

Abstract
Theiler's murine encephalomyelitis virus (TMEV) and other neurotropic virus infections result in degeneration of each component of the neuron: apoptosis of the cell body, axonal (Wallerian) degeneration, and dendritic and synaptic pathology. In general, axonal degeneration is detrimental for hosts. However, axonal degeneration can be beneficial in the case of infection with neurotropic viruses that spread in the CNS using axonal transport. C57BL/WldS (WldS, Wallerian degeneration slow mutant) mice are protected from axonal degeneration. WldS mice infected with the neurovirulent GDVII strain of TMEV are more resistant to virus infection than wild-type mice, suggesting that axonal preservation contributes to the resistance. By contrast, infection with the less virulent Daniels strain of TMEV results in high levels of virus propagation in the CNS, suggesting that prolonged survival of axons in WldS mice favors virus spread. Thus, axonal degeneration might be a beneficial self-destruct mechanism that limits the spread of neurotropic viruses, in the case of less virulent virus infection. We hypothesize that neurons use ‘built-in’ self-destruct protection machinery (compartmental neurodegeneration) against neurotropic virus infection, since the CNS is an immunologically privileged site. Early induction of apoptosis in the neuronal cell body limits virus replication. Wallerian degeneration of the axon prevents axonal transport of virus. Dendritic and synaptic degeneration blocks virus transmission at synapses. Thus, the balance between neurodegeneration and virus propagation may be taken into account in the future design of neuroprotective therapy.
Keywords: axonal injury, CNS autoimmune demyelinating diseases, compartmentalized self-destruction, Inside-Out model, multiple sclerosis, Picornaviridae infections, synaptosis, Theiler's virus, Wallerian degeneration, Wld mice
Neurons are the transmitting cells of the nervous system and communicate by chemical and electrical means. A neuron can be divided into different components: cell body (soma), a longer cell process called an axon, branching processes called dendrites and synapses. A neuron transmits information via the axon, which terminates at a synapse for conveying ongoing signals, while dendrites integrate incoming signals. Since axons as well as their neuronal cell bodies do not usually regenerate in the CNS, axonal damage often results in permanent neurological deficits. Axonal degeneration in the CNS can be seen in several neurological conditions, such as neurodegenerative diseases (e.g., amyotrophic lateral sclerosis and neuroaxonal dystrophy) [1], traumatic lesions and infections [2-5].
Several viruses have been shown to use axonal transport to spread in the CNS [6-8]. Different virus families (Herpesviridae, Rhabdoviridae, Flaviviridae, Bornaviridae and Picornaviridae) include important human neurotropic viruses, such as herpes simplex virus, rabies virus, West Nile virus, poliovirus and others [8]. Neurotropic viruses often cause apoptosis of neurons [7,9,10] and axonal degeneration [11], which results in death of hosts or permanent neurological deficits [12]. For example, both poliovirus and West Nile virus have been shown to cause apoptosis of neurons [9,13,14] and axonal degeneration [15,16] in the CNS.
Theiler's murine encephalomyelitis virus (TMEV) is a neurotropic virus in mice. TMEV spreads in the nervous system using axonal transport and causes axonal degeneration and demyelination [17-20]. Therefore, TMEV is used as an animal model for the human demyelinating disease, multiple sclerosis (MS). In MS, axonal degeneration has been observed in the CNS, and clinical disability of MS patients is associated with axonal degeneration [18,21,22]. Although its etiology is unknown, MS is considered to be an immune-mediated disease [18,23,24], occurring in genetically susceptible individuals, precipitated by one or more environmental agents, particularly viruses [25-28].
In this review, we will use TMEV as a model system for neurotropic virus infection that causes axonal degeneration as well as apoptosis. We will discuss whether axonal degeneration and apoptosis are ‘built-in’ beneficial self-destructive machineries within neurons used to combat neurotropic virus infection. TMEV induces axonal degeneration and apoptosis, which potentially suppress virus spread in the axon and virus propagation in the neuronal cell body, respectively. Both axonal degeneration and apoptosis are observed during physiological conditions, particularly during development [29]. Thus, neurons may use these mechanisms as self-destructive suicidal pathways to suppress virus spread in the CNS. These potential innate antivirus pathways may be necessary for the ‘immunologically privileged’ CNS. Also in this review, we will introduce a relatively new concept, compartmental neurodegeneration, an idea originally proposed by Gillingwater and Richester [30], to describe the processes of Wallerian degeneration in the axon, apoptosis in the cell body, and dendritic and synaptic degeneration. We propose that neuronal degeneration can be a self-destructive mechanism against neurotropic virus infection. The balance between neurodegeneration and virus propagation should be taken into account in the future design of neuroprotective therapy.
TMEV
TMEV belongs to the family Picornaviridae, genus Cardiovirus, species Theilovirus [19]. Serological studies indicate that Mus musculus is the natural host of TMEV [31]. TMEV is divided into two subgroups, GDVII and Theiler's original (TO), according to neurovirulence and demyelination within the CNS [17,20]. The two subgroups are 95% identical at the amino acid level, and studies using recombinant viruses between the two subgroups have revealed the multigenic nature of neurovirulence and demyelination [20]. Although TMEV is a natural enteric pathogen in mice, only intracerebral inoculation of virus induces CNS disease efficiently. The GDVII subgroup viruses, including the GDVII and FA strains, cause acute fatal polioencephalomyelitis in mice [32]. The less virulent TO subgroup viruses, including Daniels (DA) and BeAn strains, cause acute nonfatal polioencephalomyelitis 1–2 weeks postinfection (acute phase), and chronic inflammatory demyelinating disease approximately 1 month after infection (chronic phase) [33]. A similar biphasic inflammatory disease has been described in infection with coxsackievirus B3, which belongs to the family Picornaviridae [34]. Persistence of picornavirus has also been shown in poliovirus infection in cell culture and animal models, as well as in immunodeficient humans. Portions of poliovirus genomes may persist for years in the CNS of post-polio syndrome patients [35].
The mechanism(s) of TMEV-induced demyelination appear to be multifactorial, although immune-mediated damage to myelin has been emphasized [19]. There is substantial evidence that all major immune cells, including CD4+ and CD8+ T cells, antibodies and macrophages, can contribute to demyelination. Virus-specific CD4+ Th1-cell responses are associated with demyelination, while myelin-specific CD4+ T cells have been detected during the late chronic phase (epitope spreading) [36-38]. CD8+ T cells play an important role in virus clearance, while virus-specific as well as autoreactive CD8+ T cells have been suggested to contribute to demyelination [17,23,36,37,39,40]. Similarly, anti-TMEV humoral immune responses can help virus clearance, while some antiviral antibodies cross-react with host myelin molecules, including galactocerebroside [38]. However, it should be noted that TMEV can cause demyelination in the organotypic culture in vitro in the absence of immune cells. In addition, immune-deficient mice, including nude mice and MHC class I or II deficient mice, also develop demyelination [20]. A transgenic mouse model (DA/Cre) that had tamoxifen-inducible expression of a subgenomic segment of DA virus RNA in oligodendrocytes and Schwann cells developed demyelination in the sciatic nerve, when DA/Cre mice were treated with tamoxifen [41]. Demyelination was not accompanied by inflammation in the DA/Cre mice. Here, viral RNA and/or protein, but not the immune system, seemed to cause demyelination.
Axonal degeneration precedes inflammatory demyelination: Inside-Out model
In neurovirulent GDVII virus infection, severe axonal degeneration in the white matter of the spinal cord can be detected within 1 week postinfection, in the absence of inflammation or demyelination, by immunohistochemistry against nonphosphorylated neurofilament (NFP) [22,42]. In DA virus infection, mild axonal degeneration can also be detected in the spinal cord white matter, as early as 1 week post-infection The number and extent of swelling of NFP+ axons increased over time. During the subclinical phase, 2–3 weeks postinfection, NFP+ damaged axons were detected in normal-appearing myelin sheaths containing myelin basic protein at the single fiber level, using double immunofluorescence confocal microscopy. Three weeks postinfection, beaded, fragmented and degenerating axons were detected. Significant axonal swelling occurred and NFP+ fragmented and degenerating axons were seen 1 month postinfection. On the other hand, significant demyelination and perivascular mononuclear cell infiltration were not evident until 1 month postinfection Early onset axonal injury was also confirmed in mice infected with BeAn virus (TO subgroup), 14 days postinfection [43]. Therefore, axonal degeneration precedes demyelination in TMEV infection.
Here, the lesion develops from the inside axon to the outside myelin (Inside-Out model) [5]. This is opposite to the conventional theory for lesion development in demyelinating diseases accompanying axonal degeneration, where the inside axon is believed to be damaged secondarily to outside myelin damage (Outside-In model). In some patients with MS and in a neurotropic coronavirus-induced MS model, there is evidence that axonal damage can precede demyelination [5,44]. In an autoimmune model for MS, experimental allergic (autoimmune) encephalomyelitis (EAE), autoantibodies against neurofascin, which were detected in MS patients, have been shown to bind the node of Ranvier and induce axonal damage and disease exacerbation in the absence of demyelination or inflammation [45]. Interestingly, predominant primary axonal damage has also been reported in a mouse model for peripheral nervous demyelinating disease, experimental autoimmune neuritis, induced by the P0106-125 peptide [46].
Axonal degeneration has also been observed in vitro in cerebellar explant culture infected with DA virus [47]. By 72 h postinfection, electron micrographic analysis indicated that DA virus induced apoptosis of neuronal cells as well as axonal degeneration within the intact myelin sheath. In this cerebellar explant culture, myelin damage was not seen. Thus, TMEV can induce axonal degeneration both in vivo and in vitro in the absence of lymphocytes or demyelination.
Preservation of axons is beneficial in neurovirulent GDVII virus infection
When an axon is cut, the distal part of the axon, which is disconnected from the neuronal cell body, breaks up into fragments; this classical type of axonal degeneration is called Wallerian degeneration insult [29]. Wallerian degeneration is a fundamental reaction of the nervous system to loss of continuity of axons as a result of traumatic, ischemic, toxic, metabolic or other injury. Wallerian degeneration slow mutant mice, C57BL/WldS (WldS) mice, are a substrain of C57BL/6 (B6) mice [48]. WldS mice have prolonged survival of the distal stumps of transected axons [30,49,50] both in the peripheral nervous system and the CNS [51]. Transected axons from WldS mice survive for up to 4 weeks, support action potentials for at least 2 weeks, and continue anterograde and retrograde transport of proteins for similar amounts of time [52-54]. WldS mice are protected from Wallerian degeneration by overexpression of a chimeric gene (Wld), which encodes the ubiquitin assembly protein Ufd2a fused to nicotinamide mononucleotide adenylyl transferase (Nmnat) [55]. Nmnat is responsible for preservation of axons, and SIRT1 is the downstream effector of increased Nmnat activity that leads to axonal protection [56].
Using WldS mice, we investigated the role of axonal degeneration in mice infected with the neurovirulent GDVII virus. GDVII virus causes acute fatal polioencephalitis in mice, regardless of the strain of mouse. Most infected mice die within 10 days. In the brain, high levels of apoptosis of neurons and viral replication are observed, which is associated with a failure in both the cellular and humoral immune response against virus [12,32]. GDVII virus was propagated in baby hamster kidney (BHK)-21 cells, and virus titer was determined by a plaque assay [57]. We inoculated different doses of GDVII virus intracerebrally into wild-type B6 and WldS mice. Between days 4 and 5 postinfection, the mice started to show signs of encephalitis, such as weight loss, ruffled fur and a hunched posture. Mice receiving higher dosages of virus lost weight more rapidly than those receiving lower dosages. The B6 mice had more rapid weight loss and impairment of righting reflex [28] than the WldS mice. Although most infected mice died within 1 week postinfection, more WldS mice survived than B6 mice, particularly at low infectious doses (Figure 1A). Using the Reed and Muench calculation of the 50% end point [58], we found that the median lethal dose (LD50) titer of B6 mice was 1.03 plaque forming unit (PFU) and that of WldS mice was 23.3 PFU (the LD50 titer is defined as the dose that will kill 50% of the animals receiving that dose). The survival period was also longer in WldS mice than in B6 mice (Figure 1B).
Figure 1. Mean survival percentages (A) and periods (B) of mice infected with the GDVII strain of Theiler's murine encephalomyelitis virus.

C57BL/6 mice (B6, triangle) and C57BL/WldS (Wallerian degeneration slow mutant) mice (WldS, circle) were infected intracerebrally with GDVII virus at different PFUs. WldS mice were more resistant to GDVII virus infection and survived longer than B6 mice. Shown are means of a group of 5–14 mice per dose (the total number of mice inoculated; B6, 47; WldS, 45).
PFU: Plaque-forming units.
Histologically, WldS mice developed a similar polioencephalitis as seen in B6 mice 1 week postinfection; apoptotic and/or viral antigen-positive neurons were observed in the gray matter of the brain. In the spinal cord, both mouse groups had small numbers of viral antigen-positive anterior horn cells in the gray matter, but not white matter, of the spinal cord (Figure 2A & B). The white matter appeared normal by conventional staining in both mouse groups. However, using immunohistochemistry against NFP, substantial numbers of damaged axons were detected in the white matter of the spinal cord in B6 mice (Figure 2C). By contrast, axonal degeneration was not observed in the spinal cord white matter of WldS mice (Figure 2D). Thus, axons were preserved in infected WldS mice, which likely contributed to the relative resistance of WldS mice to GDVII virus infection. Therefore, axonal preservation can play a beneficial role in neurotropic virus infection. The beneficial role of preservation of axons has also been seen in other neurodegenerative models using WldS mice, including EAE [59,60].
Figure 2. Viral antigen-positive cells and axonal damage in the spinal cord of wild-type B6 mice and WldS mice infected with GDVII virus.
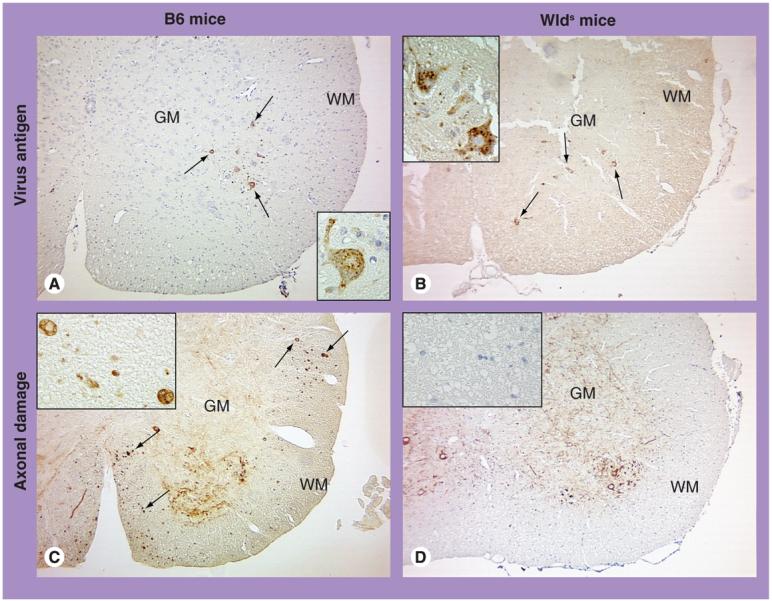
(A & B) In both B6 and WldS mice, viral antigen (arrow) was seen in the cytoplasm of anterior horn cells of the GM, using immunohistochemistry against a viral antigen. The WM contained no viral antigen or inflammation, and appeared normal in both mouse groups. (C & D) Immunohistochemistry against nonphosphorylated neurofilament (NFP) visualizes normal cell bodies and dendrites of neurons, while it visualizes damaged axons, but not normal axons. In both mouse groups, NFP+ normal cell bodies and dendrites of neurons were seen in the GM. However, damaged axons (arrow) in the WM were detected only in B6 mice (C), but not in WldS mice. Higher magnification showed NFP+ axonal swelling in B6 mice, while no NFP+ axon was seen in WldS mice.
Magnification: ×42; inset: ×190.
GM: Gray matter; WM: White matter.
Preservation of axons is detrimental in less virulent DA virus infection
The above results contrast to what has been reported in less virulent DA virus infection of B6 and WldS mice [60]. B6 mice are resistant to persistent DA virus infection and can clear DA virus from the CNS; no mice develop chronic paralytic disease. By contrast, DA virus infection in WldS mice causes chronic limb paralysis in infected mice. Histologically, infected WldS mice had prolonged inflammation with larger numbers of viral antigen-positive cells in the CNS, compared with B6 mice. WldS mice also had higher lymphoproliferative responses against TMEV than B6 mice. During the late chronic phase, however, neither virus persistence nor demyelination was observed in WldS or B6 mice, while WldS mice developed muscle pathology, similar to what is seen in post-polio syndrome.
Since TMEV can be transported by axonal flow [61,62], prolonged survival of axons in WldS mice would favor virus spread in the CNS, leading to increased levels of inflammation and virus propagation. Recently, axonal degeneration has been suggested to be an active program of self-destruction in many physiological and pathological settings, such as axonal branch elimination in development and neuronal insult [29,56]. We speculated that axonal degeneration in B6 mice infected with TMEV can be a beneficial self-destructive mechanism that limits the spread of virus in the CNS. In addition, transection of infected axons would efficiently limit transport and spread of viruses from the gray matter to the white matter. This could help explain why GDVII virus, unlike DA virus, does not persistently infect cells in the white matter of the spinal cord in rare survivor mice [63].
Apoptosis in TMEV infection
TMEV induces apoptosis in neurons during the acute phase of infection and in oligodendrocytes (and possibly macrophages) during the chronic phase of infection in vivo [12]. Terminal transferase dUTP nick-end labeling (TUNEL)+ apoptotic nuclei can be seen in both TMEV antigen-positive and -negative cells. TMEV also induces apoptosis in vitro in cell lines, including cells of monocyte/macrophage and neuronal lineages [12,19,64]. Induction of apoptosis in vitro is inversely associated with cell permissiveness for infection: TMEV replication is restricted in cells that undergo apoptosis. In contrast, permissive cells, which are productively infected with TMEV, predominantly die by necrosis.
GDVII virus induces a higher level of apoptosis of neurons than DA virus in the CNS [12]. This could be, at least partly, related to the absence of L* synthesis in the GDVII infection. The initiating AUG for the L* reading frame is present in all TO subgroup genomes, but not in the GDVII subgroup. GDVII virus and a DA virus mutant that lacks L* are more efficient at inducing apoptosis of cultured macrophages than the wild-type DA virus, suggesting that the L* protein has an antiapoptotic effect in macrophages [65]. On the other hand, in primary peritoneal macrophages infected with BeAn virus in vitro, one-third of the cells were apoptotic but negative for virus RNA and antigens [64]. This suggests that apoptosis was induced in BeAn virus antigen-negative cells by bystander killing, such as TNF-α production or cell-to-cell contact, but not by direct virus infection. Regardless of its mechanism, early induction of apoptosis in virus-infected cells as well as adjacent uninfected cells can limit virus spread both in vivo and in vitro.
Theoretically, however, some viruses might use the apoptotic machinery to enter phagocytic cells. In the late stage of vaccinia virus infection, a host cell undergoes apoptosis and externalizes phosphatidylserine on its plasma membrane [66]. A virus, budding from apoptotic cells, acquires an envelope that exposes phosphatidylserine, a signal for a phagocytic cell to engulf apoptotic cells [67]. This results in viral uptake by noninfected phagocytes through a mechanism similar to macropinocytosis. Potentially, non-enveloped viruses, including TMEV, can use a similar mechanism to infect phagocytes, if the viruses can induce apoptosis in infected cells.
Wallerian degeneration & neuronal apoptosis do not induce lymphocyte infiltration
One histological hallmark of tissues displaying apoptosis is a lack of lymphocyte infiltration. GDVII virus causes apoptosis in a large number of neurons, while recruitment of T and B cells is minimal. In contrast, less virulent DA virus causes apoptosis in fewer neurons, but recruits a large number of lymphocytes into the CNS. The number of apoptotic neurons is negatively correlated with the extent of lymphocyte infiltrate [68]. The lack of lymphocyte recruitment in GDVII virus infection is correlated with induction of TGF-β in neurons. However, it should be noted that substantial macrophage/microglia infiltration is seen in both GDVII and DA virus infections.
In contrast to apoptosis, necrosis induces inflammation. The difference in induction of inflammation between apoptosis and necrosis can be attributed to a release of certain intra-cellular molecules, known as damage-associated molecular patterns (DAMPs), only from necrotic cells [69]. DAMPs are normally hidden in the interior of cells and are only revealed after necrosis. Candidate molecules for DAMPs include heat-shock proteins, uric acid, high-mobility group box 1 protein (HMGB1) and genomic dsDNA. Pathogen-associated molecular patterns (PAMPs) from microorganisms, such as viral RNAs, are other molecules that play an important role in the induction of inflammation. In virus infection, phagocytosis of virus-infected apoptotic cells by resident macrophages should minimize the release of PAMPs extracellularly, compared with necrosis of infected cells. In addition, in some virus infections, necrosis of infected cells resulted from active virus replication, while apoptosis of infected cells resulted in abortive infection [64]. This also contributes to overall differences in the amount of PAMPs between apoptotic versus necrotic cell death in virus infection, leading to a lack of inflammation in the former and induction of inflammation in the latter. In addition, since DAMPs are known to function as an adjuvant [69], a lack of release of DAMPs from apoptotic neurons in GDVII virus infection may contribute to a relative lack of antiviral immune responses. Furthermore, phagocytosis of apoptotic cells can lead to the production of anti-inflammatory cytokines [70].
Executive summary.
Theiler's murine encephalomyelitis virus infection
■ Theiler's murine encephalomyelitis virus (TMEV) is a neurotropic virus and causes axonal degeneration and demyelination, similar to multiple sclerosis (MS).
■ TMEV is divided into the neurovirulent GDVII subgroup and the less virulent Theiler's original subgroup (Daniels [DA] and BeAn viruses), the latter of which cause demyelination by immune-mediated damage and/or direct virus infection.
Axonal degeneration in TMEV infection
■ Axonal degeneration precedes demyelination in TMEV infection; the lesion develops from the inside axon to the outside myelin – the Inside-Out model.
■ Axonal degeneration is detrimental to hosts infected with the neurovirulent GDVII virus, while it is beneficial to hosts infected with the less virulent DA virus, preventing virus spread via axonal transport in the CNS.
Apoptosis in TMEV infection
■ TMEV causes apoptosis of neurons and oligodendrocytes in the CNS in vivo. In vitro TMEV replication is restricted in apoptotic cells, while TMEV productively propagates in permissive cells, which die by necrosis.
■ Apoptosis of virus-infected cells as well as adjacent uninfected cells can be a self-destructive defense mechanism to prevent virus spread in the CNS.
Compartmental neurodegeneration
■ Axonal degeneration and apoptosis can be innate antivirus machineries against neurotropic virus infection in the immunologically privileged CNS.
■ The neuron can be divided into compartments: cell body (soma), axon, dendrite and synapses. Each compartment of the neuron can be degenerated independently: apoptosis of the cell body, axonal (Wallerian) degeneration and dendritic or synaptic degeneration (synaptosis).
■ Compartmental neurodegeneration can be a self-destructive defense mechanism against neurotropic virus infection.
Future perspective
■ The balance between neurodegeneration and virus replication can be important for the outcome in neurotropic virus infection.
■ Caution is needed in future neuroprotection therapies targeting axonal degeneration.
It is unknown whether DAMPs or PAMPs are released in any conditions accompanied by Wallerian degeneration. DAMPs should be released in Wallerian degeneration caused by mechanical damage, such as CNS trauma. DAMPs would contribute to the induction of chemokines and upregulation of adhesion molecules on CNS endothelial cells, both of which can contribute to CNS lymphocyte infiltration. It should be noted, however, that recruitment of lymphocytes requires activation of lymphocytes in the periphery, which results in upregulation of adhesion molecules on lymphocytes [71]. This can explain why we do not see lymphocyte infiltration in stroke and CNS trauma, in which there is no activation of peripheral lymphocytes, despite the fact that CNS tissue destruction may result in DAMP release.
However, in the presence of activated lymphocytes in the periphery, lymphocyte recruitment can occur in CNS tissues with Wallerian degeneration. The distribution of axonal degeneration observed during the early stage of DA virus infection corresponded to regions where subsequent demyelination occurs during the chronic phase [42]. This suggests that axonal degeneration plays a role in recruiting inflammatory cells and targeting sites of demyelination. If Wallerian degeneration was artificially induced during the early chronic phase of DA virus infection when virus-specific activated T cells are present in the periphery, T cells were recruited to the site of Wallerian degeneration [72]. Thus, CNS Wallerian degeneration itself can recruit activated T cells from the periphery into the CNS. It has also been reported in the adoptive transfer EAE model that activated T cells are recruited from the periphery to the site of Wallerian degeneration in the CNS [73].
There are similarities between apoptosis in the CNS and Wallerian degeneration. Both induce microglia activation (also astrocyte activation in some instances). When there are no activated lymphocytes in the periphery, apoptosis or Wallerian degeneration alone does not recruit lymphocytes into the CNS. However, in the presence of activated (encephalitogenic) lymphocytes in the periphery, Wallerian degeneration can recruit lymphocytes into the site of the degenerated tract by as yet unknown mechanisms (upregulation of MHC molecules has been suggested to play a role in the lymphocyte recruitment [73]).
Two self-destruct programs in neurons: Wallerian degeneration & apoptosis
Raff et al. suggested that neurons have two self-destruct programs: apoptosis and axonal degeneration [29]. Apoptosis can be induced by a self-destructive mechanism (physiological cell death), and apoptosis of TMEV-infected cells could be beneficial if it occurs prior to intracellular virus assembly [12]. Similarly, axonal degeneration has been shown to be an active program of self-destruction in many physiological and pathological settings, such as axonal branch elimination in development and neuronal insult [29,56,74]. In a physiologic condition, it may be appropriate to call such an axonal change a ‘physiological axotomy’ rather than axonal degeneration (‘axotomy’ or ‘axonotmesis’ is defined as a nerve lesion producing discontinuity of axons, with preservation of the supporting nerve structures). Axonal injury may occur through a local self-destruct program designed to disconnect the axon from its target cell, thereby conserving energy or preventing spread of toxic substances, including virus, via axonal transport.
The self-destructive axotomy can be beneficial by preventing DA virus spread in the CNS. In this scenario, slower axonal degeneration in WldS mice would favor virus spread to the white matter of the spinal cord. This could result in virus infection in glial cells, including oligodendrocytes, in the white matter. Transfer of TMEV from the axon to the myelin sheath has been shown in the optic nerve of WldS mice [62]. Thus, the lack of axonal degeneration in WldS mice can be a double-edged sword: it would contribute to survival of mice, yet result in virus spread in the white matter.
As we discussed in the previous section, apoptosis and Wallerian degeneration are not accompanied by lymphocyte infiltration in the CNS, particularly when there are no activated lymphocytes in the periphery. Since both apoptosis and Wallerian degeneration can suppress virus propagation in the CNS, it seems reasonable to hypothesize that the neuron has apoptosis and Wallerian degeneration as built-in innate antivirus machineries in an environment where there is no antiviral adoptive immune cell infiltration. In addition, the CNS is an immunologically privileged site:
■ The CNS lacks a ‘conventional’ lymphatic drainage system
■ The blood–brain barrier restricts and regulates passage of immune cells and selective molecules
■ Most CNS cells, including neurons, do not express MHC class I or II molecules, which are important for association with lymphocytes
Thus, the neuron needs to have its own machinery to combat virus infection.
Compartmental neurodegeneration
Portions of some cells have been shown to maintain their morphology and function independent of their nucleus. The brief application of heat to human polymorphonuclear leukocytes induced the formation of fragments of former protopods (leading fronts), which become uncoupled from the cell body and leave the cell body [75]. These anucleate fragments can respond chemotactically and retain their functional capacities for over 24 h, longer than intact PMNs. Conversely, one compartment of cells can degenerate locally without causing death of the entire cell. In a mouse model for slow-channel congenital myasthenic syndrome, apoptosis was induced in postsynaptic nuclei of the muscle fiber at the neuromuscular junction, with localized caspase activation [76]. However, this did not spread or lead to death of the entire muscle fiber syncytium, suggesting that the apoptotic pathway of the muscle fiber at the neuromuscular junction is circumscribed and reversible.
The major structural and functional compartments of the neurons are well defined: the cell body (soma), dendrites, axon and synaptic terminals. There has been little debate on a possible compartmentalization of the neuron with regard to pathophysiology. In some neurological diseases, axons, dendrites and synapses often degenerate well before degeneration of the neuronal soma. The distinctive nature of apoptosis of the neuronal cell body, Wallerian degeneration of distal axons, and dendritic or synapse-specific degeneration suggests that different mechanisms are embedded in neurons for executing these processes (compartmental neurodegeneration; Figure 3) [30].
Figure 3. Compartmental neurodegeneration.

[30]. The neuron can be divided into compartments, structurally and functionally: the cell body (soma), axon, dendrite and synapse. During development and neurological diseases, each compartment often degenerates well before or independently of the other compartments. Degeneration of the cell body and the axon occurs through apoptosis and axonal (Wallerian) degeneration, respectively, which can be self-destructive in certain conditions [29,56]. Although localized degeneration of the dendrite and synapse has also been demonstrated, precise mechanisms of dendritic and synaptic pathology (synaptosis) are not known. The myelin sheath is not a compartment of the neuron, but of the oligodendrocyte. Degeneration of the myelin sheath is demyelination, which can occur independently of the above compartmental neurodegenerative mechanisms or destruction of oligodendrocyte cell bodies.
For example, in vitro experiments using compartment chambers showed that a low dose of vincristine affected axons, but not cell bodies of neurons [77]. In this experiment, a toxic signal was not transmitted centrally to the cell bodies (which were cultured in the central chamber) when vincristine was administered only to axons that grew in the lateral chamber. This suggests that vincristine-induced axonal degeneration results from the direct vulnerability of distal axons. This is in contrast with a conventional ‘dying back’ theory of distal axonal degeneration in vincristine neuropathy, in which the distal axon was explained to be vulnerable because it resides farthest for the source of its nourishment when the cell body of the neuron is damaged.
The experiments using WldS mice add to a growing literature identifying the independence of the axon from the cell body (soma) and provide another piece of evidence that factors involved in axonal survival and death are distinct from those of the cell body. In WldS mice, nerve growth factor deprivation induces apoptosis of the soma of neurons, but the neurites persist and remain viable; the Wlds mutation maintains neuritic viability while not affecting programmed neuronal death of the neuronal soma [78].
There is evidence that retinal ganglion cells (RGCs) undergo compartmentalized self-degeneration in experimental models for glaucoma [79,80]. Like other neurons, RGCs are composed of distinct compartments, including the cell body (soma), axon, dendrite and synapse. In explants from anti-apoptotic Bax−/−Bak−/− mice, the axons degenerated within 2 days, but the RGCs were protected from apoptosis [81]. By contrast, in explants from WldS mice, the axons were protected from degeneration, whereas the RGCs were not protected from apoptosis. Thus, Bax−/−Bak−/− RGCs were protected from apoptosis but not from axonal degeneration, whereas the reverse was the case for WldS RGCs.
Dendritic & synaptic degeneration
Dendrites and synapses have also been demonstrated to degenerate, independent of apoptosis or axonal degeneration [82], although little information is available regarding whether dendritic or synaptic degeneration can be self-destructive [80]. Synaptic degeneration is delayed in WldS mice [83]. Some features of synaptic withdrawal (degeneration) in axotomized nerve terminals in WldS mice resemble synapse elimination in neonatal muscle, which is termed ‘synaptosis’ or ‘synapoptosis’ [30]. Early changes in dendrites and synapses have been described in some neurological diseases, including glaucoma and EAE [84,85]. In Alzheimer's disease and Huntington disease, dendritic and synaptic pathology has been shown to precede neuronal loss, suggesting that attempts to prevent neuronal cell death might intervene too late in the pathologic process to be of any therapeutic benefit [82]. In a transgenic model of inherited prion disease, the loss of Bax prevents neuronal loss but does not prevent synaptic degeneration or development of neurological signs [86].
During development, the formation of mature neural circuits requires the selective elimination of inappropriate synaptic connections [74,87,88]. This ‘physiological’ synaptic elimination is mediated by the complement cascade. Thus, the CNS seems to have a built-in complement-mediated synaptic elimination pathway during development. In pathologic conditions, such as glaucoma, this pathway may play a detrimental role, while this local destruction mechanism might be used as an arm of the innate immune response to block trans-synaptic spread of the pathogen during neurotropic virus infection.
Synaptic pathology has been reported in neurotropic virus infection. Downregulation of the expression of synaptic markers, such as GAP-43 and/or synaptophysin, has been observed in the absence of destruction of neurons in the brains of those infected with lymphocytic choriomeningitis virus [89] and Borna disease virus [90]. In TMEV infection, axo-somatic and axodendritic synapses were completely destroyed in spinal cord slice cultures infected with the WW strain (TO subgroup) or the GDVII strain of TMEV [91]. In this study, virions were observed in electron-opaque cells, but not within axons. Thus, synaptic pathology may occur without the presence of virions in axons or in the absence of immune cells. Immunostaining for microtubule-associated protein (MAP)-2, a marker for dendrites [85], showed dendritic pathology in the hippocampus of wild-type mice infected with GDVII virus [92]. We found that WldS mice infected with GDVII virus also developed pathology in dendrites; marked loss of MAP-2 reactivity was seen in apical dendrites of the hippocampus (Figure 4B, arrows), and some dendrites were beaded (Figure 4D, arrows). These results demonstrate that dendritic pathology can occur independently of Wallerian degeneration. It will be worth investigating whether dendritic or synaptic pathology can be induced independent of neuronal apoptosis.
Figure 4. Dendritic pathology of WldS mice in TMEV infection.

Dendrites of neurons were visualized using immunostaining for microtubule-associated protein (MAP)-2 [92]. (A) In the hippocampus of uninfected mice, MAP-2-positive dendrites were seen in the gray matter. (B) 1 week after DA virus infection, WldS mice developed pathology in dendrites; marked loss of MAP-2 reactivity was seen in apical dendrites of the CA1 area of the hippocampus (arrows). (C) Higher magnification of (A) showed MAP-2-positive apical dendrites along with the pyramidal cell layer of the hippocampus. (D) Higher magnification of (B) showed some dendrites were beaded (arrows).
Magnification: (A & B) ×21; (C & D) ×318.
TMEV: Theiler's murine encephalomyelitis virus.
Induction of apoptosis of neuronal soma and dendritic pathology in GDVII virus infection in WldS mice suggest that compartmental neurodegeneration can occur in virus infection. Compartmental neurodegeneration has been described in other virus infections. For example, in an in vitro model of HIV-associated sensory neuropathy, HIV envelope protein gp120 directly induced local axonal degeneration through binding to chemokine receptors and local activation of caspases in the axon, which was independent of the cell body in compartmentalized cultures of sensory neurons [93].
In the CNS, compartmentalized degeneration can be seen not only in neurons but also in some other cell types; myelin degeneration could be regarded as compartmentalized degeneration of oligodendrocytes. Roussarie et al. [62] showed that, in WldS mice, TMEV infection in the optic nerve resulted in virus transport from the axon to the myelin. Thus, preservation of the axon may favor virus spread not only within the axons (from the axon to the dendrite or from one neuron to the other) but also into the adjacent myelin/oligodendrocytes. However, if the myelin sheath surrounding an infected axon degenerates before virus infects the myelin sheath, this early degeneration (demyelination) can block centripetal transfer of virus from the myelin sheath to the cell body of the oligodendrocyte. In this context, demyelination might represent the oligodendrocytes' self-destructive attempt to suppress virus spread, although it ultimately results in potentially irreversible myelin damage [38]. Indeed, loss of myelin (demyelination) has been shown to occur in the presence of surviving cell bodies of oligodendrocytes in subtypes of MS and animal models for MS [43,94]. This might represent demyelination as a compartmentalized degeneration of oligodendrocytes (we will not discuss this issue further, since it is beyond the scope of this review).
Conclusion & future perspective
The precise mechanism of axonal degeneration is not known in many neurological diseases and their animal models. The nature and site of initial injury to the neuron are often unknown; it is unclear if the process starts in the axon itself or within the neuronal cell body. In neurotropic virus infection, direct insult by virus can induce cell destruction, while infected neurons (and perhaps neighboring neurons also) may operate self-destructive machinery to combat neuro-tropic virus infection. The infected neuronal cell body can signal the axon to initiate the localized self-destruct process. Alternatively, the axon may sense danger signals locally and determine whether the axon should remain connected or degenerate. The candidate factors for axonal degeneration include influx of calcium, activation of m-calpain, caspase and/or gelsolin, and the ubiquitin–proteasome system [80]. Axonal degeneration can be induced by disruption of cross-talk between myelin sheaths and axons [5]. Here, elucidation of the precise mechanism of axonal degeneration in different neurological diseases is necessary for the development of a new therapeutic modality. Although there is no efficient treatment targeting axonal degeneration, several drugs, such as sodium and calcium channel antagonists, proteasome inhibitors and resveratrol (a red wine component that enhances SIRT1 activity), have been tested for their potential axonal preservation property [56,95,96].
Preservation of axons will allow delivery of target-derived neurotrophic signals from the axon to the cell body as well as delivery of maintenance factors, including neuronal electrical activity, from the cell body to the axon [80]. As shown in animal models for spinal cord injury and MS, axonal degeneration can result in secondary myelin and oligodendrocyte damage by several mechanisms, such as disruption of axon–myelin cross-talk, leading to expansion of neurodegeneration [5]. Thus, preservation of the axon could be a way of ‘buying time’ to interfere with the cascade reaction consisting of axonal and myelin degeneration until effector mechanisms, such as direct virus attack or antiviral inflammatory responses, subside in the CNS.
However, as we discussed in this review, axonal degeneration can be a self-destructive defense mechanism to prevent the spread of neurotropic virus (or other toxic substances) (Figure 5). In theory, neurons have individual self-destructive defense mechanisms in each compartment. Apoptosis of infected cell bodies can suppress virus replication, resulting in abortive infection. Dendritic and synaptic degeneration can block virus transmission trans-synaptically or at the neuromuscular junction. Compartmental neurodegeneration is a reasonable price to pay to restrict virus spread in the CNS (Figure 5). One example of this can be DA virus clearance in wild-type B6 mice, compared with virus persistence in WldS mice during the early chronic phase of infection [60]. On the other hand, it is much more costly when this defense mechanism is not effective enough to combat highly neurovirulent virus infection. In GDVII virus infection, axonal preservation in WldS mice could be crucial for better survival of hosts, compared with infected B6 mice with axonal degeneration.
Figure 5. The hypothetical balance between neurodegeneration versus virus propagation in neurotropic virus infection.

Since the CNS is an immunologically privileged site, the neuron itself has built-in self-destruct protection machinery – compartmental neurodegeneration. Early induction of apoptosis in the neuronal cell body limits virus replication intracellularly. Wallerian degeneration of the axon prevents axonal transport of virus, preventing ‘intraneural’ spread of virus inside the neuron (which can be more than 1-meter long in large animals). Dendritic and synaptic degeneration blocks virus transmission at synapses between neurons and at neuromuscular junctions, which prevents virus spread from one neuron to the other (‘interneural’ spread) or between the neuron and periphery organs, such as muscle, which are innervated by the neuron. The overall balance between neurodegeneration and viral propagation determines the outcome of infection.
Local inhibition of axonal transport at the site of neurotropic virus infection can be a future antiviral therapeutic strategy, particularly if the inhibition is reversible. Although such a strategy has not been tested clinically, there are several reversible pharmaceutical drugs that target cytoskeletal proteins involved in axonal transport [97]. There are also several methods that inhibit motor proteins, using function-blocking antibody, antisense or siRNAs.
We propose that the balance between neuro-degeneration and virus propagation determines whether the overall outcome favors the host or not (Figure 5). In the case of a highly neuro-virulent virus that can induce severe neurode-generation, neuroprotection strategies should be prioritized to prevent the fatal outcome. In infection with a less virulent virus that propagates slowly and/or infects persistently in the host CNS, self-destructive neurodegeneration machinery should efficiently suppress virus propagation in the CNS, and benefit the host, particularly if neurodegeneration is compartmentalized in small regions (Figure 5). Theoretically, compartmental neurodegeneration in the axon, dendrite or synapses allows the neuronal cell body to survive [89]. Then, after clearance of virus, the neuron might restore its function by regenerating its neuronal processes, although regeneration of neuronal processes in the CNS is generally not possible (in spite of some successful experimental reports [98,99]), in stark contrast to the peripheral nervous system. Future neuroprotection therapeutic strategies in neurotropic virus infection require careful consideration of the balance between neurodegeneration and viral propagation in the CNS.
Acknowledgements
The author thanks Robert S Fujinami, PhD, Jane E Libbey, MS, Eiji Morita, PhD, Tomoko Tanaka, MD, and Nathan J Young, BS, for helpful discussions, along with Reina Yamaji, MD, Nikki J Kirkman, BS, Andy Luu and Faris Hasanovic for excellent technical assistance. The author is grateful to Kathleen Borick for preparation of the manuscript.
Financial & competing interests disclosure
This work was supported by the National Multiple Sclerosis Society (PP1499) and the NIH (R21NS059724). The author has no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
■ of interest
■■ of considerable interest
- 1.Cochran EJ. Neurodegenerative diseases. In: Prayson RA, Goldblum JR, editors. Neuropathology. A Volume in the Series, Foundations in Diagnostic Pathology. Elsevier Churchill Livingstone; Philadelphia, USA: 2005. pp. 223–286. [Google Scholar]
- 2.An SF, Giometto B, Groves M, et al. Axonal damage revealed by accumulation of β-APP in HIV-positive individuals without AIDS. J. Neuropathol. Exp. Neurol. 1997;56:1262–1268. doi: 10.1097/00005072-199711000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Beattie MS, Li Q, Bresnahan JC. Cell death and plasticity after experimental spinal cord injury. Prog. Brain Res. 2000;128:9–21. doi: 10.1016/S0079-6123(00)28003-5. [DOI] [PubMed] [Google Scholar]
- 4.Chowdhury D, Arora A. Axonal Guillain–Barre syndrome: a critical review. Acta Neurol. Scand. 2001;103:267–277. doi: 10.1034/j.1600-0404.2001.103005267.x. [DOI] [PubMed] [Google Scholar]
- 5■■.Tsunoda I, Fujinami RS. Inside-Out versus Outside-In models for virus induced demyelination: axonal damage triggering demyelination. Springer Semin. Immunopathol. 2002;24:105–125. doi: 10.1007/s00281-002-0105-z. A novel hypothesis for pathogenesis of multiple sclerosis (MS) is proposed, where the lesion develops from the inside axon to the outside myelin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gromeier M, Wimmer E. Mechanism of injury-provoked poliomyelitis. J. Virol. 1998;72:5056–5060. doi: 10.1128/jvi.72.6.5056-5060.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park C-H, Kondo M, Inoue S, et al. The histopathogenesis of paralytic rabies in six-week-old C57BL/6J mice following inoculation of the CVS-11 strain into the right triceps surae muscle. J. Vet. Med. Sci. 2006;68:589–595. doi: 10.1292/jvms.68.589. [DOI] [PubMed] [Google Scholar]
- 8.Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc. Natl Acad. Sci. USA. 2007;104:17140–17145. doi: 10.1073/pnas.0705837104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Girard S, Couderc T, Destombes J, Thiesson D, Delpeyroux F, Blondel B. Poliovirus induces apoptosis in the mouse central nervous system. J. Virol. 1999;73:6066–6072. doi: 10.1128/jvi.73.7.6066-6072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weissenböck H, Hornig M, Hickey WF, Lipkin WI. Microglial activation and neuronal apoptosis in Bornavirus infected neonatal Lewis rats. Brain Pathol. 2000;10:260–272. doi: 10.1111/j.1750-3639.2000.tb00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minguetti G, Hofmeister RM, Hayashi Y, Montano JA. Ultrastructure of cranial nerves of rats inoculated with rabies virus. Arq. Neuropsiquiatr. 1997;55:680–686. doi: 10.1590/s0004-282x1997000500002. [DOI] [PubMed] [Google Scholar]
- 12■.Tsunoda I, Kurtz CIB, Fujinami RS. Apoptosis in acute and chronic central nervous system disease induced by Theiler's murine encephalomyelitis virus. Virology. 1997;228:388–393. doi: 10.1006/viro.1996.8382. One of the first papers to describe how neurotropic virus infection leads to apoptosis of neurons and oligodendrocytes, suggesting a potential protective role for apoptosis against virus infection. [DOI] [PubMed] [Google Scholar]
- 13.Shrestha B, Gottlieb D, Diamond MS. Infection and injury of neurons by West Nile encephalitis virus. J. Virol. 2003;77:13203–13213. doi: 10.1128/JVI.77.24.13203-13213.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao S-Y, Guzman H, Zhang H, Travassos da Rosa APA, Tesh RB. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg. Infect. Dis. 2001;7:714–721. doi: 10.3201/eid0704.010420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodian D. The virus, the nerve cell, and paralysis. A study of experimental poliomyelitis in the spinal cord. Bull. Johns Hopkins Hosp. 1948;83:1–107. [PubMed] [Google Scholar]
- 16.Burton JM, Kern RZ, Halliday W, et al. Neurological manifestations of West Nile virus infection. Can. J. Neurol. Sci. 2004;31:185–193. doi: 10.1017/s0317167100053828. [DOI] [PubMed] [Google Scholar]
- 17.Drescher KM, Sosnowska D. Being a mouse in a man's world: what TMEV has taught us about human disease. Front Biosci. 2008;13:3775–3785. doi: 10.2741/2966. [DOI] [PubMed] [Google Scholar]
- 18.Howe CL, Rodriguez M. Remyelination as neuroprotection. In: Waxman SG, editor. Multiple Sclerosis as a Neuronal Disease. Elsevier Academic Press; Burlington, MA, USA: 2005. pp. 389–419. [Google Scholar]
- 19.Lipton HL, Kumar ASM, Hertzler S. Cardioviruses: encephalomyocarditis virus and Theiler's murine encephalomyelitis virus. In: Fox J, Barthold S, Davisson MI, editors. The Mouse in Biomedical Research (2nd Edition) Elsevier/Academic Press; San Diego, USA: 2007. pp. 311–323. [Google Scholar]
- 20.Tsunoda I, Fujinami RS. Theiler's murine encephalomyelitis virus. In: Ahmed R, Chen ISY, editors. Persistent Viral Infections. John Wiley & Sons, Ltd.; Chichester, West Sussex, UK: 1999. pp. 517–536. [Google Scholar]
- 21.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120:393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 22.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. N. Engl. J. Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 23.Lassmann H. Models of multiple sclerosis: new insights into pathophysiology and repair. Curr. Opin. Neurol. 2008;21:242–247. doi: 10.1097/WCO.0b013e3282fee94a. [DOI] [PubMed] [Google Scholar]
- 24.Johnson AJ, Suidan GL, McDole J, Pirko I. The CD8 T cell in multiple sclerosis: suppressor cell or mediator of neuropathology? Int. Rev. Neurobiol. 2007;79:73–97. doi: 10.1016/S0074-7742(07)79004-9. [DOI] [PubMed] [Google Scholar]
- 25.Olson JK, Ercolini AM, Miller SD. A virus-induced molecular mimicry model of multiple sclerosis. Curr. Top. Microbiol. Immunol. 2005;296:39–53. doi: 10.1007/3-540-30791-5_3. [DOI] [PubMed] [Google Scholar]
- 26.Nicholas AP, Gupta KB, McInnis C, Benjamin WH, Jr, Williams SL, Kern ER. Evidence for Epstein–Barr virus autoantibodies against human brain in demyelinating encephalitis. Neuropathol. Appl. Neurobiol. 2002;28:498–501. doi: 10.1046/j.1365-2990.2002.t01-1-00429.x. [DOI] [PubMed] [Google Scholar]
- 27.Posnett DN. Herpesviruses and autoimmunity. Curr. Opin. Investig. Drugs. 2008;9:505–514. [PubMed] [Google Scholar]
- 28.Tsunoda I, Libbey JE, Fujinami RS. Sequential polymicrobial infections lead to CNS inflammatory disease: possible involvement of bystander activation in heterologous immunity. J. Neuroimmunol. 2007;188:22–33. doi: 10.1016/j.jneuroim.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29■■.Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science. 2002;296:868–871. doi: 10.1126/science.1068613. Review proposing apoptosis and axonal degeneration as two self-destruct programs. [DOI] [PubMed] [Google Scholar]
- 30■■.Gillingwater TH, Ribchester RR. Compartmental neurodegeneration and synaptic plasticity in the Wlds mutant mouse. J. Physiol. 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. Suggests synaptic degeneration, synaptosis, as compartmental neurodegeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipton HL, Kim BS, Yahikozawa H, Nadler CF. Serological evidence that Mus musculus is the natural host of Theiler's murine encephalomyelitis virus. Virus Res. 2001;76:79–86. doi: 10.1016/s0168-1702(01)00256-8. [DOI] [PubMed] [Google Scholar]
- 32.Tsunoda I, Iwasaki Y, Terunuma H, Sako K, Ohara Y. A comparative study of acute and chronic diseases induced by two subgroups of Theiler's murine encephalomyelitis virus. Acta Neuropathol. (Berl) 1996;91:595–602. doi: 10.1007/s004010050472. [DOI] [PubMed] [Google Scholar]
- 33.Carlson NG, Hill KE, Tsunoda I, Fujinami RS, Rose JW. The pathologic role for COX-2 in apoptotic oligodendrocytes in virus induced demyelinating disease: implications for multiple sclerosis. J. Neuroimmunol. 2006;174:21–31. doi: 10.1016/j.jneuroim.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Richer MJ, Poffenberger MC, Horwitz MS. Early inflammatory responses direct chronic autoimmunity development in the heart following coxsackievirus infection. Future Virol. 2007;2:283–291. [Google Scholar]
- 35.Baj A, Monaco S, Zanusso G, Dall'ora E, Bertolasi L, Toniolo A. Virology of the post-polio syndrome. Future Virol. 2007;2:183–192. [Google Scholar]
- 36.Brahic M, Bureau J-F, Michiels T. The genetics of the persistent infection and demyelinating disease caused by Theiler's virus. Annu. Rev. Microbiol. 2005;59:279–298. doi: 10.1146/annurev.micro.59.030804.121242. [DOI] [PubMed] [Google Scholar]
- 37.Oleszak EL, Chang JR, Friedman H, Katsetos CD, Platsoucas CD. Theiler's virus infection: a model for multiple sclerosis. Clin. Microbiol. Rev. 2004;17:174–207. doi: 10.1128/CMR.17.1.174-207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tsunoda I, Fujinami RS. TMEV and neuroantigens: myelin genes and proteins, molecular mimicry, epitope spreading, and autoantibody-mediated remyelination. In: Lavi E, Constantinescu CS, editors. Experimental Models of Multiple Sclerosis. Springer; New York, USA: 2005. pp. 593–616. [Google Scholar]
- 39.Tsunoda I, Kuang L-Q, Kobayashi-Warren M, Fujinami RS. Central nervous system pathology caused by autoreactive CD8+ T cell clones following virus infection. J. Virol. 2005;79:14640–14646. doi: 10.1128/JVI.79.23.14640-14646.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsunoda I, Libbey JE, Kobayashi-Warren M, Fujinami RS. IFN-γ production and astrocyte recognition by autoreactive T cells induced by Theiler's virus infection: role of viral strains and capsid proteins. J. Neuroimmunol. 2006;172:85–93. doi: 10.1016/j.jneuroim.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 41.Baida G, Popko B, Wollmann RL, et al. A subgenomic segment of Theiler's murine encephalomyelitis virus RNA causes demyelination. J. Virol. 2008;82:5879–5886. doi: 10.1128/JVI.02432-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42■.Tsunoda I, Kuang L-Q, Libbey JE, Fujinami RS. Axonal injury heralds virus-induced demyelination. Am. J. Pathol. 2003;162:1259–1269. doi: 10.1016/S0002-9440(10)63922-3. First evidence to support the ‘Inside-Out’ model in MS pathogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ulrich R, Seeliger F, Kreutzer M, Germann P-G, Baumgärtner W. Limited remyelination in Theiler's murine encephalomyelitis due to insufficient oligodendroglial differentiation of nerve/glial antigen 2 (NG2)-positive putative oligodendroglial progenitor cells. Neuropathol. Appl. Neurobiol. 2008 doi: 10.1111/j.1365-2990.2008.00956.x. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 44.Dandekar AA, Wu GF, Pewe L, Perlman S. Axonal damage is T cell mediated and occurs concomitantly with demyelination in mice infected with a neurotropic coronavirus. J. Virol. 2001;75:6115–6120. doi: 10.1128/JVI.75.13.6115-6120.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mathey EK, Derfuss T, Storch MK, et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J. Exp. Med. 2007;204:2363–2372. doi: 10.1084/jem.20071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brunn A, Utermöhlen O, Carstov M, et al. CD4 T cells mediate axonal damage and spinal cord motor neuron apoptosis in murine P0106-125-induced experimental autoimmune neuritis. Am. J. Pathol. 2008;173:93–105. doi: 10.2353/ajpath.2008.071101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson R, Harting E, Frey MS, Leibowitz JL, Miranda RC. Theiler's murine encephalomyelitis virus induces rapid necrosis and delayed apoptosis in myelinated mouse cerebellar explant cultures. Brain Res. 2000;868:259–267. doi: 10.1016/s0006-8993(00)02338-6. [DOI] [PubMed] [Google Scholar]
- 48.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 49.Coleman MP, Perry VH. Axon pathology in neurological disease: a neglected therapeutic target. Trends Neurosci. 2002;25:532–537. doi: 10.1016/s0166-2236(02)02255-5. [DOI] [PubMed] [Google Scholar]
- 50.Perry VH, Lunn ER, Brown MC, Cahusac S, Gordon S. Evidence that the rate of Wallerian degeneration is controlled by a single autosomal dominant gene. Eur. J. Neurosci. 1990;2:408–413. doi: 10.1111/j.1460-9568.1990.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 51.Ludwin SK, Bisby MA. Delayed wallerian degeneration in the central nervous system of Ola mice: an ultrastructural study. J. Neurol. Sci. 1992;109:140–147. doi: 10.1016/0022-510x(92)90160-m. [DOI] [PubMed] [Google Scholar]
- 52.Glass JD, Griffin JW. Neurofilament redistribution in transected nerves: evidence for bidirectional transport of neurofilaments. J. Neurosci. 1991;11:3146–3154. doi: 10.1523/JNEUROSCI.11-10-03146.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glass JD, Griffin JW. Retrograde transport of radiolabeled cytoskeletal proteins in transected nerves. J. Neurosci. 1994;14:3915–3921. doi: 10.1523/JNEUROSCI.14-06-03915.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watson DF, Glass JD, Griffin JW. Redistribution of cytoskeletal proteins in mammalian axons disconnected from their cell bodies. J. Neurosci. 1993;13:4354–4360. doi: 10.1523/JNEUROSCI.13-10-04354.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mack TGA, Reiner M, Beirowski B, et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 56.Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi: 10.1126/science.1098014. [DOI] [PubMed] [Google Scholar]
- 57.Tsunoda I, Wada Y, Libbey JE, Cannon TS, Whitby FG, Fujinami RS. Prolonged gray matter disease without demyelination caused by Theiler's murine encephalomyelitis virus with a mutation in VP2 puff B. J. Virol. 2001;75:7494–7505. doi: 10.1128/JVI.75.16.7494-7505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burleson FG, Chambers TM, Wiedbrauk DL. Virology. A Laboratory Manual. Academic Press, Inc.; San Diego, USA: 1992. Introduction to quantal virus assays; pp. 53–57. [Google Scholar]
- 59.Kaneko S, Wang J, Kaneko M, et al. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J. Neurosci. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60■■.Tsunoda I, Tanaka T, Terry EJ, Fujinami RS. Contrasting roles for axonal degeneration in an autoimmune versus viral model for multiple sclerosis: when can axonal injury be beneficial? Am. J. Pathol. 2007;170:214–226. doi: 10.2353/ajpath.2007.060683. Describes a potentially beneficial role for axonal degeneration in neurotropic virus infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinat C, Jarousse N, Prévost M-C, Brahic M. The GDVII strain of Theiler's virus spreads via axonal transport. J. Virol. 1999;73:6093–6098. doi: 10.1128/jvi.73.7.6093-6098.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roussarie J-P, Ruffié C, Edgar JM, Griffiths I, Brahic M. Axon myelin transfer of a non-enveloped virus. PLoS. ONE. 2007;2:e1331. doi: 10.1371/journal.pone.0001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lipton HL. Persistent Theiler's murine encephalomyelitis virus infection in mice depends on plaque size. J. Gen. Virol. 1980;46:169–177. doi: 10.1099/0022-1317-46-1-169. [DOI] [PubMed] [Google Scholar]
- 64.Jelachich ML, Reddi HV, Trottier MD, Schlitt BP, Lipton HL. Susceptibility of peritoneal macrophages to infection by Theiler's virus. Virus Res. 2004;104:123–127. doi: 10.1016/j.virusres.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 65.Guest ST, Roos RP. Molecular determinants of TMEV pathogenesis. In: Lavi E, Constantinescu CS, editors. Experimental Models of Multiple Sclerosis. Springer; New York, USA: 2005. pp. 673–683. [Google Scholar]
- 66.Fairn GD, Grinstein S. A one-sided signal. Science. 2008;320:458–460. doi: 10.1126/science.1158173. [DOI] [PubMed] [Google Scholar]
- 67.Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 68.Tsunoda I, Libbey JE, Fujinami RS. TGF-β1 suppresses T cell infiltration and VP2 puff B mutation enhances apoptosis in acute polioencephalitis induced by Theiler's virus. J. Neuroimmunol. 2007;190:80–89. doi: 10.1016/j.jneuroim.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69■.Kono H, Rock KL. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 2008;8:279–289. doi: 10.1038/nri2215. Excellent review on damage-associated molecular patterns, with simplified figures and comprehensive tables. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tsunoda I, Libbey JE, Kuang L-Q, Terry EJ, Fujinami RS. Massive apoptosis in lymphoid organs in animal models for primary and secondary progressive multiple sclerosis. Am. J. Pathol. 2005;167:1631–1646. doi: 10.1016/S0002-9440(10)61247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tsunoda I, Terry EJ, Marble BJ, Lazarides E, Woods C, Fujinami RS. Modulation of experimental autoimmune encephalomyelitis by VLA-2 blockade. Brain Pathol. 2007;17:45–55. doi: 10.1111/j.1750-3639.2006.00042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tsunoda I, Tanaka T, Saijoh Y, Fujinami RS. Targeting inflammatory demyelinating lesions to sites of Wallerian degeneration. Am. J. Pathol. 2007;171:1563–1575. doi: 10.2353/ajpath.2007.070147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Konno H, Yamamoto T, Suzuki H, et al. Targeting of adoptively transferred experimental allergic encephalitis lesion at the sites of wallerian degeneration. Acta Neuropathol. (Berl) 1990;80:521–526. doi: 10.1007/BF00294613. [DOI] [PubMed] [Google Scholar]
- 74.Low LK, Cheng H-J. A little nip and tuck: axon refinement during development and axonal injury. Curr. Opin. Neurobiol. 2005;15:549–556. doi: 10.1016/j.conb.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 75.Malawista SE, De Boisfleury Chevance A. The cytokineplast: purified, stable, and functional motile machinery from human blood polymorphonuclear leukocytes. J. Cell Biol. 1982;95:960–973. doi: 10.1083/jcb.95.3.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vohra BPS, Groshong JS, Zayas R, Wollmann RL, Gomez CM. Activation of apoptotic pathways at muscle fiber synapses is circumscribed and reversible in a slow-channel syndrome model. Neurobiol. Dis. 2006;23:462–470. doi: 10.1016/j.nbd.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 77.Silva A, Wang Q, Wang M, Ravula SK, Glass JD. Evidence for direct axonal toxicity in vincristine neuropathy. J. Peripher. Nerv. Syst. 2006;11:211–216. doi: 10.1111/j.1529-8027.2006.0090.x. [DOI] [PubMed] [Google Scholar]
- 78.Deckwerth TL, Johnson EM., Jr Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis) Dev. Biol. 1994;165:63–72. doi: 10.1006/dbio.1994.1234. [DOI] [PubMed] [Google Scholar]
- 79■.Nickells RW. Ganglion cell death in glaucoma: from mice to men. Vet. Ophthalmol. 2007;10(Suppl 1):88–94. doi: 10.1111/j.1463-5224.2007.00564.x. Reviews compartmentalized self-destruction of retinal ganglion cells in animal models for glaucoma. [DOI] [PubMed] [Google Scholar]
- 80■■.Whitmore AV, Libby RT, John SWM. Glaucoma: thinking in new ways – a role for autonomous axonal self-destruction and other compartmentalised processes? Prog. Retin. Eye Res. 2005;24:639–662. doi: 10.1016/j.preteyeres.2005.04.004. Comprehensive review on compartmentalized self-destruction in retinal ganglion cells in glaucoma. [DOI] [PubMed] [Google Scholar]
- 81.Whitmore AV, Lindsten T, Raff MC, Thompson CB. The proapoptotic proteins Bax and Bak are not involved in Wallerian degeneration. Cell Death. Differ. 2003;10:260–261. doi: 10.1038/sj.cdd.4401147. [DOI] [PubMed] [Google Scholar]
- 82.Wishart TM, Parson SH, Gillingwater TH. Synaptic vulnerability in neurodegenerative disease. J. Neuropathol. Exp. Neurol. 2006;65:733–739. doi: 10.1097/01.jnen.0000228202.35163.c4. [DOI] [PubMed] [Google Scholar]
- 83.Gillingwater TH, Ingham CA, Parry KE, et al. Delayed synaptic degeneration in the CNS of Wlds mice after cortical lesion. Brain. 2006;129:1546–1556. doi: 10.1093/brain/awl101. [DOI] [PubMed] [Google Scholar]
- 84.Ishimaru H, Casamenti F, Uéda K, Maruyama Y, Pepeu G. Changes in presynaptic proteins, SNAP-25 and synaptophysin, in the hippocampal CA1 area in ischemic gerbils. Brain Res. 2001;903:94–101. doi: 10.1016/s0006-8993(01)02439-8. [DOI] [PubMed] [Google Scholar]
- 85.Zhu B, Luo L, Moore GRW, Paty DW, Cynader MS. Dendritic and synaptic pathology in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2003;162:1639–1650. doi: 10.1016/S0002-9440(10)64298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chiesa R, Piccardo P, Dossena S, et al. Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc. Natl Acad. Sci. USA. 2005;102:238–243. doi: 10.1073/pnas.0406173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stevens B, Allen NJ, Vazquez LE, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 88.Fourgeaud L, Boulanger LM. Synapse remodeling, compliments of the complement system. Cell. 2007;131:1034–1036. doi: 10.1016/j.cell.2007.11.031. [DOI] [PubMed] [Google Scholar]
- 89.de la Torre JC, Mallory M, Brot M, et al. Viral persistence in neurons alters synaptic plasticity and cognitive functions without destruction of brain cells. Virology. 1996;220:508–515. doi: 10.1006/viro.1996.0340. [DOI] [PubMed] [Google Scholar]
- 90.Gonzalez-Dunia D, Watanabe M, Syan S, Mallory M, Masliah E, de la Torre JC. Synaptic pathology in Borna disease virus persistent infection. J. Virol. 2000;74:3441–3448. doi: 10.1128/jvi.74.8.3441-3448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shahar A, Frankel G, David Y, Friedmann A. In vitro cytotoxicity and demyelination induced by Theiler viruses in cultures of spinal cord slices. J. Neurosci. Res. 1986;16:671–681. doi: 10.1002/jnr.490160408. [DOI] [PubMed] [Google Scholar]
- 92.Tsunoda I, Fujinami RS. Neuropathogenesis of Theiler's murine encephalomyelitis virus infection. In: Zhang X, editor. Encyclopedia of Bioscience, “RNA viruses and CNS diseases: Mechanisms Pathogenesis and Persistence”. Frontiers in Bioscience; 2008. In Press. [Google Scholar]
- 93.Melli G, Keswani SC, Fischer A, Chen W, Höke A. Spatially distinct and functionally independent mechanisms of axonal degeneration in a model of HIV-associated sensory neuropathy. Brain. 2006;129:1330–1338. doi: 10.1093/brain/awl058. [DOI] [PubMed] [Google Scholar]
- 94.Lucchinetti CF, Parisi J, Bruck W. The pathology of multiple sclerosis. Neurol. Clin. 2005;23:77–105. doi: 10.1016/j.ncl.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 95.Kapoor R, Davies M, Blaker PA, Hall SM, Smith KJ. Blockers of sodium and calcium entry protect axons from nitric oxide-mediated degeneration. Ann. Neurol. 2003;53:174–180. doi: 10.1002/ana.10443. [DOI] [PubMed] [Google Scholar]
- 96.Shindler KS, Ventura E, Rex TS, Elliott P, Rostami A. SIRT1 activation confers neuroprotection in experimental optic neuritis. Invest. Ophthalmol. Vis. Sci. 2007;48:3602–3609. doi: 10.1167/iovs.07-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Döhner K, Sodeik B. The role of the cytoskeleton during viral infection. Curr. Top. Microbiol. Immunol. 2004;285:67–108. doi: 10.1007/3-540-26764-6_3. [DOI] [PubMed] [Google Scholar]
- 98.Li Y, Field PM, Raisman G. Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science. 1997;277:2000–2002. doi: 10.1126/science.277.5334.2000. [DOI] [PubMed] [Google Scholar]
- 99.Schnell L, Schneider R, Kolbeck R, Barde Y-A, Schwab ME. Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature. 1994;367:170–173. doi: 10.1038/367170a0. [DOI] [PubMed] [Google Scholar]