

INTRODUCTION
Recombinant human insulin-like growth factor (rhIGF)-I, singly or in combination with its binding protein (recombinant human IGF binding protein [rhIGFBP]-3), was recently approved by the U.S. Food and Drug Administration for the treatment of severe short stature (height <–3 standard deviations) caused by primary IGF-I deficiency. In addition to its role as the principal mediator of somatic growth in humans (together with growth hormone [GH]), IGF-I exerts multiple metabolic and organ-specific effects. IGF-I acts by binding the type 1 IGF receptor (IGF1R), an α2β2 transmembrane tyrosine kinase receptor, leading to phosphorylation cascades involving the mitogen-activated protein (MAP) kinase pathway and the phosphoinositide 3 (PI3) kinase/Akt pathway and, through the latter, the mammalian target of rapamycin (mTOR) pathway. Growing appreciation for the pleiotropic actions of IGF-I has expanded its potential therapeutic usefulness beyond height enhancement. The ongoing trials to assess the role and efficacy of this agent and the current state of investigation regarding IGF-I in experimental models are shown in the Table. We herein review the highlights of some pertinent trials of the potential non-growth uses of rhIGF-I.
Table.
Potential Non-growth Uses of rhIGF-I and Their Level of Development
| Potential indication | Preclinical data |
Non- randomized trials |
Randomized controlled trials |
|---|---|---|---|
| Enhancing insulin signaling | |||
| Type 1 Diabetes mellitus | ✓ | ✓ | |
| Type 2 Diabetes mellitus | ✓ | ✓ | |
| Type A Insulin Resistance | ✓ | ✓ | |
| Rabson Mendenhall Syndrome | ✓ | ✓ | |
| Lipodystrophy | ✓ | ✓ | |
| Diseases of the Central Nervous System | |||
| Dementia | ✓ | ||
| Hearing loss | ✓ | ||
| Spinal cord injury | ✓ | ||
| Cardiovascular disease | ✓ | ||
| Osteoporosis | ✓ | ✓ | |
ENHANCING INSULIN SIGNALING
The effects of rhIGF-I and rhIGF-I/rhIGFBP-3 have been studied in patients with type 1 diabetes (T1DM)1-5 and type 2 diabetes (T2DM).6-9 The rationale for the use of these agents in diabetes is based on the disruptions of the GH/IGF axis associated with diabetes mellitus (Figure). In T1DM, GH levels are elevated,10,11 yet IGF-I is low,12,13 indicating a potentially impaired hepatic response to GH. Portal insulinopenia is thought to contribute to this impairment of GH signaling.14 Decreased delivery of insulin to the liver also produces an increase in IGFBP-1 synthesis, a phenomenon observed in both T1DM and T2DM.12,15-17 In turn, elevated IGFBP-1 concentrations decrease the bioavailability of IGF-I, further diminishing IGF-I signaling. The loss of negative feedback by IGF-I on GH secretion results in even greater GH secretion, which itself is known to cause insulin resistance.18-21 Low IGF-I exacerbates hyperglycemia by increasing hepatic glucose output.22 Given the evidence that GH therapy can cause or worsen diabetes,23 trials of rhIGF-I and rhIGF-I/rhIGFBP-3 have been carried out in patients with T1DM and T2DM. The results of the larger randomized clinical trials of rhIGF-I and rhIGF-1/rhIGFBP-3 are discussed here.
Figure.
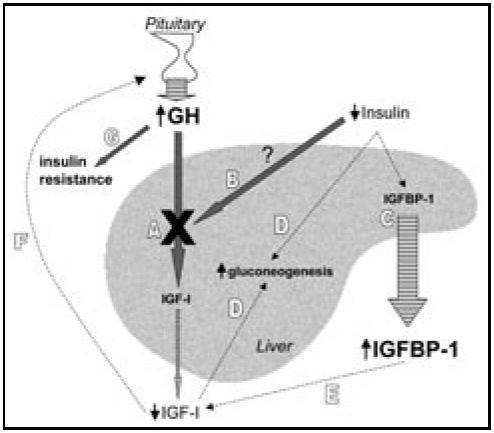
Disrupted GH/IGF-I signaling in diabetes exacerbates hyperglycemia. Stimulatory pathways are indicated by thick arrows, and inhibitory paths by dashed lines. Striped arrows indicate secretion. A. In diabetes, IGF-I is low relative to the GH hypersecretion, ie, a state of GH resistance (indicated by X). B. GH resistance may be related to deficient insulin delivery to the liver. C. Insulinopenia directly causes increased IGFBP-1 production. D. Low IGF-I levels have a permissive effect on hepatic gluconeogenesis, as does insulinopenia itself. E. Elevated IGFBP-1 levels decrease the bioavailability and hence activity of IGF-I. F. Reduced IGF-I synthesis and bioavailability produce less negative feedback inhibition on GH secretion from the pituitary. G. Resultant elevation of GH itself promotes insulin resistance.
Type 1 Diabetes
Acerini et al2 reported on 53 young adults with T1DM randomized to 24 weeks of placebo, or 20 or 40 μg/kg daily of rhIGF-I administered as a single evening injection in addition to their usual multiple-injection insulin regimen. Patients receiving 40 μg/kg/day had an approximately 0.5% lower HbA1c at the end of the 24-week treatment period compared to placebo. There was no difference in retinopathy, hypoglycemia, or any other adverse event.
Thrailkill et al3 found a similar degree of benefit in a randomized, placebo-controlled trial of 223 subjects with T1DM aged 11 to 66 years. For 12 weeks, patients received 2 injections a day of placebo, or rhIGF-I at doses of 40/40, 80/40, or 80/60 μg/kg (AM dose/PM dose). All patients continued their usual split/mix insulin therapy. At the end of 12 weeks, the HbA1c was 0.5% lower in groups treated with rhIGF-I compared to placebo treatment. Treated groups also experienced a reduction of their daily insulin requirements. The number of hypoglycemic events per person per day increased with increasing dose although the differences were not statistically significant (0.14 and 0.23 episodes per subject per day in the placebo and highest dose groups, respectively). These episodes were defined as a blood glucose ≤60mg/dL, or symptoms of hypoglycemia without a blood glucose measurement. Treated groups had more frequent edema, peripheral edema, jaw pain, headache, and arthralgia, which occurred in a dose-related fashion. Although these were considered minor side effects, they were cited as the cause for the higher drop-out rates in the highest dose groups (21% and 29% in the 80/40 and 80/60 groups, respectively, compared to 15% in the placebo group). Also of concern was that 16 of 199 subjects studied had worsening of diabetic retinopathy, and 13 of these 16 were in the 2 highest dose rhIGF-I groups. Furthermore, 17 patients developed new optic disk swelling, an appearance which can be caused by diabetes or by pseudotumor cerebri.
A 2001 subgroup analysis of the 1999 Thrailkill et al study24 focused on the younger patients (age 11-21 years) and found a similar degree of HbA1c lowering (about −0.7%, P<0.05). Again, insulin requirements were reduced in the rhIGF-I treated groups. Worsening of diabetic retinopathy was also observed in this subgroup. Overall, the reports on the 2 largest controlled trials2,3 of rhIGF-I suggest improved glycemic control albeit—particularly with higher doses—a high frequency of worsening diabetic retinopathy and other adverse effects often leading to discontinuation of treatment.
The effects of rhIGF-I/rhIGFBP-3 have also been studied in T1DM.4,5 In April 2000, Clemmons et al4 published results of their randomized cross-over study of 12 adults with T1DM randomized to 2 weeks of placebo or rhIGF-I/rhIGFBP-3 (2mg/kg/day, composed of an equimolar concentration of IGF-I and IGFBP-3, equaling a ratio of 1:4 by weight) delivered by continuous subcutaneous infusion. All subjects continued their home insulin treatment and measured 4 daily blood glucoses. After a 2-week wash-out period, the patients received the opposite therapy. Investigators and subjects were masked to assignment. At the end of the trial, insulin dose decreased 49% in the treatment group compared to placebo, and mean glucose decreased 23%. The HbA1c did not change during the short trial. There was no difference in hypoglycemic events. Edema, arthralgias, and jaw pain did not occur. Retinal exams were not conducted. The authors concluded that rhIGF-I/rhIGFBP-3 may provide improvements in glycemic control without the adverse effects associated with rhIGF-I alone. However, long-term studies will need to be performed in a larger number of patients to carefully document retinal changes and other side effects, in addition to measures of efficacy.
Type 2 Diabetes and Insulin Resistance
In 2005, Clemmons et al9 reported on the short-term use of rhIGF-I/rhIGFBP-3 in patients with T2DM. They enrolled 58 adult patients with long-standing (mean 17.1 years) T2DM treated with insulin alone (44 patients) or insulin plus oral hypoglycemic agents (14 patients). The mean baseline HbA1c was 8.2%, fasting glucose was 211 mg/dL, and body mass index was 32 kg/m2. Subjects were randomized to one of 4 treatment groups: (1) continuous infusion of rhIGF-I/rhIGFBP-3 (2 mg/kg/d); (2) 6h infusion between 2000 and 0200h of 2 mg/kg/d; (3) twice daily subcutaneous injection of 1 mg/kg; or (4) a single evening injection of 1 mg/kg. Patients were hospitalized for the entire 2-week intervention, and all continued their usual home regimen of injected insulin and any oral agents. If needed, insulin was adjusted to “maintain glycemic control.” There was no placebo group, and assignment was not masked. Diets were standardized for protein and caloric content, and physical activity was limited to less than 1 mile per day of walking. The mean insulin dose for days 10-14 was reduced in all groups by 54% to 82%. The decrease was significant compared to baseline for all arms, but did not differ from each other, so no difference by delivery method or dose was observed. The mean fasting glucose for days 2-14 decreased by 32% to 37% in all groups. Side effects were frequent, including headache in 19%, hypoglycemia in 10%, back pain in 15%, nausea in 12%, and jaw pain in 4%. Bell's palsy occurred in 1 patient. Six patients dropped out. No retinal exams were conducted. The authors concluded that combination therapy with rhIGF-I/rhIGFBP-3 improves glycemic control in T2DM. The brevity of the study may have reduced the apparent frequency of adverse side effects. However, the major concerns regarding this study are the absence of a placebo group and lack of masking. It is possible, and perhaps likely, that the stringent in-patient protocol consisting of frequent blood glucose monitoring, ensuring compliance with medications, and strict control of diet, by itself would lead to reductions of fasting glucose and insulin dosage in any patient with suboptimal diabetes control.
Other Insulin-Resistant States
Recombinant IGF-I has been studied in metabolic diseases characterized by markedly impaired insulin signaling, including type A insulin resistance,25-27 Rabson Mendenhall syndrome,28 and congenital lipodystrophy.27 These reports uniformly show improvements in parameters of glucose homeostasis, although the numbers of patients studied have been small. Recombinant IGF-I is a logical candidate therapy to enhance glucose homeostasis in these conditions because it acts primarily through the type 1 IGF receptor. Thus, it may provide an “alternate pathway,” circumventing a defective or impaired insulin receptor and improving glucose homeostasis through the mechanisms described previously.
Lipodystrophy associated with human immunodeficiency virus (HIV) anti-retrovirals is characterized by insulin resistance, abnormal fat distribution (central fat accumulation and peripheral lipoatrophy), and dyslipidemia. This syndrome is associated with impaired GH and IGF-I secretion.29,30 Clinical trials have shown that rhGH can induce hyperglycemia while reducing visceral and subcutaneous fat,31-33 although low doses of rhGH may be a less diabetogenic option.34 In contrast, rhIGF-I could enhance glucose homeostasis while also improving fat distribution, given evidence that IGF-I may be adipogenic.35,36 The latter effect could benefit patients with the most severe peripheral lipoatrophy. Currently, the use of rhIGF-I or rhIGF-I/rhIGFBP-3 in the setting of HIV-associated lipodystrophy awaits further study.
THE CENTRAL NERVOUS SYSTEM
IGF-I is essential for normal central nervous system development37 and function throughout the lifespan, including neuronal plasticity and neuroprotection against potentially pathological disturbances.38 IGF-I may serve as a regenerative agent in the central nervous system, due to its mitogenic and anti-apoptotic actions, which stimulate progenitor cell proliferation and the formation and survival of new neurons, oligodendrocytes, and blood vessels.39 Because IGF-I and insulin are both actively transported from the circulation across the blood-brain barrier,40 intracranial administration of rhIGF-I, as performed in some experiments, may not be required. Clinical efficiacy of rhIGF-I treatment, systemic or otherwise, remains to be established.
Dementia
Alzheimer disease (AD), the most common form of age-related dementia, is characterized by: (1) extensive brain atrophy from neuronal loss; (2) accumulation of neuritic plaques (deposits of amyloid beta protein); (3) neurofibrillary tangles (aggregates of hyperphosphorylated tau, which misfolds and dissociates from the microtubules); and (4) neuroinflammation surrounding the plaques and tangles. IGF-I has been implicated in affecting all 4 components.41 Analysis of frontal lobe tissue from brains of AD patients and age-matched controls found that as clinical severity of AD increased, there was decreased expression of insulin, IGF-I, IGF-II, their receptors, tau and Hu D (a neuronal RNA-binding protein that inhibits the decay of labile mRNAs that contain AU-rich elements and affects their nuclear export and translation), but increased expression of disease-related amyloid beta protein precursor and glial fibrillary acidic protein. Choline acetyltransferase expression in insulin receptor and IGF1R positive neurons was also reduced in AD, and increased with insulin or IGF-I stimulation.42
The neurodegenerative changes of AD were replicated in vivo by intracerebral streptozotocin (ic-STZ) injection in rats. Without altering peripheral glucose, insulin, or pancreatic status, ic-STZ chemically depleted insulin and IGF signaling and induced oxidative injury within the brain. Brains of rats treated with ic-STZ had reduced size, immunohistochemical changes of neurodegeneration (cell loss, gliosis, and increases in p53, active glycogen synthase kinase [GSK]-3β, phosphorylated tau, and amyloid beta), and changes in gene expression profiles consistent with disease activity.43 The neurodegenerative changes of AD were also replicated in vivo by blocking IGF1R in the choroid plexus. These rats developed cognitive disturbances, gliosis, synaptic protein loss, brain amyloidosis, and deposits of hyperphosphorylated tau. Restoring IGF1R function mostly corrected these disturbances, and blocking IGF1R exacerbated AD-related pathology in older, already affected mutant mice.44
Mechanistically, megalin/low-density lipoprotein receptor-related protein-2 (LRP2) is a multicargo transporter expressed by the choroid plexus that is involved in IGF-I transport into the brain and mediates IGF-induced clearance of brain amyloid beta. Levels of choroid plexus megalin/LRP2 in normal animals were reduced by aging and increased by physical exercise.45
Premature cerebral accumulation of amyloid beta was found in mice with hepatic-specific IGF1 gene deletion, while subcutaneous chronic infusion of IGF-I to aged rats promoted amyloid beta levels to decrease in the brain parenchyma and increase in the cerebrospinal fluid.46 IGF-I treatment was also able to reduce amyloid beta levels in the brains of mice over-expressing mutant amyloid.46 In addition to megalin/LRP2, albumin and transthyretin have been implicated in IGF-mediated clearance of brain amyloid beta through the choroid plexus, and this function was inhibited by intracarotid injection of tumor necrosis factor alpha (TNF-α), a proinflammatory cytokine involved in neurodegeneration.46 Regarding neurofibrillary tangles, IGF-I can reduce tau phosphorylation directly or indirectly through its effects on amyloid beta.41 IGF-I also promotes neurogenesis and neuronal survival in adult brains.47
The administration of IGF-I showed promising results in several animal models of dementia. Over-expression of mutant amyloid beta precursor protein (APP) and presenilin (PS)2 in transgenic mice results in AD-like cognitive deficits and severe brain amyloidosis. Systemic, slow-release IGF-I treatment of one-year-old, neurologically affected APP/PS2 mice improved cognitive performance, increased levels of synaptic proteins, and decreased brain amyloid beta load and its associated gliosis.44 A 14-day infusion of amyloid beta 25-35 into the cerebroventricles of rats decreased somatostatinergic signaling in the temporal cortex (a system commonly affected in AD), decreased levels of phosphorylated Akt and increased cell death; these changes were prevented by simultaneous subcutaneous infusion of IGF-I.48
Brain atrophy and dementia have also been associated with the catabolic state of diabetes. Like AD, it has been associated with lower IGF-I levels. Subcutaneous infusion of IGF-I in rats with 12 weeks of uncontrolled STZ-induced diabetes partially prevented the loss of brain protein content (by 27.3%), despite ongoing hyperglycemia.49 Subcutaneous IGF-I infusion in diabetic rats also improved learning/memory performance without ameliorating the hyperglycemia, catabolism or reductions in both total brain and hippocampal weights induced by subcutaneous STZ injection.50
Hearing Loss
In addition to short stature and neurodevelopmental delays, sensorineural deafness has been reported in individuals with IGF1 gene mutation51 or homozygous partial deletion.52 Homozygous IGF1−/− mice had all-frequency bilateral sensorineural hearing loss and delayed response to acoustic stimuli that increased along the auditory pathway, thereby indicating involvement of both cochlear and central nervous system function.53 A biodegradable hydrogel, immersed with rhIGF-I or saline for control, was applied to the round window membranes of Sprague-Dawley rats, 3 days before 2 hours of exposure to 120 dB of white noise. The local rhIGF-I administration significantly blunted the noise-induced elevation of threshold on auditory brain stem response testing (a marker of cochlear function) one week and one month later, and significantly prevented loss of outer hair cells in the temporal bones.54 These preliminary results suggest IGF-I may be protective for the hearing apparatus against noise-induced damage. These studies also raise speculation about the possible contribution of the age-related decline in circulating IGF-I levels to presbycusis.
Spinal Cord Injury
Moderate voluntary physical exercise can be induced in rats through enriched housing, wherein water and food are placed on opposite sides of the cage and additional attributes, such as running wheels, climbing frames and tubes, are provided.55 Enriched housing also stimulates the recovery of locomotion after spinal cord injury in rats by inducing voluntary locomotor training. Locomotor training in turn provides locomotor-specific sensory feedback to the central pattern generators that stimulate remodeling of the central nervous system pathways. Subcutaneous rhIGF-I treatment improved locomotor recovery after spinal cord injury in rats, compared to control rats receiving saline infusion, while neutralization of circulating IGF-I with a chronic infusion of anti-IGF-I serum inhibited the benefits of enriched environment on functional recovery.56
CARDIOVASCULAR DISEASE
The importance of the GH/IGF axis for cardiovascular health was first indicated by the increased cardiovascular mortality of individuals with GH deficiency.57 In vitro mechanistic studies further suggested potential benefits from rhIGF-I administration in myocardial disease. IGF-I treatment of cardiac myocytes in culture was shown to attenuate apoptosis induced by hyperosmotic stress; the protective effects of IGF-I required the CREB transcription factor, which was itself activated through the MAP kinase, PI3 kinase, calcium/calmodulin kinase, and calcineurin systems.58 IGF-I was also shown to protect adult rat ventricular myocytes from high glucose-induced contractile impairments; this required the PI3 kinase/Akt/mTOR pathways but not the calcineurin system.59
While GH/IGF-I deficiency portends a worsened cardiovascular prognosis, so too does GH/IGF-I excess; ventricular hypertrophy is a recognized complication of acromegaly60 and pituitary gigantism.61 Thus, the conceptual approach of rhIGF-I treatment for cardiovascular remodeling and regeneration, such as the use of rhIGF-I as a myocyte survival and mitogenic factor after myocardial infarction, may be limited by dose-response or situation-dependent toxicities. For example, hypertrophy can be physiologic and adaptive (response to aerobic exercise) or pathologic (response to pressure or volume overload). An intriguing checkpoint has recently been identified. In cultured rat neonatal cardiomyocytes exposed to cyclic mechanical stretch, IGF-I was shown to mediate the induction of myostatin through the stress-activated p38 MAP kinase.62 A member of the transforming growth factor (TGF)-β superfamily, myostatin is a negative regulator of myocyte growth. Thus, IGF-I and myostatin form a negative feedback loop to regulate cardiac tissue size.63 Understanding how this balance is achieved and how it can be manipulated will be important for developing effective and safe therapeutic strategies.
Studies of GH, IGF-I, or GH-releasing peptides to treat cardiomyopathies are summarized elsewhere.64 While there have been clinical trials of rhGH, hexarelin, and ghrelin, studies of rhIGF-I, either singly or in combination with rhGH, are limited to rats following left coronary artery ligation, a common experimental model of post-myocardial infarction heart failure. These studies have shown that in rats with prior coronary artery ligation, rhIGF-I can increase ventricular mass and cardiac output, and lower systemic vascular resistance.65-68 Results in humans, however, have not been published.
Another role of IGF-I may be to stimulate cardiac tissue regeneration in the setting of stem cell transplantation. Heterologous bone marrow cells were transplanted into the myocardial scars of Lewis rats 3 weeks following experimental ischemia; co-transfection of the donor cells with the genes for IGF-I and vascular endothelial growth factor (VEGF) resulted in better transplanted cell survival, lower apoptosis, and greater left ventricular ejection fraction than cells transfected with either gene singly or control transplantation with cell-free medium.69 Cardiomyocytes derived from human embryonic stem cells proliferate in vitro, slowing gradually with increasing differentiation. In vitro proliferation of human stem cell-derived cardiomyocytes was inhibited by IGF1R-neutralizing antibodies and dose-dependently enhanced by IGF-I or IGF-II treatment.70
OSTEOPOROSIS
IGF-I plays a role in promoting bone anabolism.71 A pilot, randomized, double-blind, placebo-controlled trial of short-term, continuous subcutaneous infusion of rhIGF-I/rhIGFBP-3 was performed in older women (65-90 years of age) recovering from recent hip fracture. The infusion, administered via portable mini-pump, was initiated within 72 hours of the fracture event and continued for 8 weeks after hip fracture surgery.72 Thirty patients were randomized 1:1:1 to higher dose (1 mg/kg/day), lower dose (0.5 mg/kg/day) or placebo infusions, and evaluated 6 months post-operatively (ie, 4 months after discontinuation of the infusion). Following the immediate post-operative loss of hip bone density (measured in the contralateral side), the high dose group regained femoral bone density while the placebo group remained with a deficit at 6 months' follow-up; changes from baseline were −2.6% (P = 0.53) in the former and −6.1% (P<0.05) in the latter. The high dose group also benefited from an 11.4% increase in grip strength (P<0.05), while the placebo group lost 11.6% (P = 0.16), which further contributed to their functional recovery. The rhIGF-I/rhIGFBP-3 was tolerated well at both doses.72
Studies of rhGH and/or rhIGF-I, alone or in combination with anti-resorptive drugs, in the treatment of osteoporosis were recently summarized.73 Controlled trials establishing an effect on fracture incidence are still needed. Also, with multiple options available for the treatment of osteoporosis, cost-effectiveness analyses of using rhIGF-I will need to be considered. For example, a similar short-term, randomized, double-blind, placebo-controlled study was done to address the protein malnutrition that is frequently found in osteoporotic, elderly individuals.74 Subjects with recent osteoporotic hip fracture (n =82, mean age 80.7 ± 7.4 years) received 550 mg/day calcium supplementation and a single 200 000 IU dose of vitamin D at baseline, and were then randomized to receive 20 gm/day protein supplementation or an isocaloric placebo for 6 months. The protein-supplemented group had significant increases in their serum IGF-I levels (85.6 ± 14.8%, vs 34.1 ± 7.2% among controls; P<0.005) at 6 months, less proximal femoral bone mineral loss at 12 months (−2.29 ± 0.75% vs −4.71 ± 0.77% among controls; P<0.05), and shorter median stay in rehabilitation wards (33 vs 54 days; P<0.05). Thus, there may be less expensive interventions than rhIGF-I or rhIGF-I/rhIGFBP-3 to achieve similar therapeutic goals.
NEGATIVE CLINICAL TRIALS OF rhIGF-I
Despite promising animal data or theoretical appeal of rhIGF-I for certain conditions, the results of some clinical trials have shown no benefit. For example, rhIGF-I was shown to enhance recovery in a rat model of acute renal failure.75 However, a clinical trial of rhIGF-I showed no benefit in human subjects with delayed graft function following cadaveric renal transplantation.76 Another trial of rhIGF-I was conducted based on the association of aging with lower IGF-I levels. In this study, 16 healthy, non-obese, post-menopausal women (mean age 71 years) were randomized to 1 year of rhIGF-I at a dose of 15 μg/kg given twice daily. The mean circulating IGF-I increased from 66 ng/ml at baseline to 298 ng/ml at 12 months. However, at the end of the study there were no significant differences in bone mineral density, muscle mass, or cognitive function.77 These studies highlight the critical importance of rigorous testing of all potential uses of rhIGF-I and rhIGF-I/rhIGFBP-3 by randomized, controlled trials.
POTENTIAL SAFETY ISSUES
The adverse effect most commonly encountered in trials of rhIGF-I is hypoglycemia. This can be mitigated by taking the medicine with food. The greatest theoretical risk of rhIGF-I treatment is that of cancer. Not only have higher circulating IGF-I levels been associated with increased risk of multiple cancers, but mechanistically, IGF signaling can contribute to all stages of the neoplastic process.78 Current evidence supports a permissive—not causal—role for either GH or IGF-I in cancer development.79 This has been borne out in formal carcinogenicity studies; over-expression of IGF-I in animals or the administration of rhIGF-I increased food intake, body size, and the growth rate of existing tumors, but did not increase tumor incidence.80 One must keep this effect in mind when considering IGF-I as potential treatment for mature individuals at ages in which cancer incidence is highest to begin with. Careful screening and patient selection, as well as a thorough risk-benefit analysis for each patient, are warranted.
SUMMARY
Recombinant human IGF-I therapy, singly or in combination with rhIGFBP-3, remains experimental for non-growth indications (Table). In diabetes, the best studied of the non-growth clinical indications, rhIGF-I clearly showed enhanced glycemic control in T1DM, but the benefit is offset by a worsening of diabetic retinopathy and a high frequency of other adverse effects including jaw pain, arthralgias, and edema. This is especially concerning at higher doses. In limited studies, low dose rhIGF-I therapy (40μg/kg daily or twice daily) appears to improve glycemic control to a similar extent as higher doses but with fewer side effects. Given the risks of high dose rhIGF-I therapy, robustly designed long-term clinical trials of low-dose rhIGF-I therapy would be required to ensure its efficacy and safety. Combination rhIGF-I/rhIGFBP-3 therapy appears to improve glycemic control in patients with T1DM, although its efficacy in T2DM remains somewhat undefined. The safety of rhIGF-I/rhIGFBP-3 when used for more than 2 weeks is not clear and requires further study. Longer-term, placebo-controlled, double-masked efficacy studies need to be conducted before rhIGF-I is considered for either T1DM or T2DM. Any such studies should include careful interim assessments of microvascular complications and other adverse effects. Additionally, these agents are particularly appealing for insulin-resistant conditions such as lipodystrophy and when there is defective insulin receptor signaling. IGF-I may play a role in other conditions where undesired lean mass catabolism occurs with insulin resistance, as in low-calorie diets for obesity, and where there is severe systemic stress, such as renal disease, burns, cachexia, and other critical illnesses. However, there are no published studies available to date.
Potential use of rhIGF-I for central nervous system disease is promising, especially in light of the paucity of current treatment options. The fact that IGF-I stimulates clearance of brain amyloid beta, thereby correcting what is believed to be the primary pathogenic event, makes it an especially appealing candidate treatment for AD. However, current evidence is still limited to preliminary preclinical data. While there are a few favorable trials of rhIGF-I in animal models of ischemic myocardial disease, much work needs to be done to determine the patient characteristics and treatment parameters that optimize efficacy and safety. Administration of rhIGF-I improves bone mineral density in some studies, but impact on fracture incidence and cost-effectiveness still needs to be resolved. Other conditions for which rhIGF-I has been found to be unhelpful are in the recovery of renal function after ischemia and as an anti-aging medicine. Potential benefits of IGF-I therapy must always be weighed against the potential risks of raising IGF-I levels. Thus, clinical use of rhIGF-I, singly or in combination with rhIGFBP-3, for non-growth indications should be limited to well-designed clinical studies until adequate evidence supporting efficacy and safety can be collected.
Acknowledgments
Disclosures: The National Institute of Diabetes and Digestive and Kidney Diseases (grant 5K08 DK64352) provides support to A.Grimberg. The Genentech Center for Clinical Research in Endocrinology has provided grant support to R.J.Kim. A.Grimberg is an investigator for an Insmed-sponsored, growth-related clinical trial of rhIGF-I/rhIGFBP-3.
References
- 1.Cheetham TD, Dunger DB. Diabet Med. 1995;12:885–92. doi: 10.1111/j.1464-5491.1995.tb00391.x. [DOI] [PubMed] [Google Scholar]
- 2.Acerini CL, Patton CM, Savage MO, et al. Lancet. 1997;350:1199–1204. doi: 10.1016/S0140-6736(97)06467-2. [DOI] [PubMed] [Google Scholar]
- 3.Thrailkill KM, Quattrin T, Baker L, et al. Diabetes Care. 1999;22:585–92. doi: 10.2337/diacare.22.4.585. [DOI] [PubMed] [Google Scholar]
- 4.Clemmons DR, Moses AC, McKay MJ, et al. J Clin Endocrinol Metab. 2000;85:1518–24. doi: 10.1210/jcem.85.4.6559. [DOI] [PubMed] [Google Scholar]
- 5.Saukkonen T, Amin R, Williams RM, et al. J Clin Endocrinol Metab. 2004;89:4634–41. doi: 10.1210/jc.2004-0243. [DOI] [PubMed] [Google Scholar]
- 6.Zenobi PD, Jaeqqi-Groisman SE, Riesen WF, et al. J Clin Invest. 1992;90:2234–41. doi: 10.1172/JCI116109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moses AC, Young SC, Morrow LA, et al. Diabetes. 1996;45:91–100. doi: 10.2337/diab.45.1.91. [DOI] [PubMed] [Google Scholar]
- 8.Cusi K, DeFronzo R. J Clin Endocrinol Metab. 2000;85:3077–84. doi: 10.1210/jcem.85.9.6827. [DOI] [PubMed] [Google Scholar]
- 9.Clemmons DR, Moses AC, Sommer A, et al. Growth Hormone IGF Res. 2005;15:265–74. doi: 10.1016/j.ghir.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 10.Zadik Z, Kayne R, Kappy M, et al. Diabetes. 1980;29:655–58. doi: 10.2337/diab.29.8.655. [DOI] [PubMed] [Google Scholar]
- 11.Edge JA, Dunger DB, Matthews DR, et al. J Clin Endocrinol Metab. 1990;71:1356–62. doi: 10.1210/jcem-71-5-1356. [DOI] [PubMed] [Google Scholar]
- 12.Bereket A, Lang CH, Blethen SL, et al. J Clin Endocrinol Metab. 1995;80:1312–7. doi: 10.1210/jcem.80.4.7536205. [DOI] [PubMed] [Google Scholar]
- 13.Radetti G, Paganini G, Antoniazzi F, et al. Horm Res. 1997;47:110–5. doi: 10.1159/000185444. [DOI] [PubMed] [Google Scholar]
- 14.Thrailkill K, Quattrin T, Baker L, et al. J Clin Endocrinol Metab. 1997;82:1181–7. doi: 10.1210/jcem.82.4.3881. [DOI] [PubMed] [Google Scholar]
- 15.Frystyk J, Skjaerbaek C, Vestbo E, et al. Diabetes Metab Res Rev. 1999;15:314–22. doi: 10.1002/(sici)1520-7560(199909/10)15:5<314::aid-dmrr56>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 16.Bang P, Brismar K, Rosenfeld RG, Hall K. J Clin Endocrinol Metab. 1994;78:960–7. doi: 10.1210/jcem.78.4.7512573. [DOI] [PubMed] [Google Scholar]
- 17.Clauson PG, Brismar K, Hall K, et al. Scand J Clin Lab Invest. 1998;58:353–60. doi: 10.1080/00365519850186544. [DOI] [PubMed] [Google Scholar]
- 18.Takeda S, Podskalny JM, Gorden P. Metabolism. 1984;33:658–61. doi: 10.1016/0026-0495(84)90066-0. [DOI] [PubMed] [Google Scholar]
- 19.Johansson Jo, Fowelin J, Landin K, et al. Metabolism. 1995;44:1126–9. doi: 10.1016/0026-0495(95)90004-7. [DOI] [PubMed] [Google Scholar]
- 20.Aman J, Kroon M, Karlsson I, et al. Acta Paediatr. 1996;85:31–7. doi: 10.1111/j.1651-2227.1996.tb13886.x. [DOI] [PubMed] [Google Scholar]
- 21.Smith TR, Elmendorf JS, David TS, et al. Am J Physiol Endocrinol Metab. 1997;272:E1071–9. doi: 10.1152/ajpendo.1997.272.6.E1071. [DOI] [PubMed] [Google Scholar]
- 22.Simpson HL, Jackson NC, Shojaee-Moradie F, et al. J Clin Endocrinol Metab. 2004;89:425–32. doi: 10.1210/jc.2003-031274. [DOI] [PubMed] [Google Scholar]
- 23.Cutfield WS, Wilton P, Bennmarker H, et al. Lancet. 2000;355:610–13. doi: 10.1016/S0140-6736(99)04055-6. [DOI] [PubMed] [Google Scholar]
- 24.Quattrin T, Thrailkill K, Baker L, et al. J Pediatr Endocrinol Metab. 2001;14:267–77. doi: 10.1515/jpem.2001.14.3.267. [DOI] [PubMed] [Google Scholar]
- 25.Schoenle EJ, Zenobi PD, Torresani T, et al. Diabetologia. 1991;34:675–9. doi: 10.1007/BF00400998. [DOI] [PubMed] [Google Scholar]
- 26.Morrow LA, O’Brien MB, Moller DE, et al. J Clin Endocrinol Metab. 1994;79:205–10. doi: 10.1210/jcem.79.1.8027228. [DOI] [PubMed] [Google Scholar]
- 27.Kuzuya H, Matsuura N, Sakamoto M, et al. Diabetes. 1993;42:696–705. doi: 10.2337/diab.42.5.696. [DOI] [PubMed] [Google Scholar]
- 28.Quin JD, Fisher BM, Paterson KR, et al. N Engl J Med. 1990;323:1425–6. doi: 10.1056/NEJM199011153232016. [DOI] [PubMed] [Google Scholar]
- 29.Rietschel P, Hadigan C, Corcoran C, et al. J Clin Endocrinol Metab. 2001;86:504–10. doi: 10.1210/jcem.86.2.7175. [DOI] [PubMed] [Google Scholar]
- 30.Vigano A, Mora S, Brambilla P, et al. AIDS. 2003;17:1435–41. doi: 10.1097/00002030-200307040-00003. [DOI] [PubMed] [Google Scholar]
- 31.Wanke C, Gerrior J, Kantaros J, et al. AIDS. 1999;13:2099–103. doi: 10.1097/00002030-199910220-00013. [DOI] [PubMed] [Google Scholar]
- 32.Schwarz JM, Mulligan K, Lee J, et al. J Clin Endocrinol Metab. 2002;87:942. doi: 10.1210/jcem.87.2.8391. [DOI] [PubMed] [Google Scholar]
- 33.Engelson ES, Glesby MJ, Mendez D, et al. J Acquir Immune Defic Syndr. 2002;30:379–91. doi: 10.1097/00042560-200208010-00002. [DOI] [PubMed] [Google Scholar]
- 34.Lo JC, Mulligan K, Noor MA, et al. Clin Infect Dis. 2004;39:732–5. doi: 10.1086/422725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bluher S, Kratzsch J, Kiess W. Best Pract Res Clin Endocrinol Metab. 2005;19:577–87. doi: 10.1016/j.beem.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 36.Laron Z, Ginsberg S, Lilos P, et al. Growth Horm IGF Res. 2006;16:61–4. doi: 10.1016/j.ghir.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 37.Popken GJ, Dechert-Zeger M, Ye P, et al. Adv Exp Med Biol. 2005;567:187–220. doi: 10.1007/0-387-26274-1_8. [DOI] [PubMed] [Google Scholar]
- 38.Aleman Torres., I Adv Exp Med Biol. 2005;567:243–58. doi: 10.1007/0-387-26274-1_10. [DOI] [PubMed] [Google Scholar]
- 39.Aberg ND, Brywe KG, Isgaard J. ScientificWorldJournal. 2006;6:53–80. doi: 10.1100/tsw.2006.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu Y, Kastin AJ, Pan W. Endocrinology. 2006;147:2611–5. doi: 10.1210/en.2006-0020. [DOI] [PubMed] [Google Scholar]
- 41.Gasparini L, Xu H. Trends Neurosci. 2003;26:404–6. doi: 10.1016/S0166-2236(03)00163-2. [DOI] [PubMed] [Google Scholar]
- 42.Rivera EJ, Goldin A, Fulmer N, et al. J Alzheimers Dis. 2005;8:247–68. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- 43.Lester-Coll N, Rivera EJ, Soscia SJ, et al. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- 44.Carro E, Trejo JL, Gerber A, et al. Neurobiol Aging. 2005;27:1250–7. doi: 10.1016/j.neurobiolaging.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 45.Carro E, Spuch C, Trejo JL, et al. J Neurosci. 2005;25:10884–93. doi: 10.1523/JNEUROSCI.2909-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carro E, Trejo JL, Gomez-Isla T, et al. Nat Med. 2002;8:1390–7. doi: 10.1038/nm1202-793. [DOI] [PubMed] [Google Scholar]
- 47.Trejo Jl, Carro E, Lopez-Lopez C, et al. Growth Horm IGF Res. 2004;14(Suppl A):S39–43. doi: 10.1016/j.ghir.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 48.Aguado-Llera D, Arilla-Ferreiro E, Campos-Barros A, et al. J Neurochem. 2005;92:607–15. doi: 10.1111/j.1471-4159.2004.02889.x. [DOI] [PubMed] [Google Scholar]
- 49.Lupien SB, Bluhm EJ, Ishii DN. Neurobiol Dis. 2006;21:487–95. doi: 10.1016/j.nbd.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 50.Lupien SB, Bluhm EJ, Ishii DN. J Neurosci Res. 2003;74:512–23. doi: 10.1002/jnr.10791. [DOI] [PubMed] [Google Scholar]
- 51.Bonapace G, Concolino D, Formicola S, et al. J Med Genet. 2003;40:913–7. doi: 10.1136/jmg.40.12.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woods KA, Camado-Hubner C, Barter D, et al. Acta Paediatr Suppl. 1997;423:39–45. doi: 10.1111/j.1651-2227.1997.tb18367.x. [DOI] [PubMed] [Google Scholar]
- 53.Cediel R, Riquelme R, Contreras J, et al. Eur J Neurosci. 2006;23:587–90. doi: 10.1111/j.1460-9568.2005.04584.x. [DOI] [PubMed] [Google Scholar]
- 54.Iwai K, Nakagawa T, Endo T, et al. Laryngoscope. 2006;116:529–33. doi: 10.1097/01.mlg.0000200791.77819.eb. [DOI] [PubMed] [Google Scholar]
- 55.Lankhorst AJ, ter Laak MP, van Laar TJ, et al. J Neurotrauma. 2001;18:203–15. doi: 10.1089/08977150150502622. [DOI] [PubMed] [Google Scholar]
- 56.Koopmans GC, Brans M, Gomez-Pinilla F, et al. Eur J Neurosci. 2006;23:1035–46. doi: 10.1111/j.1460-9568.2006.04627.x. [DOI] [PubMed] [Google Scholar]
- 57.Colao A, Di Somma C, Savanelli MC, et al. Growth Horm IGF Res. 2006;16(Suppl):41–8. doi: 10.1016/j.ghir.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 58.Maldonado C, Cea P, Adasme T, et al. Biochem Biophys Res Commun. 2005;336:1112–8. doi: 10.1016/j.bbrc.2005.08.245. [DOI] [PubMed] [Google Scholar]
- 59.Li SY, Fang CX, Aberle NS, 2nd, et al. J Endocrinol. 2005;186:491–503. doi: 10.1677/joe.1.06168. [DOI] [PubMed] [Google Scholar]
- 60.Colao A, Pivonello R, Marzullo P, et al. J Endocrinol Invest. 2005;28:65–77. [PubMed] [Google Scholar]
- 61.Bondanelli M, Bonadonna S, Ambrosio MR, et al. Metabolism. 2005;54:1174–80. doi: 10.1016/j.metabol.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 62.Shyu KG, Ko WH, Yang WS, et al. Cardiovasc Res. 2005;68:405–14. doi: 10.1016/j.cardiores.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 63.Gaussin V, Depre C. Cardiovasc Res. 2005;68:347–9. doi: 10.1016/j.cardiores.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 64.Marleau S, Mulumba M, Lamontagne D, et al. Cardiovasc Res. 2006;69:26–35. doi: 10.1016/j.cardiores.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 65.Duerr Rl, Huang S, Mirakliakbar HR, et al. J Clin Invest. 1995;95:619–27. doi: 10.1172/JCI117706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Duerr Rl, McKirnan MD, Glim RD, et al. Circulation. 1996;93:2188–96. doi: 10.1161/01.cir.93.12.2188. [DOI] [PubMed] [Google Scholar]
- 67.Tivesten A, Caidhal K, Kujacic V, et al. Growth Horm IGF Res. 2001;11:187–95. doi: 10.1054/ghir.2001.0202. [DOI] [PubMed] [Google Scholar]
- 68.Tivesten A, Bollano E, Andersson I, et al. Endocrinology. 2002;143:4235–42. doi: 10.1210/en.2002-220524. [DOI] [PubMed] [Google Scholar]
- 69.Yau TM, Kim C, Li G, et al. Circulation. 2005;112:I123–8. doi: 10.1161/CIRCULATIONAHA.104.525147. [DOI] [PubMed] [Google Scholar]
- 70.Mcdevitt TC, Laflamme MA, Murry CE. J Mol Cell Cardiol. 2005;39:865–73. doi: 10.1016/j.yjmcc.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niu T, Rosen CJ. Gene. 2005;361:38–56. doi: 10.1016/j.gene.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 72.Boonen S, Rosen C, Bouillion R. J Clin Endocrinol Metab. 2002;87:1593–9. doi: 10.1210/jcem.87.4.8426. [DOI] [PubMed] [Google Scholar]
- 73.Agnusdei D, Gentiella R. J Endocrinol Invest. 2005;28:32–36. [PubMed] [Google Scholar]
- 74.Schurch MA, Rizolli R, Slosman D, et al. Ann Intern Med. 1998;128:801–9. doi: 10.7326/0003-4819-128-10-199805150-00002. [DOI] [PubMed] [Google Scholar]
- 75.Ding H, Kopple JD, Cohen A, et al. J Clin Invest. 1993;91:2281–7. doi: 10.1172/JCI116456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hladunewich MA, Corrigan G, Derby GC, et al. Kidney Int. 2003;64:593–602. doi: 10.1046/j.1523-1755.2003.00100.x. [DOI] [PubMed] [Google Scholar]
- 77.Friedlander Al, Butterfield CE, Moynihan S, et al. J Clin Endocrinol Metab. 2001;86:1496–503. doi: 10.1210/jcem.86.4.7377. [DOI] [PubMed] [Google Scholar]
- 78.Grimberg A. Cancer Biol Ther. 2003;2:630–5. [PMC free article] [PubMed] [Google Scholar]
- 79.Grimberg A. Cancer. Adv Exp Med Biol. 2005;567:305–39. doi: 10.1007/0-387-26274-1_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Clark RG. Horm Res. 2004;62(Suppl 1):93–100. doi: 10.1159/000080766. [DOI] [PubMed] [Google Scholar]