial aspects of the host-pathogen interaction that are relevant to the process of bacteria-induced PCD and development of immunity. One of these aspects is cellular tropism, or predilection for particular host cell types. Cellular tropism for PCD is demonstrated by a number of the pathogens discussed in this review. For example, the S. aureus proapoptotic influence toward lymphocytes and epithelial cells and the prosurvival influence of staphylococcal exotoxins toward eosinophils [69]. Fundamental differences exist in the susceptibility of different cell types to staphylococcal enterotoxin A-triggered PCD [75, 211, 402]. Listeria listeriolysin O induces PCD in T cells and B cells but not in macrophages. Cellular tropism may be influenced by key differences in the ability of the host cell to phagocytose the organism, to produce antimicrobial substances, and to activate second messenger pathways. It is also influenced by the ability of the pathogen to produce bioactive ARMs that can hijack components of the host PCD machinery in a particular cell type. An additional level of complexity in cellular tropism exists when considering that different immune cells exhibit inherent differences in susceptible to apoptosis. For example, NKT cells are relatively resistant to PCD compared to other cell types [403]. Postproliferative T lymphocytes are more susceptible to PCD than are nonantigen-differentiated T cells. Monocytes die by PCD after phagocytosis of various pathogenic bacteria, whereas death of polymorphonuclear leukocytes (PMNLs) is delayed after internalizing the same microbes [404]. These cellular tropisms or “PCD restrictions” may dictate the role of bacteria-induced PCD regulatory activity in the overall immune response during these infections.
The second aspect of the host-pathogen interaction related to PCD activity and immunity is the functional relevance that a particular host cell type has in the overall immune response to the organism. Simply because a pathogen induces (or prevents) PCD under experimental conditions does not mean such an observation reflects a biologically relevant response that occurs during the disease process in vivo. It is important to understand the instigator of the PCD deregulation; is it the pathogen or the host that is primarily responsible for triggering the PCD event? In the example of listeriosis, the role of T lymphocytes is to restrict microbial growth by killing bacteria-infected host cells and provide help for antimicrobial sentinels such as macrophages. If PCD in CD8+ T cells during listeriosis is driven proactively by listeriolysin O rather than by the host immune response, our hypothesis would be that PCD hinders adaptive immunity, because CD8+ T cells are key effectors of bacterial clearance. The outcome of these interactions could include deletion of Listeria-specific T cells required for immunity and downregulation of the immune system by generation of an immunosuppressive milieu (e.g., less IFN-γ, more TGF-β, and a shift to the Th2 response) [163]. Apoptotic bodies from toxin-killed T lymphocytes could also inhibit the proliferation of antigen-stimulated naïve T cells, which might dampen immune responsiveness [80, 405]. Finally, there is the possibility that AICD in T lymphocytes that were triggered by ARMs may result in some surviving T cells with TCRs that become anergic (i.e., lose their capacity to proliferate in response to the antigen in vitro), and this could render T-lymphocyte populations suboptimally responsive [51]. T-cell unresponsiveness resulting from interruption of the normal physical interaction between T cells and antigen-presenting cells by apoptotic bodies has also been described; S. aureus SEB represents an example of this [406]. On the other hand, T cells would be deleted at the end of active microbial disease, as the host immune-driven homeostatic response returns expanded pools of T-lymphocyte subsets to appropriate levels. Using the example of listeriosis, host immune-driven PCD in infected hepatocytes would have different implications for immunity compared with the affects described for lymphocytes, because these two cell types play very different roles in the infectious process (i.e., T cells drive immunity and are targeted by the pathogen for elimination, whereas hepatocytes are used as a niche for bacterial survival and are targeted for destruction by the host immune system). Thus, the functional relevance of individual cell types in immunity to a given pathogen bears influence on the potential role of PCD in disease. This must be considered in the context of what comprises a successful immune response to a given organism.
Another potentially important aspect of bacteria-modulated PCD is the immune status of the host and whether different underlying immunologic conditions affect the ability of a pathogen to modulate PCD pathways. To date, few studies have investigated this and little is known about the impact of genetic defects on PCD responses. Studies of immune-deficient animal models, however, have provided some clues. In listeriosis for example, deficient TNF receptor signaling influences the ability of the bacterium to modulate PCD.
From the pathogen’s perspective, different bacterial strains within the same species may produce a more or less pronounced PCD event [204]. As a result, our understanding of the significance of PCD regulatory activity of a given species may be incomplete, because some strains trigger (or prevent) the PCD event, while others do not. It is intriguing to speculate that differences in strain virulence or production of ARMs may be responsible for these differential PCD effects. Future studies will help to clarify this. A greater understanding of strain differences could also provide the framework for defining the influence of PCD on immunity in the overall disease process, because more virulent bacterial strains cause more severe disease.
Once these aspects of the host-pathogen interaction are defined in more detail, the implications of PCD on immunity can be addressed. Against the background of these variables, the emerging underlying theme of bacteria-modulated PCD is that induction or prevention of PCD can benefit either the host or the pathogen, depending on the factors discussed herein. In the broader context, the in vivo relevance of most findings to date remain unclear, because the vast majority of studies have not addressed the fundamental and more difficult question of the role of these processes in development of disease and immunity in vivo.
A BACTERIAL CALL TO ARMS: NOVEL TARGETS FOR THERAPY?
Because bacterial modulation of PCD may alter immunity, bacterial ARMs and their actions on apoptosis may represent novel therapeutic targets [407]. Indeed, it is clear that, at least experimentally, pathogen-induced PCD can be manipulated by pharmacological intervention to alter the outcome of infection. During pneumococcal infection, for example, inhibition of caspase activation brought about by pneumolysin and H2O2 reduces neuropathology in an animal model of pneumococcal meningitis [276]. On the other hand, prevention of PCD in alveolar macrophages in a murine model of lower respiratory tract infection impeded immune responses, thereby benefiting the microbe [273]. In listeriosis, the importance of suppressing FASL expression by T cells during early infection to prevent premature termination of the cell-mediated immune response has been emphasized [319], suggesting a possible immunomodulatory target. Inhibiting TRAIL in listeriosis appears to augment host immune responses, indicating another potential therapeutic strategy [408]. “Blanket” prevention of apoptosis, however, may ultimately abrogate protective immunity, because some aspects of the PCD response in listeriosis are important for immunity to infection [319]. In anthrax, antecedent exposure to sublethal doses of the lethal toxin may desensitize macrophages to toxin-triggered PCD; this process may be amenable to interventions designed to reduce the severity of anthrax infection [305]. Heat shock protein 72 [142] and epidermal growth factor [409, 410] can protect against C. difficile toxin-A induced cytotoxicity through stabilizing actin, protecting mitochondria, and preventing caspase-9 activation. Overexpression of Rho proteins can also limit C. difficile toxin B-induced cell death [411]. In animal models, deliberate colonization with Saccharomyces boulardii is an appealing probiotic approach to prevent C. difficile diarrhea. Yeast protease degrades bacterial toxin components, which reduces tissue damage [412, 413]. Passive immunization with IgA to neutralize C. difficile toxic activity represents another therapeutic avenue [414]. Collectively, these examples illustrate that any therapeutic approach needs to be based on a sound understanding of the role of PCD in the overall disease process.
Studies have demonstrated that apoptosis activities in some cell types (e.g., T cells) can promote the intracellular growth of microbes in other cell types (e.g., macrophages) by impairing antimicrobial function [415, 416]. More finely targeted therapeutic schemes directed at bacterial toxigenic activity related to PCD represent a logical avenue for further investigation. Future research detailing the molecular events that occur in particular disease states and ultimately alter PCD pathways will pave the way for a better understanding of possible interventional strategies.
Fig. (2).
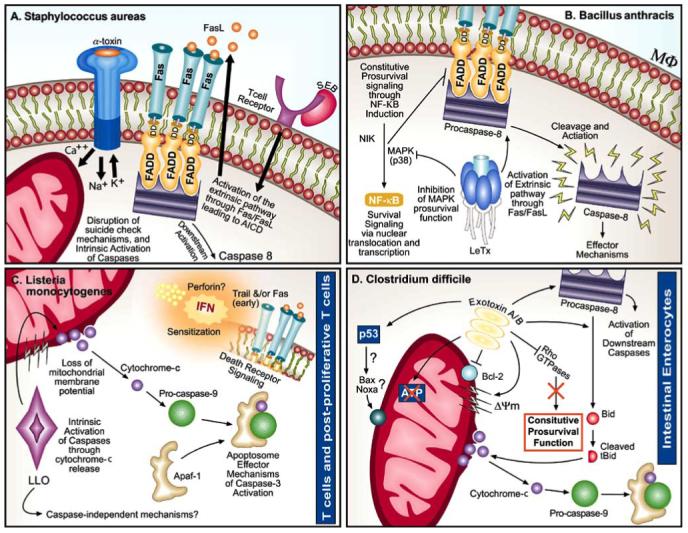
Programmed cell death (PCD) regulatory activities of gram-positive bacterial apoptosis regulatory molecules (ARMs). Numerous bacterial ARMs commandeer different elements of the intrinsic and extrinsic PCD pathways to seize control of the apoptotic machinery of the host cell; an activity that may improve the microbe’s survival and persistence. In S. aureus infection (A), α-toxin causes pores to form in the host cell membrane, which disrupts ionic balance, and alters steady state intrinsic mitochondrial PCD activity. Additionally, SEB triggers activation-induced cell death in T cells by a mechanism centered on extrinsic PCD factors involving FAS-FASL signaling. LeTx produced by B. anthracis (B) inhibits MAPK activity in macrophages and triggers PCD by a mechanism that involves activation of caspase-8. PCD in activated T cells in response to L. monocytogenes listeriolysin O (C) is characterized by a variety of alterations in both the intrinsic and extrinsic PCD pathways. Perforin and IFN-γ appear to contribute to these alterations. Finally, PCD in intestinal enterocytes triggered by C. difficile exotoxin (D) is executed by inactivation of Rho GTPases and involves toxin-induced alterations in both the intrinsic and extrinsic PCD pathways.
ACKNOWLEDGEMENTS
This work was supported by grants A140918 (E.E.A.) and CA21765 from the National Institutes of Health and by the American Lebanese Syrian Associated Charities (ALSAC). The authors thank Klo Spelshouse for assistance with digital art.
REFERENCES
- [1].Chen Y, Zychlinsky A. Apoptosis induced by bacterial pathogens. Microb Pathog. 1994;17(4):203–12. doi: 10.1006/mpat.1994.1066. [DOI] [PubMed] [Google Scholar]
- [2].Weinrauch Y, Zychlinsky A. The induction of apoptosis by bacterial pathogens. Annu Rev Microbiol. 1999;53:155–87. doi: 10.1146/annurev.micro.53.1.155. [DOI] [PubMed] [Google Scholar]
- [3].Behnia M, Robertson KA, Martin WJ., 2nd Lung infections: role of apoptosis in host defense and pathogenesis of disease. Chest. 2000;117(6):1771–7. doi: 10.1378/chest.117.6.1771. [DOI] [PubMed] [Google Scholar]
- [4].Navarre WW, Zychlinsky A. Pathogen-induced apoptosis of macrophages: a common end for different pathogenic strategies. Cell Microbiol. 2000;2(4):265–73. doi: 10.1046/j.1462-5822.2000.00056.x. [DOI] [PubMed] [Google Scholar]
- [5].Dockrell DH. Apoptotic cell death in the pathogenesis of infectious diseases. J Infect. 2001;42(4):227–34. doi: 10.1053/jinf.2001.0836. [DOI] [PubMed] [Google Scholar]
- [6].Hasnain SE, Begum R, Ramaiah KV, et al. Host-pathogen interactions during apoptosis. J Biosci. 2003;28(3):349–58. doi: 10.1007/BF02970153. [DOI] [PubMed] [Google Scholar]
- [7].Winau F, Kaufmann SH, Schaible UE. Apoptosis paves the detour path for CD8 T cell activation against intracellular bacteria. Cell Microbiol. 2004;6(7):599–607. doi: 10.1111/j.1462-5822.2004.00408.x. [DOI] [PubMed] [Google Scholar]
- [8].Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358(6382):167–9. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- [9].Dockrell DH, Lee M, Lynch DH, Read RC. Immune-mediated phagocytosis and killing of Streptococcus pneumoniae are associated with direct and bystander macrophage apoptosis. J Infect Dis. 2001;184(6):713–22. doi: 10.1086/323084. [DOI] [PubMed] [Google Scholar]
- [10].Rojas M, Barrera LF, Puzo G, Garcia LF. Differential induction of apoptosis by virulent Mycobacterium tuberculosis in resistant and susceptible murine macrophages: role of nitric oxide and mycobacterial products. J Immunol. 1997;159(3):1352–61. [PubMed] [Google Scholar]
- [11].Ahr B, Robert-Hebmann V, Devaux C, Biard-Piechaczyk M. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology. 2004;1(1):12. doi: 10.1186/1742-4690-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zychlinsky A, Sansonetti P. Perspectives series: host/pathogen interactions. Apoptosis in bacterial pathogenesis. J Clin Invest. 1997;100(3):493–5. doi: 10.1172/JCI119557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Moss JE, Aliprantis AO, Zychlinsky A. The regulation of apoptosis by microbial pathogens. Int Rev Cytol. 1999;187:203–59. doi: 10.1016/s0074-7696(08)62419-5. [DOI] [PubMed] [Google Scholar]
- [14].Dao DN, Kremer L, Guerardel Y, et al. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect Immun. 2004;72(4):2067–74. doi: 10.1128/IAI.72.4.2067-2074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Guerardel Y, Maes E, Briken V, et al. Lipomannan and lipoarabinomannan from a clinical isolate of Mycobacterium kansasii: novel structural features and apoptosis-inducing properties. J Biol Chem. 2003;278(38):36637–51. doi: 10.1074/jbc.M305427200. [DOI] [PubMed] [Google Scholar]
- [16].Briken V, Porcelli SA, Besra GS, Kremer L. Mycobacterial lipoarabinomannan and related lipoglycans: from biogenesis to modulation of the immune response. Mol Microbiol. 2004;53(2):391–403. doi: 10.1111/j.1365-2958.2004.04183.x. [DOI] [PubMed] [Google Scholar]
- [17].Prescott LM, Harley JP, Klein DA. Microbiology. 5th ed. McGraw-Hill; Boston: 2002. [Google Scholar]
- [18].Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;284(6):L899–914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- [19].Yokochi T, Morikawa A, Kato Y, Sugiyama T, Koide N. Apoptotic cell death in response to LPS. Prog Clin Biol Res. 1998;397:235–42. [PubMed] [Google Scholar]
- [20].Mangan DF, Welch GR, Wahl SM. Lipopolysaccharide, tumor necrosis factor-alpha, and IL-1 beta prevent programmed cell death (apoptosis) in human peripheral blood monocytes. J Immunol. 1991;146(5):1541–6. [PubMed] [Google Scholar]
- [21].Colotta F, Re F, Polentarutti N, Sozzani S, Mantovani A. Modulation of granulocyte survival and programmed cell death by cytokines and bacterial products. Blood. 1992;80(8):2012–20. [PubMed] [Google Scholar]
- [22].Nakagawa I, Nakata M, Kawabata S, Hamada S. Cytochrome c-mediated caspase-9 activation triggers apoptosis in Streptococcus pyogenes-infected epithelial cells. Cell Microbiol. 2001;3(6):395–405. doi: 10.1046/j.1462-5822.2001.00122.x. [DOI] [PubMed] [Google Scholar]
- [23].Lotz S, Aga E, Wilde I, et al. Highly purified lipoteichoic acid activates neutrophil granulocytes and delays their spontaneous apoptosis via CD14 and TLR2. J Leukoc Biol. 2004;75(3):467–77. doi: 10.1189/jlb.0803360. [DOI] [PubMed] [Google Scholar]
- [24].Wang PL, Shirasu S, Daito M, Ohura K. Streptococcus mutans lipoteichoic acid-induced apoptosis in cultured dental pulp cells from human deciduous teeth. Biochem Biophys Res Commun. 2001;281(4):957–61. doi: 10.1006/bbrc.2001.4451. [DOI] [PubMed] [Google Scholar]
- [25].Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297(5589):2048–51. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- [26].Popov SG, Villasmil R, Bernardi J, et al. Lethal toxin of Bacillus anthracis causes apoptosis of macrophages. Biochem Biophys Res Commun. 2002;293(1):349–55. doi: 10.1016/S0006-291X(02)00227-9. [DOI] [PubMed] [Google Scholar]
- [27].Carrero JA, Calderon B, Unanue ER. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J Immunol. 2004;172(8):4866–74. doi: 10.4049/jimmunol.172.8.4866. [DOI] [PubMed] [Google Scholar]
- [28].Ring A, Braun JS, Pohl J, Nizet V, Stremmel W, Shenep JL. Group B streptococcal beta-hemolysin induces mortality and liver injury in experimental sepsis. J Infect Dis. 2002;185(12):1745–53. doi: 10.1086/340818. [DOI] [PubMed] [Google Scholar]
- [29].Liu GY, Doran KS, Lawrence T, et al. Sword and shield: Linked group B streptococcal {beta}-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc Natl Acad Sci USA. 2004;101(40):14491–6. doi: 10.1073/pnas.0406143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Balachandran P, Hollingshead SK, Paton JC, Briles DE. The autolytic enzyme LytA of Streptococcus pneumoniae is not responsible for releasing pneumolysin. J Bacteriol. 2001;183(10):3108–16. doi: 10.1128/JB.183.10.3108-3116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ginsburg I. Role of lipoteichoic acid in infection and inflammation. Lancet Infect Dis. 2002;2(3):171–9. doi: 10.1016/s1473-3099(02)00226-8. [DOI] [PubMed] [Google Scholar]
- [32].Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–12. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- [33].Li H, Yuan J. Deciphering the pathways of life and death. Curr Opin Cell Biol. 1999;11(2):261–6. doi: 10.1016/s0955-0674(99)80035-0. [DOI] [PubMed] [Google Scholar]
- [34].Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- [35].Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signaling by death receptors. Eur J Biochem. 1998;254(3):439–59. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- [36].Luder CG, Gross U, Lopes MF. Intracellular protozoan parasites and apoptosis: diverse strategies to modulate parasite-host interactions. Trends Parasitol. 2001;17(10):480–6. doi: 10.1016/s1471-4922(01)02016-5. [DOI] [PubMed] [Google Scholar]
- [37].MacDonald HR, Baschieri S, Lees RK. Clonal expansion precedes anergy and death of V beta 8+ peripheral T cells responding to staphylococcal enterotoxin B in vivo. Eur J Immunol. 1991;21(8):1963–6. doi: 10.1002/eji.1830210827. [DOI] [PubMed] [Google Scholar]
- [38].Groux H, Monte D, Bourrez JM, Capron A, Ameisen JC. Activation of CD4+ T-lymphocytes in asymptomatic HIV infected patients induce the program action of lymphocyte death by apoptosis. C R Acad Sci III. 1991;312(12):599–606. [PubMed] [Google Scholar]
- [39].Groux H, Torpier G, Monte D, Mouton Y, Capron A, Ameisen JC. Activation-induced death by apoptosis in CD4+ T cells from human immunodeficiency virus-infected asymptomatic individuals. J Exp Med. 1992;175(2):331–40. doi: 10.1084/jem.175.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lin YS, Lei HY, Low TL, Shen CL, Chou LJ, Jan MS. In vivo induction of apoptosis in immature thymocytes by staphylococcal enterotoxin B. J Immunol. 1992;149(4):1156–63. [PubMed] [Google Scholar]
- [41].D’Adamio L, Awad KM, Reinherz EL. Thymic and peripheral apoptosis of antigen-specific T cells might cooperate in establishing self tolerance. Eur J Immunol. 1993;23(3):747–53. doi: 10.1002/eji.1830230327. [DOI] [PubMed] [Google Scholar]
- [42].Jenkinson EJ, Kingston R, Smith CA, Williams GT, Owen JJ. Antigen-induced apoptosis in developing T cells: a mechanism for negative selection of the T cell receptor repertoire. Eur J Immunol. 1989;19(11):2175–7. doi: 10.1002/eji.1830191132. [DOI] [PubMed] [Google Scholar]
- [43].Gonzalo JA, Baixeras E, Gonzalez-Garcia A, et al. Differential in vivo effects of a superantigen and an antibody targeted to the same T cell receptor. Activation-induced cell death vs passive macrophage-dependent deletion. J Immunol. 1994;152(4):1597–608. [PubMed] [Google Scholar]
- [44].Aroeira LS, Moreno MC, Martinez C. In vivo activation of T-cell induction into the primed phenotype and programmed cell death by staphylococcal enterotoxin B. Scand J Immunol. 1996;43(5):545–50. doi: 10.1046/j.1365-3083.1996.d01-75.x. [DOI] [PubMed] [Google Scholar]
- [45].Lin YS, Kao SF, Jan MS, et al. Changes of protein kinase C subspecies in staphylococcal enterotoxin-B-induced thymocyte apoptosis. Biochem Biophys Res Commun. 1995;213(3):1132–9. doi: 10.1006/bbrc.1995.2244. [DOI] [PubMed] [Google Scholar]
- [46].Weber AK, Wahn U, Renz H. Superantigen-induced T cell death by apoptosis: analysis on a single cell level and effect of IFN-gamma and IL-4 treatment. Int Arch Allergy Immunol. 2000;121(3):215–23. doi: 10.1159/000024320. [DOI] [PubMed] [Google Scholar]
- [47].Stohl W, Elliott JE, Lynch DH, Kiener PA. CD95 (Fas)-based, superantigen-dependent, CD4+ T cell-mediated down-regulation of human in vitro immunoglobulin responses. J Immunol. 1998;160(11):5231–8. [PubMed] [Google Scholar]
- [48].Urayama S, Kawakami A, Matsuoka N, et al. Fas/Fas ligand interaction regulates cytotoxicity of CD4+ T cells against staphylococcal enterotoxin B-pulsed endothelial cells. Biochem Biophys Res Commun. 1997;239(3):782–8. doi: 10.1006/bbrc.1997.7535. [DOI] [PubMed] [Google Scholar]
- [49].Esen M, Schreiner B, Jendrossek V, et al. Mechanisms of Staphylococcus aureus induced apoptosis of human endothelial cells. Apoptosis. 2001;6(6):431–9. doi: 10.1023/a:1012445925628. [DOI] [PubMed] [Google Scholar]
- [50].White J, Herman A, Pullen AM, Kubo R, Kappler JW, Marrack P. The V beta-specific superantigen staphylococcal enterotoxin B: stimulation of mature T cells and clonal deletion in neonatal mice. Cell. 1989;56(1):27–35. doi: 10.1016/0092-8674(89)90980-x. [DOI] [PubMed] [Google Scholar]
- [51].Kawabe Y, Ochi A. Selective anergy of V beta 8+,CD4+ T cells in Staphylococcus enterotoxin B-primed mice. J Exp Med. 1990;172(4):1065–70. doi: 10.1084/jem.172.4.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kawabe Y, Ochi A. Programmed cell death and extrathymic reduction of Vbeta8+ CD4+ T cells in mice tolerant to Staphylococcus aureus enterotoxin B. Nature. 1991;349(6306):245–8. doi: 10.1038/349245a0. [DOI] [PubMed] [Google Scholar]
- [53].Wahl C, Miethke T, Heeg K, Wagner H. Clonal deletion as direct consequence of an in vivo T cell response to bacterial superantigen. Eur J Immunol. 1993;23(5):1197–200. doi: 10.1002/eji.1830230536. [DOI] [PubMed] [Google Scholar]
- [54].Huang L, Soldevila G, Leeker M, Flavell R, Crispe IN. The liver eliminates T cells undergoing antigen-triggered apoptosis in vivo. Immunity. 1994;1(9):741–9. doi: 10.1016/s1074-7613(94)80016-2. [DOI] [PubMed] [Google Scholar]
- [55].Gonzalo JA, Martinez C, Springer TA, Gutierrez-Ramos JC. ICAM-1 is required for T cell proliferation but not for anergy or apoptosis induced by Staphylococcus aureus enterotoxin B in vivo. Int Immunol. 1995;7(10):1691–8. doi: 10.1093/intimm/7.10.1691. [DOI] [PubMed] [Google Scholar]
- [56].Renno T, Attinger A, Locatelli S, Bakker T, Vacheron S, MacDonald HR. Cutting edge: apoptosis of superantigen-activated T cells occurs preferentially after a discrete number of cell divisions in vivo. J Immunol. 1999;162(11):6312–5. [PubMed] [Google Scholar]
- [57].McLeod JD, Walker LS, Patel YI, Boulougouris G, Sansom DM. Activation of human T cells with superantigen (staphylococcal enterotoxin B) and CD28 confers resistance to apoptosis via CD95. J Immunol. 1998;160(5):2072–9. [PubMed] [Google Scholar]
- [58].Gorak-Stolinska P, Kemeny DM, Noble A. Activation-induced cell death in human T cells is a suicidal process regulated by cell density but superantigen induces T cell fratricide. Cell Immunol. 2002;219(2):98–107. doi: 10.1016/s0008-8749(02)00598-1. [DOI] [PubMed] [Google Scholar]
- [59].Dhein J, Walczak H, Baumler C, Debatin KM, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95) Nature. 1995;373(6513):438–41. doi: 10.1038/373438a0. [DOI] [PubMed] [Google Scholar]
- [60].Brunner T, Mogil RJ, LaFace D, et al. Cell-autonomous Fas (CD95)/Fas-ligand interaction mediates activation-induced apoptosis in T-cell hybridomas. Nature. 1995;373(6513):441–4. doi: 10.1038/373441a0. [DOI] [PubMed] [Google Scholar]
- [61].Singer GG, Abbas AK. The fas antigen is involved in peripheral but not thymic deletion of T lymphocytes in T cell receptor transgenic mice. Immunity. 1994;1(5):365–71. doi: 10.1016/1074-7613(94)90067-1. [DOI] [PubMed] [Google Scholar]
- [62].Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270(5239):1189–92. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- [63].Yang Y, Mercep M, Ware CF, Ashwell JD. Fas and activation-induced Fas ligand mediate apoptosis of T cell hybridomas: inhibition of Fas ligand expression by retinoic acid and glucocorticoids. J Exp Med. 1995;181(5):1673–82. doi: 10.1084/jem.181.5.1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vella AT, McCormack JE, Linsley PS, Kappler JW, Marrack P. Lipopolysaccharide interferes with the induction of peripheral T cell death. Immunity. 1995;2(3):261–70. doi: 10.1016/1074-7613(95)90050-0. [DOI] [PubMed] [Google Scholar]
- [65].Hildeman DA, Zhu Y, Mitchell TC, et al. Activated T cell death in vivo mediated by proapoptotic bcl-2 family member bim. Immunity. 2002;16(6):759–67. doi: 10.1016/s1074-7613(02)00322-9. [DOI] [PubMed] [Google Scholar]
- [66].Bras A, Rodriguez-Borlado L, Gonzalez-Garcia A, Martinez AC. Nitric oxide regulates clonal expansion and activation-induced cell death triggered by staphylococcal enterotoxin B. Infect Immun. 1997;65(10):4030–7. doi: 10.1128/iai.65.10.4030-4037.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Renno T, Hahne M, MacDonald HR. Proliferation is a prerequisite for bacterial superantigen-induced T cell apoptosis in vivo. J Exp Med. 1995;181(6):2283–7. doi: 10.1084/jem.181.6.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang JK, Zhu B, Ju ST, Tschopp J, Marshak-Rothstein A. CD4+ T cells reactivated with superantigen are both more sensitive to FasL-mediated killing and express a higher level of FasL. Cell Immunol. 1997;179(2):153–64. doi: 10.1006/cimm.1997.1159. [DOI] [PubMed] [Google Scholar]
- [69].Wedi B, Wieczorek D, Stunkel T, Breuer K, Kapp A. Staphylococcal exotoxins exert proinflammatory effects through inhibition of eosinophil apoptosis, increased surface antigen expression (CD11b, CD45, CD54, and CD69), and enhanced cytokine-activated oxidative burst, thereby triggering allergic inflammatory reactions. J Allergy Clin Immunol. 2002;109(3):477–84. doi: 10.1067/mai.2002.121702. [DOI] [PubMed] [Google Scholar]
- [70].Ettinger R, Panka DJ, Wang JK, Stanger BZ, Ju ST, Marshak-Rothstein A. Fas ligand-mediated cytotoxicity is directly responsible for apoptosis of normal CD4+ T cells responding to a bacterial superantigen. J Immunol. 1995;154(9):4302–8. [PubMed] [Google Scholar]
- [71].Sytwu HK, Liblau RS, McDevitt HO. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity. 1996;5(1):17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- [72].Russell JH, Rush B, Weaver C, Wang R. Mature T cells of autoimmune lpr/lpr mice have a defect in antigen-stimulated suicide. Proc Natl Acad Sci USA. 1993;90(10):4409–13. doi: 10.1073/pnas.90.10.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].McCormack JE, Callahan JE, Kappler J, Marrack PC. Profound deletion of mature T cells in vivo by chronic exposure to exogenous superantigen. J Immunol. 1993;150(9):3785–92. [PubMed] [Google Scholar]
- [74].Murphy DB. T cell mediated immunosuppression. Curr Opin Immunol. 1993;5(3):411–7. doi: 10.1016/0952-7915(93)90061-v. [DOI] [PubMed] [Google Scholar]
- [75].Noble A, Pestano GA, Cantor H. Suppression of immune responses by CD8 cells. I. Superantigen-activated CD8 cells induce unidirectional Fas-mediated apoptosis of antigen-activated CD4 cells. J Immunol. 1998;160(2):559–65. [PubMed] [Google Scholar]
- [76].Bratton DL, May KR, Kailey JM, Doherty DE, Leung DY. Staphylococcal toxic shock syndrome toxin-1 inhibits monocyte apoptosis. J Allergy Clin Immunol. 1999;103(5 Pt 1):895–900. doi: 10.1016/s0091-6749(99)70435-5. [DOI] [PubMed] [Google Scholar]
- [77].Hofer MF, Newell K, Duke RC, Schlievert PM, Freed JH, Leung DY. Differential effects of staphylococcal toxic shock syndrome toxin-1 on B cell apoptosis. Proc Natl Acad Sci USA. 1996;93(11):5425–30. doi: 10.1073/pnas.93.11.5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jonas D, Walev I, Berger T, Liebetrau M, Palmer M, Bhakdi S. Novel path to apoptosis: small transmembrane pores created by staphylococcal alpha-toxin in T lymphocytes evoke internucleosomal DNA degradation. Infect Immun. 1994;62(4):1304–12. doi: 10.1128/iai.62.4.1304-1312.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Haslinger B, Strangfeld K, Peters G, Schulze-Osthoff K, Sinha B. Staphylococcus aureus alpha-toxin induces apoptosis in peripheral blood mononuclear cells: role of endogenous tumour necrosis factor-alpha and the mitochondrial death pathway. Cell Microbiol. 2003;5(10):729–41. doi: 10.1046/j.1462-5822.2003.00317.x. [DOI] [PubMed] [Google Scholar]
- [80].Bantel H, Sinha B, Domschke W, Peters G, Schulze-Osthoff K, Janicke RU. alpha-Toxin is a mediator of Staphylococcus aureus-induced cell death and activates caspases via the intrinsic death pathway independently of death receptor signaling. J Cell Biol. 2001;155(4):637–48. doi: 10.1083/jcb.200105081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Menzies BE, Kourteva I. Staphylococcus aureus alpha-toxin induces apoptosis in endothelial cells. FEMS Immunol Med Microbiol. 2000;29(1):39–45. doi: 10.1111/j.1574-695X.2000.tb01503.x. [DOI] [PubMed] [Google Scholar]
- [82].Das T, Sa G, Sinha P, Ray PK. Induction of cell proliferation and apoptosis: dependence on the dose of the inducer. Biochem Biophys Res Commun. 1999;260(1):105–10. doi: 10.1006/bbrc.1999.0712. [DOI] [PubMed] [Google Scholar]
- [83].Goodyear CS, Silverman GJ. Death by a B cell superantigen: In vivo VH-targeted apoptotic supraclonal B cell deletion by a Staphylococcal Toxin. J Exp Med. 2003;197(9):1125–39. doi: 10.1084/jem.20020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Kuroda K, Yagi J, Imanishi K, et al. Implantation of IL-2-containing osmotic pump prolongs the survival of superantigen-reactive T cells expanded in mice injected with bacterial superantigen. J Immunol. 1996;157(4):1422–31. [PubMed] [Google Scholar]
- [85].Antonsson P, Wingren AG, Hansson J, Kalland T, Varga M, Dohlsten M. Functional characterization of the interaction between the superantigen staphylococcal enterotoxin A and the TCR. J Immunol. 1997;158(9):4245–51. [PubMed] [Google Scholar]
- [86].Kabelitz D, Wesselborg S. Life and death of a superantigen-reactive human CD4+ T cell clone: staphylococcal enterotoxins induce death by apoptosis but simultaneously trigger a proliferative response in the presence of HLA-DR+ antigen-presenting cells. Int Immunol. 1992;4(12):1381–8. doi: 10.1093/intimm/4.12.1381. [DOI] [PubMed] [Google Scholar]
- [87].Lee MS, Ueng SW, Shih CH, Chao CC. Primary cultures of human chondrocytes are susceptible to low inocula of Staphylococcus aureus infection and undergo apoptosis. Scand J Infect Dis. 2001;33(1):47–50. doi: 10.1080/003655401750064068. [DOI] [PubMed] [Google Scholar]
- [88].Nuzzo I, Sanges MR, Folgore A, Carratelli CR. Apoptosis of human keratinocytes after bacterial invasion. FEMS Immunol Med Microbiol. 2000;27(3):235–40. doi: 10.1111/j.1574-695X.2000.tb01435.x. [DOI] [PubMed] [Google Scholar]
- [89].Mempel M, Schnopp C, Hojka M, et al. Invasion of human keratinocytes by Staphylococcus aureus and intracellular bacterial persistence represent haemolysin-independent virulence mechanisms that are followed by features of necrotic and apoptotic keratinocyte cell death. Br J Dermatol. 2002;146(6):943–51. doi: 10.1046/j.1365-2133.2002.04752.x. [DOI] [PubMed] [Google Scholar]
- [90].Tucker KA, Reilly SS, Leslie CS, Hudson MC. Intracellular Staphylococcus aureus induces apoptosis in mouse osteoblasts. FEMS Microbiol Lett. 2000;186(2):151–6. doi: 10.1111/j.1574-6968.2000.tb09096.x. [DOI] [PubMed] [Google Scholar]
- [91].Fettucciari K, Rosati E, Scaringi L, et al. Group B Streptococcus induces apoptosis in macrophages. J Immunol. 2000;165(7):3923–33. doi: 10.4049/jimmunol.165.7.3923. [DOI] [PubMed] [Google Scholar]
- [92].Ulett GC, Bohnsack JF, Armstrong J, Adderson EE. Beta-hemolysin-independent induction of apoptosis of macrophages infected with serotype III group B streptococcus. J Infect Dis. 2003;188(7):1049–53. doi: 10.1086/378202. [DOI] [PubMed] [Google Scholar]
- [93].Bogdan I, Leib SL, Bergeron M, Chow L, Tauber MG. Tumor necrosis factor-alpha contributes to apoptosis in hippocampal neurons during experimental group B streptococcal meningitis. J Infect Dis. 1997;176(3):693–7. doi: 10.1086/514092. [DOI] [PubMed] [Google Scholar]
- [94].Halaas O, Liabakk NB, Vik R, et al. Monocytes stimulated with group B streptococci or interferons release tumour necrosis factor-related apoptosis-inducing ligand. Scand J Immunol. 2004;60(12):74–81. doi: 10.1111/j.0300-9475.2004.01448.x. [DOI] [PubMed] [Google Scholar]
- [95].Leib SL, Kim YS, Chow LL, Sheldon RA, Tauber MG. Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J Clin Invest. 1996;98(11):2632–9. doi: 10.1172/JCI119084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Loeffler JM, Ringer R, Hablutzel M, Tauber MG, Leib SL. The free radical scavenger alpha-phenyl-tert-butyl nitrone aggravates hippocampal apoptosis and learning deficits in experimental pneumococcal meningitis. J Infect Dis. 2001;183(2):247–252. doi: 10.1086/317921. [DOI] [PubMed] [Google Scholar]
- [97].Braun JS, Sublett JE, Freyer D, et al. Pneumococcal pneumolysin and H(2)O(2) mediate brain cell apoptosis during meningitis. J Clin Invest. 2002;109(1):19–27. doi: 10.1172/JCI12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Marriott HM, Ali F, Read RC, Mitchell TJ, Whyte MK, Dockrell DH. Nitric oxide levels regulate macrophage commitment to apoptosis or necrosis during pneumococcal infection. FASEB J. 2004;18(10):1126–8. doi: 10.1096/fj.03-1450fje. [DOI] [PubMed] [Google Scholar]
- [99].Bifrare YD, Gianinazzi C, Imboden H, Leib SL, Tauber MG. Bacterial meningitis causes two distinct forms of cellular damage in the hippocampal dentate gyrus in infant rats. Hippocampus. 2003;13(4):481–8. doi: 10.1002/hipo.10142. [DOI] [PubMed] [Google Scholar]
- [100].Ali F, Lee ME, Iannelli F, et al. Streptococcus pneumoniae-associated human macrophage apoptosis after bacterial internalization via complement and Fcgamma receptors correlates with intracellular bacterial load. J Infect Dis. 2003;188(8):1119–31. doi: 10.1086/378675. [DOI] [PubMed] [Google Scholar]
- [101].Tuomanen EI. The pneumococcus. ASM Press; Washington, DC: 2004. [Google Scholar]
- [102].Engelich G, White M, Hartshorn KL. Neutrophil survival is markedly reduced by incubation with influenza virus and Streptococcus pneumoniae: role of respiratory burst. J Leukoc Biol. 2001;69(1):50–6. [PubMed] [Google Scholar]
- [103].Zysk G, Bejo L, Schneider-Wald BK, Nau R, Heinz H. Induction of necrosis and apoptosis of neutrophil granulocytes by Streptococcus pneumoniae. Clin Exp Immunol. 2000;122(1):61–6. doi: 10.1046/j.1365-2249.2000.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Knapp S, Leemans JC, Florquin S, et al. Alveolar macrophages have a protective antiinflammatory role during murine pneumococcal pneumonia. Am J Respir Crit Care Med. 2003;167(2):171–9. doi: 10.1164/rccm.200207-698OC. [DOI] [PubMed] [Google Scholar]
- [105].Schmeck B, Gross R, N’Guessan PD, et al. Streptococcus pneumoniae-induced caspase 6-dependent apoptosis in lung epithelium. Infect Immun. 2004;72(9):4940–7. doi: 10.1128/IAI.72.9.4940-4947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Kemp K, Bruunsgaard H, Skinhoj P, Klarlund Pedersen B. Pneumococcal infections in humans are associated with increased apoptosis and trafficking of type 1 cytokine-producing T cells. Infect Immun. 2002;70(9):5019–25. doi: 10.1128/IAI.70.9.5019-5025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hakansson A, Carlstedt I, Davies J, Mossberg AK, Sabharwal H, Svanborg C. Aspects on the interaction of Streptococcus pneumoniae and Haemophilus influenzae with human respiratory tract mucosa. Am J Respir Crit Care Med. 1996;154(4 Pt 2):S187–91. doi: 10.1164/ajrccm/154.4_Pt_2.S187. [DOI] [PubMed] [Google Scholar]
- [108].Hotchkiss RS, Tinsley KW, Swanson PE, et al. Prevention of lymphocyte cell death in sepsis improves survival in mice. Proc Natl Acad Sci USA. 1999;96(25):14541–6. doi: 10.1073/pnas.96.25.14541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Hotchkiss RS, Chang KC, Swanson PE, et al. Caspase inhibitors improve survival in sepsis: a critical role of the lymphocyte. Nat Immunol. 2000;1(6):496–501. doi: 10.1038/82741. [DOI] [PubMed] [Google Scholar]
- [110].Bricker AL, Cywes C, Ashbaugh CD, Wessels MR. NAD+-glycohydrolase acts as an intracellular toxin to enhance the extracellular survival of group A streptococci. Mol Microbiol. 2002;44(1):257–69. doi: 10.1046/j.1365-2958.2002.02876.x. [DOI] [PubMed] [Google Scholar]
- [111].Tsai PJ, Lin YS, Kuo CF, Lei HY, Wu JJ. Group A Streptococcus induces apoptosis in human epithelial cells. Infect Immun. 1999;67(9):4334–9. doi: 10.1128/iai.67.9.4334-4339.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Tamura F, Nakagawa R, Akuta T, et al. Proapoptotic effect of proteolytic activation of matrix metalloproteinases by Streptococcus pyogenes thiol proteinase (Streptococcus pyrogenic exotoxin B) Infect Immun. 2004;72(8):4836–47. doi: 10.1128/IAI.72.8.4836-4847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Kuo CF, Wu JJ, Tsai PJ, et al. Streptococcal pyrogenic exotoxin B induces apoptosis and reduces phagocytic activity in U937 cells. Infect Immun. 1999;67(1):126–30. doi: 10.1128/iai.67.1.126-130.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Yoshida J, Takamura S, Ishibashi T, Nishio M. Antiproliferative and apoptosis-inducing effects of an antitumor glycoprotein from Streptococcus pyogenes. Anticancer Res. 1999;19(3A):1865–71. [PubMed] [Google Scholar]
- [115].Kobayashi SD, Braughton KR, Whitney AR, et al. Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc Natl Acad Sci USA. 2003;100(19):10948–53. doi: 10.1073/pnas.1833375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Popov SG, Villasmil R, Bernardi J, et al. Effect of Bacillus anthracis lethal toxin on human peripheral blood mononuclear cells. FEBS Lett. 2002;527(13):211–5. doi: 10.1016/s0014-5793(02)03228-3. [DOI] [PubMed] [Google Scholar]
- [117].Kim SO, Jing Q, Hoebe K, Beutler B, Duesbery NS, Han J. Sensitizing anthrax lethal toxin-resistant macrophages to lethal toxin-induced killing by tumor necrosis factor-alpha. J Biol Chem. 2003;278(9):7413–21. doi: 10.1074/jbc.M209279200. [DOI] [PubMed] [Google Scholar]
- [118].Pandey J, Warburton D. Knock-on effect of anthrax lethal toxin on macrophages potentiates cytotoxicity to endothelial cells. Microbes Infect. 2004;6(9):835–43. doi: 10.1016/j.micinf.2004.04.013. [DOI] [PubMed] [Google Scholar]
- [119].Kirby JE. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect Immun. 2004;72(1):430–9. doi: 10.1128/IAI.72.1.430-439.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Lin CG, Kao YT, Liu WT, et al. Cytotoxic effects of anthrax lethal toxin on macrophage-like cell line J774A.1. Curr Microbiol. 1996;33(4):224–7. doi: 10.1007/s002849900104. [DOI] [PubMed] [Google Scholar]
- [121].Kau JH, Lin CG, Huang HH, et al. Calyculin A sensitive protein phosphatase is required for Bacillus anthracis lethal toxin induced cytotoxicity. Curr Microbiol. 2002;44(2):106–11. doi: 10.1007/s00284-001-0059-8. [DOI] [PubMed] [Google Scholar]
- [122].Hsu LC, Park JM, Zhang K, et al. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004;428(6980):341–5. doi: 10.1038/nature02405. [DOI] [PubMed] [Google Scholar]
- [123].Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. 2004;200(4):535–40. doi: 10.1084/jem.20040769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Guzman CA, Domann E, Rohde M, et al. Apoptosis of mouse dendritic cells is triggered by listeriolysin, the major virulence determinant of Listeria monocytogenes. Mol Microbiol. 1996;20(1):119–26. doi: 10.1111/j.1365-2958.1996.tb02494.x. [DOI] [PubMed] [Google Scholar]
- [125].Menon A, Shroyer ML, Wampler JL, Chawan CB, Bhunia AK. In vitro study of Listeria monocytogenes infection to murine primary and human transformed B cells. Comp Immunol Microbiol Infect Dis. 2003;26(3):157–74. doi: 10.1016/s0147-9571(02)00039-5. [DOI] [PubMed] [Google Scholar]
- [126].O’Connell RM, Saha SK, Vaidya SA, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200(4):437–45. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Schluter D, Domann E, Buck C, et al. Phosphatidylcholine-specific phospholipase C from Listeria monocytogenes is an important virulence factor in murine cerebral listeriosis. Infect Immun. 1998;66(12):5930–8. doi: 10.1128/iai.66.12.5930-5938.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Valenti P, Greco R, Pitari G, et al. Apoptosis of Caco-2 intestinal cells invaded by Listeria monocytogenes: protective effect of lactoferrin. Exp Cell Res. 1999;250(1):197–202. doi: 10.1006/excr.1999.4500. [DOI] [PubMed] [Google Scholar]
- [129].Gunturi A, Berg RE, Forman J. Preferential survival of CD8 T and NK cells expressing high levels of CD94. J Immunol. 2003;170(4):1737–45. doi: 10.4049/jimmunol.170.4.1737. [DOI] [PubMed] [Google Scholar]
- [130].Mannering SI, Zhong J, Cheers C. T-cell activation, proliferation and apoptosis in primary Listeria monocytogenes infection. Immunology. 2002;106(1):87–95. doi: 10.1046/j.1365-2567.2002.01408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mukasa A, Lahn M, Fleming S, et al. Extensive and preferential Fas/Fas ligand-dependent death of gammadelta T cells following infection with Listeria monocytogenes. Scand J Immunol. 2002;56(3):233–47. doi: 10.1046/j.1365-3083.2002.01123.x. [DOI] [PubMed] [Google Scholar]
- [132].Jensen ER, Glass AA, Clark WR, Wing EJ, Miller JF, Gregory SH. Fas (CD95)-dependent cell-mediated immunity to Listeria monocytogenes. Infect Immun. 1998;66(9):4143–50. doi: 10.1128/iai.66.9.4143-4150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Fuse Y, Nishimura H, Maeda K, Yoshikai Y. CD95 (Fas) may control the expansion of activated T cells after elimination of bacteria in murine listeriosis. Infect Immun. 1997;65(5):1883–91. doi: 10.1128/iai.65.5.1883-1891.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Merrick JC, Edelson BT, Bhardwaj V, Swanson PE, Unanue ER. Lymphocyte apoptosis during early phase of Listeria infection in mice. Am J Pathol. 1997;151(3):785–92. [PMC free article] [PubMed] [Google Scholar]
- [135].Mahida YR, Makh S, Hyde S, Gray T, Borriello SP. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut. 1996;38(3):337–47. doi: 10.1136/gut.38.3.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Calderon GM, Torres-Lopez J, Lin TJ, et al. Effects of toxin A from Clostridium difficile on mast cell activation and survival. Infect Immun. 1998;66(6):2755–61. doi: 10.1128/iai.66.6.2755-2761.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Mahida YR, Galvin A, Makh S, et al. Effect of Clostridium difficile toxin A on human colonic lamina propria cells: early loss of macrophages followed by T-cell apoptosis. Infect Immun. 1998;66(11):5462–9. doi: 10.1128/iai.66.11.5462-5469.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Zhao J, Wang D, Zhao H, Lin W. Comparative study on apoptosis induction of SMMC7721 and Vero cells by Clostridium difficile toxin A. Ai Zheng. 2003;22(7):719–24. [PubMed] [Google Scholar]
- [139].He D, Hagen SJ, Pothoulakis C, et al. Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology. 2000;119(1):139–50. doi: 10.1053/gast.2000.8526. [DOI] [PubMed] [Google Scholar]
- [140].He D, Sougioultzis S, Hagen S, et al. Clostridium difficile toxin A triggers human colonocyte IL-8 release via mitochondrial oxygen radical generation. Gastroenterology. 2002;122(4):1048–57. doi: 10.1053/gast.2002.32386. [DOI] [PubMed] [Google Scholar]
- [141].Brito GA, Fujji J, Carneiro-Filho BA, Lima AA, Obrig T, Guerrant RL. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J Infect Dis. 2002;186(10):1438–47. doi: 10.1086/344729. [DOI] [PubMed] [Google Scholar]
- [142].Liu TS, Musch MW, Sugi K, et al. Protective role of HSP72 against Clostridium difficile toxin A-induced intestinal epithelial cell dysfunction. Am J Physiol Cell Physiol. 2003;284(4):C1073–82. doi: 10.1152/ajpcell.00134.2002. [DOI] [PubMed] [Google Scholar]
- [143].Sautin YY, Crawford JM, Svetlov SI. Enhancement of survival by LPA via Erk1/Erk2 and PI 3-kinase/Akt pathways in a murine hepatocyte cell line. Am J Physiol Cell Physiol. 2001;281(6):C2010–9. doi: 10.1152/ajpcell.00077.2001. [DOI] [PubMed] [Google Scholar]
- [144].Fiorentini C, Fabbri A, Falzano L, et al. Clostridium difficile toxin B induces apoptosis in intestinal cultured cells. Infect Immun. 1998;66(6):2660–5. doi: 10.1128/iai.66.6.2660-2665.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Le Gall M, Chambard JC, Breittmayer JP, Grall D, Pouyssegur J, Van Obberghen-Schilling E. The p42/p44 MAP kinase pathway prevents apoptosis induced by anchorage and serum removal. Mol Biol Cell. 2000;11(3):1103–12. doi: 10.1091/mbc.11.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Agarwal B, Halmos B, Feoktistov AS, et al. Mechanism of lovastatin-induced apoptosis in intestinal epithelial cells. Carcinogenesis. 2002;23(3):521–8. doi: 10.1093/carcin/23.3.521. [DOI] [PubMed] [Google Scholar]
- [147].Qa’Dan M, Ramsey M, Daniel J, et al. Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol. 2002;4(7):425–34. doi: 10.1046/j.1462-5822.2002.00201.x. [DOI] [PubMed] [Google Scholar]
- [148].Hippenstiel S, Schmeck B, N’Guessan PD, et al. Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;283(4):L830–8. doi: 10.1152/ajplung.00467.2001. [DOI] [PubMed] [Google Scholar]
- [149].Chakrabarti G, Zhou X, McClane BA. Death pathways activated in CaCo-2 cells by Clostridium perfringens enterotoxin. Infect Immun. 2003;71(8):4260–70. doi: 10.1128/IAI.71.8.4260-4270.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Chakrabarti G, McClane BA. The importance of calcium influx, calpain and calmodulin for the activation of CaCo-2 cell death pathways by Clostridium perfringens enterotoxin. Cell Microbiol. 2005;7(1):129–46. doi: 10.1111/j.1462-5822.2004.00442.x. [DOI] [PubMed] [Google Scholar]
- [151].Cohen JJ. Programmed cell death in the immune system. Adv Immunol. 1991;50:55–85. doi: 10.1016/s0065-2776(08)60822-6. [DOI] [PubMed] [Google Scholar]
- [152].Golstein P, Ojcius DM, Young JD. Cell death mechanisms and the immune system. Immunol Rev. 1991;121:29–65. doi: 10.1111/j.1600-065x.1991.tb00822.x. [DOI] [PubMed] [Google Scholar]
- [153].Allen PD, Bustin SA, Newland AC. The role of apoptosis (programmed cell death) in haemopoiesis and the immune system. Blood Rev. 1993;7(1):63–73. doi: 10.1016/0268-960x(93)90025-y. [DOI] [PubMed] [Google Scholar]
- [154].Abbas AK. Die and let live: eliminating dangerous lymphocytes. Cell. 1996;84(5):655–7. doi: 10.1016/s0092-8674(00)81042-9. [DOI] [PubMed] [Google Scholar]
- [155].Hetts SW. To die or not to die: an overview of apoptosis and its role in disease. JAMA. 1998;279(4):300–7. doi: 10.1001/jama.279.4.300. [DOI] [PubMed] [Google Scholar]
- [156].Van Parijs L, Abbas AK. Homeostasis and self-tolerance in the immune system: turning lymphocytes off. Science. 1998;280(5361):243–8. doi: 10.1126/science.280.5361.243. [DOI] [PubMed] [Google Scholar]
- [157].Blackman M, Kappler J, Marrack P. The role of the T cell receptor in positive and negative selection of developing T cells. Science. 1990;248(4961):1335–41. doi: 10.1126/science.1972592. [DOI] [PubMed] [Google Scholar]
- [158].Robey E, Fowlkes BJ. Selective events in T cell development. Annu Rev Immunol. 1994;12:675–705. doi: 10.1146/annurev.iy.12.040194.003331. [DOI] [PubMed] [Google Scholar]
- [159].Nossal GJ. Negative selection of lymphocytes. Cell. 1994;76(2):229–39. doi: 10.1016/0092-8674(94)90331-x. [DOI] [PubMed] [Google Scholar]
- [160].Marsden VS, Strasser A. Control of apoptosis in the immune system: Bcl-2, BH3-only proteins and more. Annu Rev Immunol. 2003;21:71–105. doi: 10.1146/annurev.immunol.21.120601.141029. [DOI] [PubMed] [Google Scholar]
- [161].Alderson MR, Tough TW, Davis-Smith T, et al. Fas ligand mediates activation-induced cell death in human T lymphocytes. J Exp Med. 1995;181(1):71–7. doi: 10.1084/jem.181.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ju ST, Panka DJ, Cui H, et al. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature. 1995;373(6513):444–8. doi: 10.1038/373444a0. [DOI] [PubMed] [Google Scholar]
- [163].Jendro MC, Kohler L, Kuipers JG, Zeidler H. Microbe-induced T cell apoptosis: subversion of the host defense system? FEMS Microbiol Lett. 2002;207(2):121–6. doi: 10.1111/j.1574-6968.2002.tb11039.x. [DOI] [PubMed] [Google Scholar]
- [164].Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6(5):513–9. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- [165].Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10(9):369–77. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- [166].Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora’s box opens. Nat Rev Mol Cell Biol. 2001;2(1):67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- [167].Ravagnan L, Roumier T, Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192(2):131–7. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- [168].Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–9. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- [169].Itoh N, Yonehara S, Ishii A, et al. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell. 1991;66(2):233–43. doi: 10.1016/0092-8674(91)90614-5. [DOI] [PubMed] [Google Scholar]
- [170].Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407(6805):770–6. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- [171].Catalfamo M, Henkart PA. Perforin and the granule exocytosis cytotoxicity pathway. Curr Opin Immunol. 2003;15(5):522–7. doi: 10.1016/s0952-7915(03)00114-6. [DOI] [PubMed] [Google Scholar]
- [172].Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57(10):1835–40. [PubMed] [Google Scholar]
- [173].Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–6. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- [174].Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–90. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- [175].Shi Y. Caspase activation: revisiting the induced proximity model. Cell. 2004;117(7):855–8. doi: 10.1016/j.cell.2004.06.007. [DOI] [PubMed] [Google Scholar]
- [176].Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117(5):561–74. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- [177].Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- [178].Cryns V, Yuan J. Proteases to die for. Genes Dev. 1998;12(11):1551–70. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- [179].Stroh C, Schulze-Osthoff K. Death by a thousand cuts: an ever increasing list of caspase substrates. Cell Death Differ. 1998;5(12):997–1000. doi: 10.1038/sj.cdd.4400451. [DOI] [PubMed] [Google Scholar]
- [180].Green DR, Amarante-Mendes GP. The point of no return: mitochondria, caspases, and the commitment to cell death. Results Probl Cell Differ. 1998;24:45–61. doi: 10.1007/978-3-540-69185-3_3. [DOI] [PubMed] [Google Scholar]
- [181].Ferri KF, Kroemer G. Control of apoptotic DNA degradation. Nat Cell Biol. 2000;2(4):E63–4. doi: 10.1038/35008692. [DOI] [PubMed] [Google Scholar]
- [182].Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391(6662):43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- [183].Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10(1):76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Nunez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. 1998;17(25):3237–45. doi: 10.1038/sj.onc.1202581. [DOI] [PubMed] [Google Scholar]
- [185].Fadok VA, Bratton DL, Frasch SC, Warner ML, Henson PM. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 1998;5(7):551–62. doi: 10.1038/sj.cdd.4400404. [DOI] [PubMed] [Google Scholar]
- [186].Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- [187].Fadok VA, Bratton DL, Rose DM, Pearson A, Ezekewitz RA, Henson PM. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405(6782):85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- [188].Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101(4):890–8. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- [190].Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. 2003;22(56):9030–40. doi: 10.1038/sj.onc.1207116. [DOI] [PubMed] [Google Scholar]
- [191].Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem. 1999;264(3):687–701. doi: 10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- [192].Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90(3):405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- [193].Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274(17):11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- [194].Susin SA, Daugas E, Ravagnan L, et al. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192(4):571–80. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281(5381):1322–6. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- [196].Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387(6635):773–6. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- [197].Peter ME, Krammer PH. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 2003;10(1):26–35. doi: 10.1038/sj.cdd.4401186. [DOI] [PubMed] [Google Scholar]
- [198].Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- [199].Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94(4):481–90. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- [200].Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1(1):50–8. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- [201].Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor 2 signal transducers TRAF2 and c-IAP1 are components of the tumor necrosis factor receptor 1 signaling complex. Proc Natl Acad Sci USA. 1996;93(24):13973–8. doi: 10.1073/pnas.93.24.13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Rosenberger CM, Finlay BB. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol. 2003;4(5):385–96. doi: 10.1038/nrm1104. [DOI] [PubMed] [Google Scholar]
- [203].DeLeo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis. 2004;9(4):399–413. doi: 10.1023/B:APPT.0000031448.64969.fa. [DOI] [PubMed] [Google Scholar]
- [204].Gao LY, Kwaik YA. The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol. 2000;8(7):306–13. doi: 10.1016/s0966-842x(00)01784-4. [DOI] [PubMed] [Google Scholar]
- [205].Gao L, Abu Kwaik Y. Hijacking of apoptotic pathways by bacterial pathogens. Microbes Infect. 2000;2(14):1705–19. doi: 10.1016/s1286-4579(00)01326-5. [DOI] [PubMed] [Google Scholar]
- [206](a).Menaker RJ, Jones NL. Fascination with bacteria-triggered cell death: the significance of Fas-mediated apoptosis during bacterial infection in vivo. Microbes Infect. 2003;5(12):1149–58. doi: 10.1016/j.micinf.2003.08.001. [DOI] [PubMed] [Google Scholar]; (b) Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–16. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Schmitt CK, Meysick KC, O’Brien AD. Bacterial toxins: friends or foes? Emerg Infect Dis. 1999;5(2):224–34. doi: 10.3201/eid0502.990206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Laohachai KN, Bahadi R, Hardo MB, Hardo PG, Kourie JI. The role of bacterial and non-bacterial toxins in the induction of changes in membrane transport: implications for diarrhea. Toxicon. 2003;42(7):687–707. doi: 10.1016/j.toxicon.2003.08.010. [DOI] [PubMed] [Google Scholar]
- [209].Fiorentini C, Falzano L, Travaglione S, Fabbri A. Hijacking Rho GTPases by protein toxins and apoptosis: molecular strategies of pathogenic bacteria. Cell Death Differ. 2003;10(2):147–52. doi: 10.1038/sj.cdd.4401151. [DOI] [PubMed] [Google Scholar]
- [210].Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–32. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- [211].Dinges MM, Orwin PM, Schlievert PM. Exotoxins of Staphylococcus aureus. Clin Microbiol Rev. 2000;13(1):16–34. doi: 10.1128/cmr.13.1.16-34.2000. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Johnson HM, Russell JK, Pontzer CH. Staphylococcal enterotoxin microbial superantigens. FASEB J. 1991;5(12):2706–12. doi: 10.1096/fasebj.5.12.1916093. [DOI] [PubMed] [Google Scholar]
- [213].Silverman GJ, Goodyear CS. A model B-cell superantigen and the immunobiology of B lymphocytes. Clin Immunol. 2002;102(2):117–34. doi: 10.1006/clim.2001.5143. [DOI] [PubMed] [Google Scholar]
- [214].Baran J, Weglarczyk K, Mysiak M, et al. Fas (CD95)-Fas ligand interactions are responsible for monocyte apoptosis occurring as a result of phagocytosis and killing of Staphylococcus aureus. Infect Immun. 2001;69(3):1287–97. doi: 10.1128/IAI.69.3.1287-1297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Weglarczyk K, Baran J, Zembala M, Pryjma J. Caspase-8 activation precedes alterations of mitochondrial membrane potential during monocyte apoptosis induced by phagocytosis and killing of Staphylococcus aureus. Infect Immun. 2004;72(5):2590–7. doi: 10.1128/IAI.72.5.2590-2597.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Menzies BE, Kourteva I. Internalization of Staphylococcus aureus by endothelial cells induces apoptosis. Infect Immun. 1998;66(12):5994–8. doi: 10.1128/iai.66.12.5994-5998.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Bayles KW, Wesson CA, Liou LE, Fox LK, Bohach GA, Trumble WR. Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect Immun. 1998;66(1):336–42. doi: 10.1128/iai.66.1.336-342.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Wesson CA, Deringer J, Liou LE, Bayles KW, Bohach GA, Trumble WR. Apoptosis induced by Staphylococcus aureus in epithelial cells utilizes a mechanism involving caspases 8 and 3. Infect Immun. 2000;68(5):2998–3001. doi: 10.1128/iai.68.5.2998-3001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Stevens DL. Superantigens: their role in infectious diseases. Immunol Invest. 1997;26(12):275–81. doi: 10.3109/08820139709048933. [DOI] [PubMed] [Google Scholar]
- [220].Janeway CA, Jr, Yagi J, Conrad PJ, et al. T-cell responses to Mls and to bacterial proteins that mimic its behavior. Immunol Rev. 1989;107:61–88. doi: 10.1111/j.1600-065x.1989.tb00003.x. [DOI] [PubMed] [Google Scholar]
- [221].Mollick JA, Cook RG, Rich RR. Class II MHC molecules are specific receptors for staphylococcus enterotoxin A. Science. 1989;244(4906):817–20. doi: 10.1126/science.2658055. [DOI] [PubMed] [Google Scholar]
- [222].Fraser JD. High-affinity binding of staphylococcal enterotoxins A and B to HLA-DR. Nature. 1989;339(6221):221–3. doi: 10.1038/339221a0. [DOI] [PubMed] [Google Scholar]
- [223].Karp DR, Teletski CL, Scholl P, Geha R, Long EO. The alpha 1 domain of the HLA-DR molecule is essential for high-affinity binding of the toxic shock syndrome toxin-1. Nature. 1990;346(6283):474–6. doi: 10.1038/346474a0. [DOI] [PubMed] [Google Scholar]
- [224].Kotzin BL, Leung DY, Kappler J, Marrack P. Superantigens and their potential role in human disease. Adv Immunol. 1993;54:99–166. doi: 10.1016/s0065-2776(08)60534-9. [DOI] [PubMed] [Google Scholar]
- [225].Fleischer B. Superantigens. Apmis. 1994;102(1):3–12. doi: 10.1111/j.1699-0463.1994.tb04839.x. [DOI] [PubMed] [Google Scholar]
- [226].Uchiyama T, Yan XJ, Imanishi K, Yagi J. Bacterial superantigens--mechanism of T cell activation by the superantigens and their role in the pathogenesis of infectious diseases. Microbiol Immunol. 1994;38(4):245–56. doi: 10.1111/j.1348-0421.1994.tb01772.x. [DOI] [PubMed] [Google Scholar]
- [227].Herman A, Kappler JW, Marrack P, Pullen AM. Superantigens: mechanism of T-cell stimulation and role in immune responses. Annu Rev Immunol. 1991;9:745–72. doi: 10.1146/annurev.iy.09.040191.003525. [DOI] [PubMed] [Google Scholar]
- [228].Kotb M. Bacterial pyrogenic exotoxins as superantigens. Clin Microbiol Rev. 1995;8(3):411–26. doi: 10.1128/cmr.8.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Kotb M. Superantigens of gram-positive bacteria: structure-function analyses and their implications for biological activity. Curr Opin Microbiol. 1998;1(1):56–65. doi: 10.1016/s1369-5274(98)80143-4. [DOI] [PubMed] [Google Scholar]
- [230].Moulding DA, Walter C, Hart CA, Edwards SW. Effects of staphylococcal enterotoxins on human neutrophil functions and apoptosis. Infect Immun. 1999;67(5):2312–8. doi: 10.1128/iai.67.5.2312-2318.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Lenardo M, Chan KM, Hornung F, et al. Mature T lymphocyte apoptosis--immune regulation in a dynamic and unpredictable antigenic environment. Ann Rev Immunol. 1999;17:221–53. doi: 10.1146/annurev.immunol.17.1.221. [DOI] [PubMed] [Google Scholar]
- [232].Bonfoco E, Stuart PM, Brunner T, et al. Inducible nonlymphoid expression of Fas ligand is responsible for superantigen-induced peripheral deletion of T cells. Immunity. 1998;9(5):711–20. doi: 10.1016/s1074-7613(00)80668-8. [DOI] [PubMed] [Google Scholar]
- [233].Brown SB, Savill J. Phagocytosis triggers macrophage release of Fas ligand and induces apoptosis of bystander leukocytes. J Immunol. 1999;162(1):480–5. [PubMed] [Google Scholar]
- [234].Vabulas R, Bittlingmaier R, Heeg K, Wagner H, Miethke T. Rapid clearance of the bacterial superantigen staphylococcal enterotoxin B in vivo. Infect Immun. 1996;64(11):4567–73. doi: 10.1128/iai.64.11.4567-4573.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Chvatchko Y, Valera S, Aubry JP, Renno T, Buell G, Bonnefoy JY. The involvement of an ATP-gated ion channel, P(2X1), in thymocyte apoptosis. Immunity. 1996;5(3):275–83. doi: 10.1016/s1074-7613(00)80322-2. [DOI] [PubMed] [Google Scholar]
- [236].Mountz JD, Baker TJ, Borcherding DR, Bluethmann H, Zhou T, Edwards CK., 3rd Increased susceptibility of fas mutant MRL-lpr/lpr mice to staphylococcal enterotoxin B-induced septic shock. J Immunol. 1995;155(10):4829–37. [PubMed] [Google Scholar]
- [237].Suttorp N, Seeger W, Dewein E, Bhakdi S, Roka L. Staphylococcal alpha-toxin-induced PGI2 production in endothelial cells: role of calcium. Am J Physiol. 1985;248(1 Pt 1):C127–34. doi: 10.1152/ajpcell.1985.248.1.C127. [DOI] [PubMed] [Google Scholar]
- [238].Suttorp N, Seeger W, Zucker-Reimann J, Roka L, Bhakdi S. Mechanism of leukotriene generation in polymorphonuclear leukocytes by staphylococcal alpha-toxin. Infect Immun. 1987;55(1):104–10. doi: 10.1128/iai.55.1.104-110.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Ahnert-Hilger G, Bhakdi S, Gratzl M. Minimal requirements for exocytosis. A study using PC 12 cells permeabilized with staphylococcal alpha-toxin. J Biol Chem. 1985;260(23):12730–4. [PubMed] [Google Scholar]
- [240].Ezepchuk YV, Leung DY, Middleton MH, Bina P, Reiser R, Norris DA. Staphylococcal toxins and protein A differentially induce cytotoxicity and release of tumor necrosis factor-alpha from human keratinocytes. J Invest Dermatol. 1996;107(4):603–9. doi: 10.1111/1523-1747.ep12583377. [DOI] [PubMed] [Google Scholar]
- [241].Walev I, Martin E, Jonas D, et al. Staphylococcal alpha-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun. 1993;61(12):4972–9. doi: 10.1128/iai.61.12.4972-4979.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Essmann F, Bantel H, Totzke G, et al. Staphylococcus aureus alpha-toxin-induced cell death: predominant necrosis despite apoptotic caspase activation. Cell Death Differ. 2003;10(11):1260–72. doi: 10.1038/sj.cdd.4401301. [DOI] [PubMed] [Google Scholar]
- [243].Das T, Sa G, Ray PK. Mechanisms of protein A superantigen-induced signal transduction for proliferation of mouse B cell. Immunol Lett. 1999;70(1):43–51. doi: 10.1016/s0165-2478(99)00128-5. [DOI] [PubMed] [Google Scholar]
- [244].Ray PK, Das T, Sa G, Ghosh AK, Chattopadhyay S. Protection of apoptotic cell death by protein A. Apoptosis. 2000;5(6):509–14. doi: 10.1023/a:1009633412009. [DOI] [PubMed] [Google Scholar]
- [245].Ghosh AK, Sinha P, Das T, Sa G, Ray PK. S. aureus superantigen protein A expands CD4(+)/CD8(+)/CD19(+)/CD34(+) cells in mice: a potential immunorestorer. Biochem Biophys Res Commun. 1999;256(1):142–6. doi: 10.1006/bbrc.1999.0194. [DOI] [PubMed] [Google Scholar]
- [246].Nizet V, Ferrieri P, Rubens CE. Molecular pathogenesis of Group B Streptococcal disease in newborns. In: Stevens DL, Kaplan EL, editors. Streptococcal Infections. Clinical aspects, microbiology, and molecular pathogenesis. Oxford University Press; New York: 2000. pp. 180–222. [Google Scholar]
- [247].Baker CJ. Group B Streptococcal infections. In: Stevens DL, Kaplan EL, editors. Streptococcal Infections. Clinical aspects, microbiology, and molecular pathogenesis. Oxford University Press; New York: 2000. pp. 222–37. [Google Scholar]
- [248].Schuchat A. Epidemiology of group B streptococcal disease in the United States: shifting paradigms. Clin Microbiol Rev. 1998;11(3):497–513. doi: 10.1128/cmr.11.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Sibille Y, Reynolds HY. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am Rev Respir Dis. 1990;141(2):471–501. doi: 10.1164/ajrccm/141.2.471. [DOI] [PubMed] [Google Scholar]
- [250].Franke-Ullmann G, Pfortner C, Walter P, Steinmuller C, Lohmann-Matthes ML, Kobzik L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J Immunol. 1996;157(7):3097–104. [PubMed] [Google Scholar]
- [251].Jonsson S, Musher DM, Chapman A, Goree A, Lawrence EC. Phagocytosis and killing of common bacterial pathogens of the lung by human alveolar macrophages. J Infect Dis. 1985;152(1):4–13. doi: 10.1093/infdis/152.1.4. [DOI] [PubMed] [Google Scholar]
- [252].Sherman MP, Johnson JT, Rothlein R, Hughes BJ, Smith CW, Anderson DC. Role of pulmonary phagocytes in host defense against group B streptococci in preterm versus term rabbit lung. J Infect Dis. 1992;166(4):818–26. doi: 10.1093/infdis/166.4.818. [DOI] [PubMed] [Google Scholar]
- [253].Doran KS, Nizet V. Molecular pathogenesis of neonatal group B streptococcal infection: no longer in its infancy. Mol Microbiol. 2004;54(1):23–31. doi: 10.1111/j.1365-2958.2004.04266.x. [DOI] [PubMed] [Google Scholar]
- [254].Valenti-Weigand P, Benkel P, Rohde M, Chhatwal GS. Entry and intracellular survival of group B streptococci in J774 macrophages. Infect Immun. 1996;64(7):2467–73. doi: 10.1128/iai.64.7.2467-2473.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Cornacchione P, Scaringi L, Fettucciari K, et al. Group B streptococci persist inside macrophages. Immunology. 1998;93(1):86–95. doi: 10.1046/j.1365-2567.1998.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Ring A, Braun JS, Pohl J, Nizet V, Stremmel W, Shenep JL. Group B streptococcal beta-hemolysin induces mortality and liver injury in experimental sepsis. J Infect Dis. 2002;185(12):1745–53. doi: 10.1086/340818. [DOI] [PubMed] [Google Scholar]
- [257].Lindahl G, Stalhammar-Carlemalm M, Areschoug T. Surface proteins of Streptococcus agalactiae and related proteins in other bacterial pathogens. Clin Microbiol Rev. 2005;18(1):102–27. doi: 10.1128/CMR.18.1.102-127.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Liu GY, Nizet V. Extracellular virulence factors of group B Streptococci. Front Biosci. 2004;9:1794–802. doi: 10.2741/1296. [DOI] [PubMed] [Google Scholar]
- [259].Adderson EE, Takahashi S, Bohnsack JF. Bacterial genetics and human immunity to group B streptococci. Mol Genet Metab. 2000;71(12):451–4. doi: 10.1006/mgme.2000.3025. [DOI] [PubMed] [Google Scholar]
- [260].Nizet V. Streptococcal beta-hemolysins: genetics and role in disease pathogenesis. Trends Microbiol. 2002;10(12):575–80. doi: 10.1016/s0966-842x(02)02473-3. [DOI] [PubMed] [Google Scholar]
- [261].Ulett GC, Maclean KH, Nekkalapu S, Cleveland JL, Adderson EE. Mechanisms of Group B Streptococcal-Induced Apoptosis of Murine Macrophages. J Immunol. 2005;175(4):2555–62. doi: 10.4049/jimmunol.175.4.2555. [DOI] [PubMed] [Google Scholar]
- [262].Ulett GC, Adderson EE. Nitric oxide is a key determinant of Group B Streptococcal-induced murine macrophage apoptosis. J Infect Dis. 2005;191(10):1761–70. doi: 10.1086/429693. [DOI] [PubMed] [Google Scholar]
- [263].Leib SL, Kim YS, Chow LL, Sheldon RA, Tauber MG. Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J Clin Invest. 1996;98(11):2632–9. doi: 10.1172/JCI119084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Fettucciari K, Fetriconi I, Bartoli A, Rossi R, Marconi P. Involvement of mitogen-activated protein kinases in Group B Streptococcus-induced macrophage apoptosis. Pharmacol Res. 2003;47(4):355–62. doi: 10.1016/s1043-6618(03)00004-5. [DOI] [PubMed] [Google Scholar]
- [265].Buratta S, Fettucciari K, Mambrini R, Fetriconi I, Marconi P, Mozzi R. Group B streptococcus (GBS) modifies macrophage phosphatidylserine metabolism during induction of apoptosis. FEBS Lett. 2002;520(13):68–72. doi: 10.1016/s0014-5793(02)02769-2. [DOI] [PubMed] [Google Scholar]
- [266].Musher DM. Infections caused by Streptococcus pneumoniae: clinical spectrum, pathogenesis, immunity, and treatment. Clin Infect Dis. 1992;14(4):801–7. doi: 10.1093/clinids/14.4.801. [DOI] [PubMed] [Google Scholar]
- [267].Jedrzejas MJ. Pneumococcal virulence factors: structure and function. Microbiol Mol Biol Rev. 2001;65(2):187–207. doi: 10.1128/MMBR.65.2.187-207.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [268].Rubins JB, Janoff EN. Pneumolysin: a multifunctional pneumococcal virulence factor. J Lab Clin Med. 1998;131(1):21–7. doi: 10.1016/s0022-2143(98)90073-7. [DOI] [PubMed] [Google Scholar]
- [269].Mitchell TJ, Andrew PW. Biological properties of pneumolysin. Microb Drug Resist. 1997;3(1):19–26. doi: 10.1089/mdr.1997.3.19. [DOI] [PubMed] [Google Scholar]
- [270].Rigden DJ, Galperin MY, Jedrzejas MJ. Analysis of structure and function of putative surface-exposed proteins encoded in the Streptococcus pneumoniae genome: a bioinformatics-based approach to vaccine and drug design. Crit Rev Biochem Mol Biol. 2003;38(2):143–68. doi: 10.1080/713609215. [DOI] [PubMed] [Google Scholar]
- [271].Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825–52. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- [272].Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- [273].Dockrell DH, Marriott HM, Prince LR, et al. Alveolar macrophage apoptosis contributes to pneumococcal clearance in a resolving model of pulmonary infection. J Immunol. 2003;171(10):5380–8. doi: 10.4049/jimmunol.171.10.5380. [DOI] [PubMed] [Google Scholar]
- [274].Gilbert RJ, Jimenez JL, Chen S, et al. Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell. 1999;97(5):647–55. doi: 10.1016/s0092-8674(00)80775-8. [DOI] [PubMed] [Google Scholar]
- [275].Braun JS, Novak R, Murray PJ, et al. Apoptosis-inducing factor mediates microglial and neuronal apoptosis caused by pneumococcus. J Infect Dis. 2001;184(10):1300–9. doi: 10.1086/324013. [DOI] [PubMed] [Google Scholar]
- [276].Braun JS, Novak R, Herzog KH, Bodner SM, Cleveland JL, Tuomanen EI. Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat Med. 1999;5(3):298–302. doi: 10.1038/6514. [DOI] [PubMed] [Google Scholar]
- [277].Duane PG, Rubins JB, Weisel HR, Janoff EN. Identification of hydrogen peroxide as a Streptococcus pneumoniae toxin for rat alveolar epithelial cells. Infect Immun. 1993;61(10):4392–7. doi: 10.1128/iai.61.10.4392-4397.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Cochrane CG. Cellular injury by oxidants. Am J Med. 1991;91(3C):23S–30S. doi: 10.1016/0002-9343(91)90280-b. [DOI] [PubMed] [Google Scholar]
- [279].Bisno AL, Stevens DL. Streptococcal infections of skin and soft tissues. N Engl J Med. 1996;334(4):240–5. doi: 10.1056/NEJM199601253340407. [DOI] [PubMed] [Google Scholar]
- [280].Ahmed S, Ayoub EM. Severe, invasive group A streptococcal disease and toxic shock. Pediatr Ann. 1998;27(5):287–92. doi: 10.3928/0090-4481-19980501-08. [DOI] [PubMed] [Google Scholar]
- [281].Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13(3):470–511. doi: 10.1128/cmr.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Rasmussen M, Bjorck L. Proteolysis and its regulation at the surface of Streptococcus pyogenes. Mol Microbiol. 2002;43(3):537–44. doi: 10.1046/j.1365-2958.2002.02766.x. [DOI] [PubMed] [Google Scholar]
- [283].Bisno AL, Brito MO, Collins CM. Molecular basis of group A streptococcal virulence. Lancet Infect Dis. 2003;3(4):191–200. doi: 10.1016/s1473-3099(03)00576-0. [DOI] [PubMed] [Google Scholar]
- [284].Imanishi K, Igarashi H, Uchiyama T. Activation of murine T cells by streptococcal pyrogenic exotoxin type A. Requirement for MHC class II molecules on accessory cells and identification of V beta elements in T cell receptor of toxin-reactive T cells. J Immunol. 1990;145(10):3170–6. [PubMed] [Google Scholar]
- [285].Norrby-Teglund A, Norrby SR, Low DE. The treatment of severe group a streptococcal infections. Curr Infect Dis Rep. 2003;5(1):28–37. doi: 10.1007/s11908-003-0062-2. [DOI] [PubMed] [Google Scholar]
- [286].Nakagawa I, Nakata M, Kawabata S, Hamada S. Transcriptome analysis and gene expression profiles of early apoptosis-related genes in Streptococcus pyogenes-infected epithelial cells. Cell Microbiol. 2004;6(10):939–52. doi: 10.1111/j.1462-5822.2004.00412.x. [DOI] [PubMed] [Google Scholar]
- [287].Marouni MJ, Sela S. Fate of Streptococcus pyogenes and epithelial cells following internalization. J Med Microbiol. 2004;53(Pt 1):1–7. doi: 10.1099/jmm.0.05263-0. [DOI] [PubMed] [Google Scholar]
- [288].Watanabe-Ohnishi R, Low DE, McGeer A, et al. Ontario Streptococcal Study Project Selective depletion of V beta-bearing T cells in patients with severe invasive group A streptococcal infections and streptococcal toxic shock syndrome. J Infect Dis. 1995;171(1):74–84. doi: 10.1093/infdis/171.1.74. [DOI] [PubMed] [Google Scholar]
- [289].Kapur V, Majesky MW, Li LL, Black RA, Musser JM. Cleavage of interleukin 1 beta (IL-1 beta) precursor to produce active IL-1 beta by a conserved extracellular cysteine protease from Streptococcus pyogenes. Proc Natl Acad Sci USA. 1993;90(16):7676–80. doi: 10.1073/pnas.90.16.7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Little SF, Ivins BE. Molecular pathogenesis of Bacillus anthracis infection. Microbes Infect. 1999;1(2):131–9. doi: 10.1016/s1286-4579(99)80004-5. [DOI] [PubMed] [Google Scholar]
- [291].Mock M, Fouet A. Anthrax. Annu Rev Microbiol. 2001;55:647–71. doi: 10.1146/annurev.micro.55.1.647. [DOI] [PubMed] [Google Scholar]
- [292].Guidi-Rontani C. The alveolar macrophage: the Trojan horse of Bacillus anthracis. Trends Microbiol. 2002;10(9):405–9. doi: 10.1016/s0966-842x(02)02422-8. [DOI] [PubMed] [Google Scholar]
- [293].Hanna P. Anthrax pathogenesis and host response. Curr Top Microbiol Immunol. 1998;225:13–35. doi: 10.1007/978-3-642-80451-9_2. [DOI] [PubMed] [Google Scholar]
- [294].Dixon TC, Meselson M, Guillemin J, Hanna PC. Anthrax. N Engl J Med. 1999;341(11):815–26. doi: 10.1056/NEJM199909093411107. [DOI] [PubMed] [Google Scholar]
- [295].Mock M, Mignot T. Anthrax toxins and the host: a story of intimacy. Cell Microbiol. 2003;5(1):15–23. doi: 10.1046/j.1462-5822.2003.00253.x. [DOI] [PubMed] [Google Scholar]
- [296].Lacy DB, Collier RJ. Structure and function of anthrax toxin. Curr Top Microbiol Immunol. 2002;271:61–85. doi: 10.1007/978-3-662-05767-4_4. [DOI] [PubMed] [Google Scholar]
- [297].Barth H, Aktories K, Popoff MR, Stiles BG. Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol Mol Biol Rev. 2004;68(3):373–402. doi: 10.1128/MMBR.68.3.373-402.2004. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414(6860):225–9. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- [299].Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100(9):5170–4. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Koehler TM. Bacillus anthracis genetics and virulence gene regulation. Curr Top Microbiol Immunol. 2002;271:143–64. doi: 10.1007/978-3-662-05767-4_7. [DOI] [PubMed] [Google Scholar]
- [301].Fouet A, Mesnage S. Bacillus anthracis cell envelope components. Curr Top Microbiol Immunol. 2002;271:87–113. doi: 10.1007/978-3-662-05767-4_5. [DOI] [PubMed] [Google Scholar]
- [302].Grinberg LM, Abramova FA, Yampolskaya OV, Walker DH, Smith JH. Quantitative pathology of inhalational anthrax I: quantitative microscopic findings. Mod Pathol. 2001;14(5):482–95. doi: 10.1038/modpathol.3880337. [DOI] [PubMed] [Google Scholar]
- [303].Moayeri M, Leppla SH. The roles of anthrax toxin in pathogenesis. Curr Opin Microbiol. 2004;7(1):19–24. doi: 10.1016/j.mib.2003.12.001. [DOI] [PubMed] [Google Scholar]
- [304].Gold JA, Hoshino Y, Hoshino S, Jones MB, Nolan A, Weiden MD. Exogenous gamma and alpha/beta interferon rescues human macrophages from cell death induced by Bacillus anthracis. Infect Immun. 2004;72(3):1291–7. doi: 10.1128/IAI.72.3.1291-1297.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Sanchez AM, Bradley KA. Anthrax toxin: can a little be a good thing? Trends Microbiol. 2004;12(4):143–5. doi: 10.1016/j.tim.2004.02.003. [DOI] [PubMed] [Google Scholar]
- [306].Agrawal A, Lingappa J, Leppla SH, et al. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature. 2003;424(6946):329–34. doi: 10.1038/nature01794. [DOI] [PubMed] [Google Scholar]
- [307].Popov SG, Popova TG, Grene E, et al. Systemic cytokine response in murine anthrax. Cell Microbiol. 2004;6(3):225–33. doi: 10.1046/j.1462-5822.2003.00358.x. [DOI] [PubMed] [Google Scholar]
- [308].Shin S, Kim YB, Hur GH. Involvement of phospholipase A2 activation in anthrax lethal toxin-induced cytotoxicity. Cell Biol Toxicol. 1999;15(1):19–29. doi: 10.1023/a:1007546505528. [DOI] [PubMed] [Google Scholar]
- [309].Duesbery NS, Webb CP, Leppla SH, et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280(5364):734–7. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- [310].Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352(Pt 3):739–45. [PMC free article] [PubMed] [Google Scholar]
- [311].Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999;462(12):199–204. doi: 10.1016/s0014-5793(99)01502-1. [DOI] [PubMed] [Google Scholar]
- [312].Koo HM, VanBrocklin M, McWilliams MJ, Leppla SH, Duesbery NS, Woude GF. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc Natl Acad Sci USA. 2002;99(5):3052–7. doi: 10.1073/pnas.052707699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Fukao T. Immune system paralysis by anthrax lethal toxin: the roles of innate and adaptive immunity. Lancet Infect Dis. 2004;4(3):166–70. doi: 10.1016/S1473-3099(04)00940-5. [DOI] [PubMed] [Google Scholar]
- [314].Luschen S, Scherer G, Ussat S, Ungefroren H, Adam-Klages S. Inhibition of p38 mitogen-activated protein kinase reduces TNF-induced activation of NF-kappaB, elicits caspase activity, and enhances cytotoxicity. Exp Cell Res. 2004;293(2):196–206. doi: 10.1016/j.yexcr.2003.10.009. [DOI] [PubMed] [Google Scholar]
- [315].Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838–49. doi: 10.1038/sj.onc.1207556. [DOI] [PubMed] [Google Scholar]
- [316].Vazquez-Boland JA, Kuhn M, Berche P, et al. Listeria pathogenesis and molecular virulence determinants. Clin Microbiol Rev. 2001;14(3):584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Southwick FS, Purich DL. Intracellular pathogenesis of listeriosis. N Engl J Med. 1996;334(12):770–6. doi: 10.1056/NEJM199603213341206. [DOI] [PubMed] [Google Scholar]
- [318].Wing EJ, Gregory SH. Listeria monocytogenes: clinical and experimental update. J Infect Dis. 2002;185(Suppl 1):S18–24. doi: 10.1086/338465. [DOI] [PubMed] [Google Scholar]
- [319].Gregory SH, Liu CC. CD8+ T-cell-mediated response to Listeria monocytogenes taken up in the liver and replicating within hepatocytes. Immunol Rev. 2000;174:112–22. doi: 10.1034/j.1600-0528.2002.017405.x. [DOI] [PubMed] [Google Scholar]
- [320].Pamer EG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4(10):812–23. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- [321].Cossart P, Mengaud J. Listeria monocytogenes. A model system for the molecular study of intracellular parasitism. Mol Biol Med. 1989;6(5):463–74. [PubMed] [Google Scholar]
- [322].Dussurget O, Pizarro-Cerda J, Cossart P. Molecular determinants of Listeria monocytogenes virulence. Annu Rev Microbiol. 2004;58:587–610. doi: 10.1146/annurev.micro.57.030502.090934. [DOI] [PubMed] [Google Scholar]
- [323].Portnoy DA, Chakraborty T, Goebel W, Cossart P. Molecular determinants of Listeria monocytogenes pathogenesis. Infect Immun. 1992;60(4):1263–7. doi: 10.1128/iai.60.4.1263-1267.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Parham P, Unanue ER, editors. Immunity to L. monocytogenes: a model intracellular pathogen. Aug, 1997. pp. 1–169. 1997. [Google Scholar]
- [325].Portnoy DA, Auerbuch V, Glomski IJ. The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol. 2002;158(3):409–14. doi: 10.1083/jcb.200205009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].North RJ, Conlan JW. Immunity to Listeria monocytogenes. Chem Immunol. 1998;70:1–20. [PubMed] [Google Scholar]
- [327].Harty JT, Tvinnereim AR, White DW. CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol. 2000;18:275–308. doi: 10.1146/annurev.immunol.18.1.275. [DOI] [PubMed] [Google Scholar]
- [328].Rogers HW, Callery MP, Deck B, Unanue ER. Listeria monocytogenes induces apoptosis of infected hepatocytes. J Immunol. 1996;156(2):679–84. [PubMed] [Google Scholar]
- [329].Miura T, Nishikawa S, Sasaki S, et al. Roles of endogenous cytokines in liver apoptosis of mice in lethal Listeria monocytogenes infection. FEMS Immunol Med Microbiol. 2000;28(4):335–41. doi: 10.1111/j.1574-695X.2000.tb01495.x. [DOI] [PubMed] [Google Scholar]
- [330].Ebe Y, Hasegawa G, Takatsuka H, et al. The role of Kupffer cells and regulation of neutrophil migration into the liver by macrophage inflammatory protein-2 in primary listeriosis in mice. Pathol Int. 1999;49(6):519–32. doi: 10.1046/j.1440-1827.1999.00910.x. [DOI] [PubMed] [Google Scholar]
- [331].Kolb-Maurer A, Gentschev I, Fries HW, et al. Listeria monocytogenes-infected human dendritic cells: uptake and host cell response. Infect Immun. 2000;68(6):3680–8. doi: 10.1128/iai.68.6.3680-3688.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [332].Barsig J, Kaufmann SH. The mechanism of cell death in Listeria monocytogenes-infected murine macrophages is distinct from apoptosis. Infect Immun. 1997;65(10):4075–81. doi: 10.1128/iai.65.10.4075-4081.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Roberts AJ, Wiedmann M. Pathogen, host and environmental factors contributing to the pathogenesis of listeriosis. Cell Mol Life Sci. 2003;60(5):904–18. doi: 10.1007/s00018-003-2225-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Shen H, Tato CM, Fan X. Listeria monocytogenes as a probe to study cell-mediated immunity. Curr Opin Immunol. 1998;10(4):450–8. doi: 10.1016/s0952-7915(98)80120-9. [DOI] [PubMed] [Google Scholar]
- [335].Cousens LP, Wing EJ. Innate defenses in the liver during Listeria infection. Immunol Rev. 2000;174:150–9. doi: 10.1034/j.1600-0528.2002.017407.x. [DOI] [PubMed] [Google Scholar]
- [336].Conlan JW, North RJ. Neutrophil-mediated dissolution of infected host cells as a defense strategy against a facultative intracellular bacterium. J Exp Med. 1991;174(3):741–4. doi: 10.1084/jem.174.3.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Conlan JW, Dunn PL, North RJ. Leukocyte-mediated lysis of infected hepatocytes during listeriosis occurs in mice depleted of NK cells or CD4+ CD8+ Thy1.2+ T cells. Infect Immun. 1993;61(6):2703–7. doi: 10.1128/iai.61.6.2703-2707.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [338].Jiang J, Lau LL, Shen H. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J Immunol. 2003;171(8):4352–8. doi: 10.4049/jimmunol.171.8.4352. [DOI] [PubMed] [Google Scholar]
- [339].Kagi D, Ledermann B, Burki K, Hengartner H, Zinkernagel RM. CD8+ T cell-mediated protection against an intracellular bacterium by perforin-dependent cytotoxicity. Eur J Immunol. 1994;24(12):3068–72. doi: 10.1002/eji.1830241223. [DOI] [PubMed] [Google Scholar]
- [340].Edelson BT, Cossart P, Unanue ER. Cutting edge: paradigm revisited: antibody provides resistance to Listeria infection. J Immunol. 1999;163(8):4087–90. [PubMed] [Google Scholar]
- [341].Edelson BT, Unanue ER. Intracellular antibody neutralizes Listeria growth. Immunity. 2001;14(5):503–12. doi: 10.1016/s1074-7613(01)00139-x. [DOI] [PubMed] [Google Scholar]
- [342].Portnoy DA. Innate immunity to a facultative intracellular bacterial pathogen. Curr Opin Immunol. 1992;4(1):20–4. doi: 10.1016/0952-7915(92)90118-x. [DOI] [PubMed] [Google Scholar]
- [343].Mueller NJ, Wilkinson RA, Fishman JA. Listeria monocytogenes infection in caspase-11-deficient mice. Infect Immun. 2002;70(5):2657–64. doi: 10.1128/IAI.70.5.2657-2664.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [344].Badovinac VP, Tvinnereim AR, Harty JT. Regulation of antigen-specific CD8+ T cell homeostasis by perforin and interferon-gamma. Science. 2000;290(5495):1354–8. doi: 10.1126/science.290.5495.1354. [DOI] [PubMed] [Google Scholar]
- [345].Jiang X, Gregory SH, Wing EJ. Immune CD8+ T lymphocytes lyse Listeria monocytogenes-infected hepatocytes by a classical MHC class I-restricted mechanism. J Immunol. 1997;158(1):287–93. [PubMed] [Google Scholar]
- [346].Kagi D, Vignaux F, Ledermann B, et al. Fas and perforin pathways as major mechanisms of T cell-mediated cytotoxicity. Science. 1994;265(5171):528–30. doi: 10.1126/science.7518614. [DOI] [PubMed] [Google Scholar]
- [347].Liu CC, Walsh CM, Young JD. Perforin: structure and function. Immunol Today. 1995;16(4):194–201. doi: 10.1016/0167-5699(95)80121-9. [DOI] [PubMed] [Google Scholar]
- [348].Nakajima H, Henkart PA. Cytotoxic lymphocyte granzymes trigger a target cell internal disintegration pathway leading to cytolysis and DNA breakdown. J Immunol. 1994;152(3):1057–63. [PubMed] [Google Scholar]
- [349].Finelli A, Pamer EG. Immune and inflammatory responses to Listeria monocytogenes infection. xiii. ASM Press; Washington, D.C.: 2000. p. 735. [Google Scholar]
- [350].White DW, Harty JT. Perforin-deficient CD8+ T cells provide immunity to Listeria monocytogenes by a mechanism that is independent of CD95 and IFN-gamma but requires TNF-alpha. J Immunol. 1998;160(2):898–905. [PubMed] [Google Scholar]
- [351].Endres R, Luz A, Schulze H, et al. Listeriosis in p47(phox-/-) and TRp55-/-mice: protection despite absence of ROI and susceptibility despite presence of RNI. Immunity. 1997;7(3):419–32. doi: 10.1016/s1074-7613(00)80363-5. [DOI] [PubMed] [Google Scholar]
- [352].Cabanes D, Dehoux P, Dussurget O, Frangeul L, Cossart P. Surface proteins and the pathogenic potential of Listeria monocytogenes. Trends Microbiol. 2002;10(5):238–45. doi: 10.1016/s0966-842x(02)02342-9. [DOI] [PubMed] [Google Scholar]
- [353].Schutze S, Machleidt T, Kronke M. The role of diacylglycerol and ceramide in tumor necrosis factor and interleukin-1 signal transduction. J Leukoc Biol. 1994;56(5):533–41. doi: 10.1002/jlb.56.5.533. [DOI] [PubMed] [Google Scholar]
- [354].Kolesnick RN, Kronke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol. 1998;60:643–65. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- [355].Ferrick DA, Schrenzel MD, Mulvania T, Hsieh B, Ferlin WG, Lepper H. Differential production of interferon-gamma and interleukin-4 in response to Th1-and Th2-stimulating pathogens by gamma delta T cells in vivo. Nature. 1995;373(6511):255–7. doi: 10.1038/373255a0. [DOI] [PubMed] [Google Scholar]
- [356].Fu YX, Roark CE, Kelly K, et al. Immune protection and control of inflammatory tissue necrosis by gamma delta T cells. J Immunol. 1994;153(7):3101–15. [PubMed] [Google Scholar]
- [357].Egan PJ, Carding SR. Downmodulation of the inflammatory response to bacterial infection by gammadelta T cells cytotoxic for activated macrophages. J Exp Med. 2000;191(12):2145–58. doi: 10.1084/jem.191.12.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [358].Zheng SJ, Wang P, Tsabary G, Chen YH. Critical roles of TRAIL in hepatic cell death and hepatic inflammation. J Clin Invest. 2004;113(1):58–64. doi: 10.1172/JCI200419255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [359].Stockinger S, Materna T, Stoiber D, et al. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol. 2002;169(11):6522–9. doi: 10.4049/jimmunol.169.11.6522. [DOI] [PubMed] [Google Scholar]
- [360].Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 2003;19(1):59–70. doi: 10.1016/s1074-7613(03)00171-7. [DOI] [PubMed] [Google Scholar]
- [361].Havell EA. Augmented induction of interferons during Listeria monocytogenes infection. J Infect Dis. 1986;153(5):960–9. doi: 10.1093/infdis/153.5.960. [DOI] [PubMed] [Google Scholar]
- [362].O’Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci USA. 2002;99(21):13861–6. doi: 10.1073/pnas.202476699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Johnson EA. Clostridial toxins as therapeutic agents: benefits of nature’s most toxic proteins. Annu Rev Microbiol. 1999;53:551–75. doi: 10.1146/annurev.micro.53.1.551. [DOI] [PubMed] [Google Scholar]
- [364].Poutanen SM, Simor AE. Clostridium difficile-associated diarrhea in adults. Cmaj. 2004;171(1):51–8. doi: 10.1503/cmaj.1031189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [365].Pothoulakis C. Effects of Clostridium difficile toxins on epithelial cell barrier. Ann N Y Acad Sci. 2000;915:347–56. doi: 10.1111/j.1749-6632.2000.tb05263.x. [DOI] [PubMed] [Google Scholar]
- [366].Poxton IR, McCoubrey J, Blair G. The pathogenicity of Clostridium difficile. Clin Microbiol Infect. 2001;7(8):421–7. doi: 10.1046/j.1198-743x.2001.00287.x. [DOI] [PubMed] [Google Scholar]
- [367].Mylonakis E, Ryan ET, Calderwood SB. Clostridium difficile-associated diarrhea: A review. Arch Intern Med. 2001;161(4):525–33. doi: 10.1001/archinte.161.4.525. [DOI] [PubMed] [Google Scholar]
- [368].Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med. 1994;330(4):257–62. doi: 10.1056/NEJM199401273300406. [DOI] [PubMed] [Google Scholar]
- [369].Rupnik M. How to detect Clostridium difficile variant strains in a routine laboratory. Clin Microbiol Infect. 2001;7(8):417–20. doi: 10.1046/j.1198-743x.2001.00290.x. [DOI] [PubMed] [Google Scholar]
- [370].Warny M, Keates AC, Keates S, et al. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest. 2000;105(8):1147–56. doi: 10.1172/JCI7545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [371].Warny M, Kelly CP. Monocytic cell necrosis is mediated by potassium depletion and caspase-like proteases. Am J Physiol. 1999;276(3 Pt 1):C717–24. doi: 10.1152/ajpcell.1999.276.3.C717. [DOI] [PubMed] [Google Scholar]
- [372].Thelestam M, Chaves-Olarte E. Cytotoxic effects of the Clostridium difficile toxins. Curr Top Microbiol Immunol. 2000;250:85–96. doi: 10.1007/978-3-662-06272-2_4. [DOI] [PubMed] [Google Scholar]
- [373].Bongaerts GP, Lyerly DM. Role of toxins A and B in the pathogenesis of Clostridium difficile disease. Microb Pathog. 1994;17(1):1–12. doi: 10.1006/mpat.1994.1047. [DOI] [PubMed] [Google Scholar]
- [374].Pothoulakis C, Lamont JT. Microbes and microbial toxins: paradigms for microbial-mucosal interactions II. The integrated response of the intestine to Clostridium difficile toxins. Am J Physiol Gastrointest Liver Physiol. 2001;280(2):G178–83. doi: 10.1152/ajpgi.2001.280.2.G178. [DOI] [PubMed] [Google Scholar]
- [375].Castagliuolo I, LaMont JT, Letourneau R, et al. Neuronal involvement in the intestinal effects of Clostridium difficile toxin A and Vibrio cholerae enterotoxin in rat ileum. Gastroenterology. 1994;107(3):657–65. doi: 10.1016/0016-5085(94)90112-0. [DOI] [PubMed] [Google Scholar]
- [376].Wershil BK, Castagliuolo I, Pothoulakis C. Direct evidence of mast cell involvement in Clostridium difficile toxin A-induced enteritis in mice. Gastroenterology. 1998;114(5):956–64. doi: 10.1016/s0016-5085(98)70315-4. [DOI] [PubMed] [Google Scholar]
- [377].Qiu B, Pothoulakis C, Castagliuolo I, Nikulasson S, LaMont JT. Participation of reactive oxygen metabolites in Clostridium difficile toxin A-induced enteritis in rats. Am J Physiol. 1999;276(2 Pt 1):G485–90. doi: 10.1152/ajpgi.1999.276.2.G485. [DOI] [PubMed] [Google Scholar]
- [378].Hecht G, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest. 1988;82(5):1516–24. doi: 10.1172/JCI113760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Fiorentini C, Thelestam M. Clostridium difficile toxin A and its effects on cells. Toxicon. 1991;29(6):543–67. doi: 10.1016/0041-0101(91)90050-2. [DOI] [PubMed] [Google Scholar]
- [380].Kushnaryov VM, Redlich PN, Sedmak JJ, Lyerly DM, Wilkins TD, Grossberg SE. Cytotoxicity of Clostridium difficile toxin A for human colonic and pancreatic carcinoma cell lines. Cancer Res. 1992;52(18):5096–9. [PubMed] [Google Scholar]
- [381].Fiorentini C, Donelli G, Nicotera P, Thelestam M. Clostridium difficile toxin A elicits Ca(2+)-independent cytotoxic effects in cultured normal rat intestinal crypt cells. Infect Immun. 1993;61(9):3988–93. doi: 10.1128/iai.61.9.3988-3993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [382].Riegler M, Sedivy R, Pothoulakis C, et al. Clostridium difficile toxin B is more potent than toxin A in damaging human colonic epithelium in vitro. J Clin Invest. 1995;95(5):2004–11. doi: 10.1172/JCI117885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [383].Fiorentini C, Arancia G, Paradisi S, et al. Effects of Clostridium difficile toxins A and B on cytoskeleton organization in HEp-2 cells: a comparative morphological study. Toxicon. 1989;27(11):1209–18. doi: 10.1016/0041-0101(89)90029-9. [DOI] [PubMed] [Google Scholar]
- [384].Hecht G, Koutsouris A, Pothoulakis C, LaMont JT, Madara JL. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology. 1992;102(2):416–23. doi: 10.1016/0016-5085(92)90085-d. [DOI] [PubMed] [Google Scholar]
- [385].Gomez J, Martinez C, Giry M, Garcia A, Rebollo A. Rho prevents apoptosis through Bcl-2 expression: implications for interleukin-2 receptor signal transduction. Eur J Immunol. 1997;27(11):2793–9. doi: 10.1002/eji.1830271108. [DOI] [PubMed] [Google Scholar]
- [386].Just I, Hofmann F, Aktories K. Molecular mode of action of the large clostridial cytotoxins. Curr Top Microbiol Immunol. 2000;250:55–83. doi: 10.1007/978-3-662-06272-2_3. [DOI] [PubMed] [Google Scholar]
- [387].Ruoslahti E. Stretching is good for a cell. Science. 1997;276(5317):1345–6. doi: 10.1126/science.276.5317.1345. [DOI] [PubMed] [Google Scholar]
- [388].Fiorentini C, Gauthier M, Donelli G, Boquet P. Bacterial toxins and the Rho GTP-binding protein: what microbes teach us about cell regulation. Cell Death Differ. 1998;5(9):720–8. doi: 10.1038/sj.cdd.4400412. [DOI] [PubMed] [Google Scholar]
- [389].Anderson RJ, Ray CJ, Popoff MR. Evidence for Rho protein regulation of renal tubular epithelial cell function. Kidney Int. 2000;58(5):1996–2006. doi: 10.1111/j.1523-1755.2000.00372.x. [DOI] [PubMed] [Google Scholar]
- [390].Leverrier Y, Ridley AJ. Requirement for Rho GTPases and PI 3-kinases during apoptotic cell phagocytosis by macrophages. Curr Biol. 2001;11(3):195–9. doi: 10.1016/s0960-9822(01)00047-1. [DOI] [PubMed] [Google Scholar]
- [391].Linseman DA, Laessig T, Meintzer MK, et al. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J Biol Chem. 2001;276(42):39123–31. doi: 10.1074/jbc.M103959200. [DOI] [PubMed] [Google Scholar]
- [392].Moorman JP, Bobak DA, Hahn CS. Inactivation of the small GTP binding protein Rho induces multinucleate cell formation and apoptosis in murine T lymphoma EL4. J Immunol. 1996;156(11):4146–53. [PubMed] [Google Scholar]
- [393].Na S, Chuang TH, Cunningham A, et al. D4-GDI, a substrate of CPP32, is proteolyzed during Fas-induced apoptosis. J Biol Chem. 1996;271(19):11209–13. doi: 10.1074/jbc.271.19.11209. [DOI] [PubMed] [Google Scholar]
- [394].Henning SW, Galandrini R, Hall A, Cantrell DA. The GTPase Rho has a critical regulatory role in thymus development. EMBO J. 1997;16(9):2397–407. doi: 10.1093/emboj/16.9.2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [395].Lombet A, Zujovic V, Kandouz M, et al. Resistance to induced apoptosis in the human neuroblastoma cell line SK-N-SH in relation to neuronal differentiation. Role of Bcl-2 protein family. Eur J Biochem. 2001;268(5):1352–62. doi: 10.1046/j.1432-1327.2001.02002.x. [DOI] [PubMed] [Google Scholar]
- [396].McClane BA. The complex interactions between Clostridium perfringens enterotoxin and epithelial tight junctions. Toxicon. 2001;39(11):1781–91. doi: 10.1016/s0041-0101(01)00164-7. [DOI] [PubMed] [Google Scholar]
- [397].Matsushita O, Okabe A. Clostridial hydrolytic enzymes degrading extracellular components. Toxicon. 2001;39(11):1769–80. doi: 10.1016/s0041-0101(01)00163-5. [DOI] [PubMed] [Google Scholar]
- [398].Lang P, Guizani L, Vitte-Mony I, et al. ADP-ribosylation of the ras-related, GTP-binding protein RhoA inhibits lymphocyte-mediated cytotoxicity. J Biol Chem. 1992;267(17):11677–80. [PubMed] [Google Scholar]
- [399].Tweten RK. Clostridium perfringens beta toxin and Clostridium septicum alpha toxin: their mechanisms and possible role in pathogenesis. Vet Microbiol. 2001;82(1):1–9. doi: 10.1016/s0378-1135(01)00372-8. [DOI] [PubMed] [Google Scholar]
- [400].Flores-Diaz M, Alape-Giron A. Role of Clostridium perfringens phospholipase C in the pathogenesis of gas gangrene. Toxicon. 2003;42(8):979–86. doi: 10.1016/j.toxicon.2003.11.013. [DOI] [PubMed] [Google Scholar]
- [401].Petit P, Breard J, Montalescot V, et al. Lethal toxin from Clostridium sordellii induces apoptotic cell death by disruption of mitochondrial homeostasis in HL-60 cells. Cell Microbiol. 2003;5(11):761–71. doi: 10.1046/j.1462-5822.2003.00309.x. [DOI] [PubMed] [Google Scholar]
- [402].Ramsdell F, Seaman MS, Miller RE, Picha KS, Kennedy MK, Lynch DH. Differential ability of Th1 and Th2 T cells to express Fas ligand and to undergo activation-induced cell death. Int Immunol. 1994;6(10):1545–53. doi: 10.1093/intimm/6.10.1545. [DOI] [PubMed] [Google Scholar]
- [403].Seino K, Harada M, Taniguchi M. NKT cells are relatively resistant to apoptosis. Trends Immunol. 2004;25(5):219–21. doi: 10.1016/j.it.2004.03.001. [DOI] [PubMed] [Google Scholar]
- [404].Baran J, Guzik K, Hryniewicz W, Ernst M, Flad HD, Pryjma J. Apoptosis of monocytes and prolonged survival of granulocytes as a result of phagocytosis of bacteria. Infect Immun. 1996;64(10):4242–8. doi: 10.1128/iai.64.10.4242-4248.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [405].Lin YT, Wang CT, Hsu CT, et al. Differential susceptibility to staphylococcal superantigen (SsAg)-induced apoptosis of CD4+ T cells from atopic dermatitis patients and healthy subjects: the inhibitory effect of IL-4 on SsAg-induced apoptosis. J Immunol. 2003;171(2):1102–8. doi: 10.4049/jimmunol.171.2.1102. [DOI] [PubMed] [Google Scholar]
- [406].Nakamura K, Yuh K, Sugyo S, Kuroki M, Shijo H, Tamura K. Unresponsiveness of peripheral T cells induced by apoptotic bodies derived from autologous T cells. Cell Immunol. 1999;193(2):147–54. doi: 10.1006/cimm.1999.1459. [DOI] [PubMed] [Google Scholar]
- [407].Critchfield JM, Lenardo MJ. Antigen-induced programmed T cell death as a new approach to immune therapy. Clin Immunol Immunopathol. 1995;75(1):13–9. doi: 10.1006/clin.1995.1046. [DOI] [PubMed] [Google Scholar]
- [408].Zheng SJ, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J Immunol. 2004;173(9):5652–8. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]
- [409].Riegler M, Sedivy R, Sogukoglu T, et al. Epidermal growth factor attenuates Clostridium difficile toxin A- and B-induced damage of human colonic mucosa. Am J Physiol. 1997;273(5 Pt 1):G1014–22. doi: 10.1152/ajpgi.1997.273.5.G1014. [DOI] [PubMed] [Google Scholar]
- [410].Lawrence JP, Brevetti L, Obiso RJ, Wilkins TD, Kimura K, Soper R. Effects of epidermal growth factor and Clostridium difficile toxin B in a model of mucosal injury. J Pediatr Surg. 1997;32(3):430–3. doi: 10.1016/s0022-3468(97)90598-4. [DOI] [PubMed] [Google Scholar]
- [411].Giry M, Popoff MR, von Eichel-Streiber C, Boquet P. Transient expression of RhoA, -B, and -C GTPases in HeLa cells potentiates resistance to Clostridium difficile toxins A and B but not to Clostridium sordellii lethal toxin. Infect Immun. 1995;63(10):4063–71. doi: 10.1128/iai.63.10.4063-4071.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [412].Castagliuolo I, Riegler MF, Valenick L, LaMont JT, Pothoulakis C. Saccharomyces boulardii protease inhibits the effects of Clostridium difficile toxins A and B in human colonic mucosa. Infect Immun. 1999;67(1):302–7. doi: 10.1128/iai.67.1.302-307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [413].Buts JP, Corthier G, Delmee M. Saccharomyces boulardii for Clostridium difficile-associated enteropathies in infants. J Pediatr Gastroenterol Nutr. 1993;16(4):419–25. doi: 10.1097/00005176-199305000-00013. [DOI] [PubMed] [Google Scholar]
- [414].Stubbe H, Berdoz J, Kraehenbuhl JP, Corthesy B. Polymeric IgA is superior to monomeric IgA and IgG carrying the same variable domain in preventing Clostridium difficile toxin A damaging of T84 monolayers. J Immunol. 2000;164(4):1952–60. doi: 10.4049/jimmunol.164.4.1952. [DOI] [PubMed] [Google Scholar]
- [415].Nunes MP, Andrade RM, Lopes MF, DosReis GA. Activation-induced T cell death exacerbates Trypanosoma cruzi replication in macrophages cocultured with CD4+ T lymphocytes from infected hosts. J Immunol. 1998;160(3):1313–9. [PubMed] [Google Scholar]
- [416].Lopes MF, Freire-de-Lima CG, DosReis GA. The macrophage haunted by cell ghosts: a pathogen grows. Immunol Today. 2000;21(10):489–94. doi: 10.1016/s0167-5699(00)01713-8. [DOI] [PubMed] [Google Scholar]