

Summary
The PPAR-gamma (PPAR-γ) activating thiazolidinedione (TZD) medications are a class of drugs used to improve lipid and glucose metabolism in type-2 diabetes. In addition to their known insulin sensitization action, these drugs have been shown to suppress tumor development in several in vitro and in vivo models. Among the proposed mechanisms for the anti-tumor effects of TZDs, apoptosis induction, cell cycle arrest, and differentiation have been extensively reported. Interestingly, some of the observed anti-tumor effects are independent of PPAR-γ activation. The following review will discuss studies employing TZDs as anti-cancer therapies for the most common types of cancers including, lung, breast, and colon and will explore the principal PPAR-γ-dependent and -independent mechanisms by which TZDs exert their anti-tumor effects.
Keywords: Apoptosis, breast cancer, colon cancer, growth/cell cycle arrest mechanism of action in neoplasia, PPAR-γ, tumor suppression, TZDs, lung cancer
I. Introduction
Thiazolidinediones (TZDs) are a class of antidiabetic drugs which include pioglitazone, rosiglitazone, ciglitazone and troglitazone, although troglitazone was removed from the market in 2000 because of hepatoxicity. These drugs improve insulin sensitivity through mechanisms that have yet to be clearly defined and therefore, constitute an area of active investigation. TZDs bind with high affinity to the PPAR-gamma (PPAR-γ) subtype of peroxisome-proliferator activated receptors (PPARs). PPARs are members of the steroid receptor superfamily of ligand-activated transcription factors. Upon activation, either by synthetic ligands such as TZDs, or endogenous ligands such as natural lipophilic ligands (e.g. fatty acids), PPAR-γ forms a heterodimer with the retinoid X receptor (RXR) and binds to PPAR response elements (PPRE) that regulate the transcription of select target genes. Although more than seventy PPAR target genes with PPREs have been identified (Yu et al, 2005), it is likely that this list of PPAR regulated genes will continue to grow. Further, the complexity of PPAR-γ -regulated gene expression is enhanced by various co-activators and co-repressors that are recruited to the transcriptional complex by the activated PPAR-γ heterodimer.
The pattern of PPAR-γ expression is relatively well-characterized. Two PPAR-γ isoforms exist that are derived from the alternate promoters, PPAR-γ1 and PPAR- γ2. The PPAR-γ2 isoform is 30 amino acids longer than PPAR-γ1 (Mueller et al, 2002) and is less abundant. PPAR-γ2 is predominantly expressed in adipose tissue where it exerts pleiotropic effects on metabolism, insulin sensitization, and inflammation. PPAR-γ2 is also expressed in vascular endothelium, suggesting a role for this protein in vascular biology as well as in alveolar macrophages (Gervois et al, 2007; Staels et al, 1998; Standiford et al, 2005). In contrast, PPAR-γ1 is expressed in a broad variety of tissues including large intestine, kidney, liver (Fajas et al, 1997), and particularly relevant to this article, PPAR-γ has been detected in cancer cells (Vamecq and Latruffe, 1999). Several reports have demonstrated that PPAR-γ activation has anti-cancer properties. For example, TZDs suppress tumor development in several animal models, and PPAR-γ activation arrests malignant cell growth (Sato et al, 2000; Takashima et al, 2001). In addition, treatment of cancer cells with PPAR-γ-activating TZDs induces cell differentiation and apoptosis (Lu et al, 2005a). These events are depicted schematically in Figure 1.
Figure 1. Activation of PPAR-γ by TZDs causes apoptosis, growth arrest, and differentiation in the cancer cell.
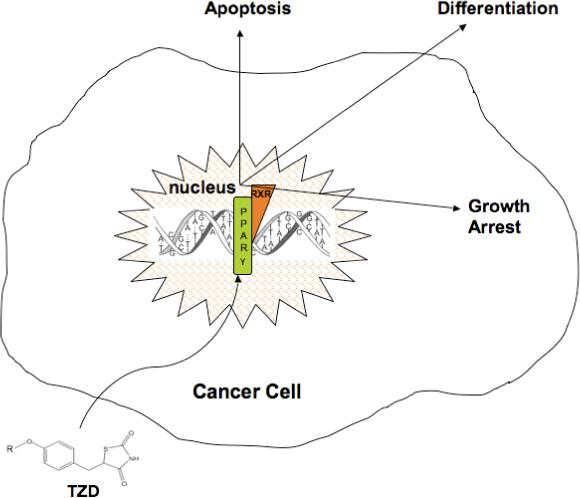
TZDs activate PPAR-γ, resulting in various anti-neoplastic effects in the cancer cell including apoptosis, growth arrest, and differentiation.
Investigation into the molecular mechanisms that underlie PPAR-γ-induced anti-cancer effects constitutes an area of active research. Nevertheless, the role of PPAR-γ in carcinogenesis remains controversial due in part to anti-neoplastic effects of TZDs that are independent of PPAR-γ. This review will examine current research evaluating TZDs as anti-tumor compounds in lung, breast and colon carcinoma, 3 of the most common cancers in the U.S., and will describe the proposed mechanisms by which TZDs exert their anti-cancer properties.
II. TZDs and lung cancer
Non small cell lung carcinoma (NSCLC), the most common cancer in the U.S. is associated with a poor prognosis, indicating that novel therapeutic approaches are urgently needed. In a recent retrospective database analysis from 10 Veterans Affairs medical centers, lung cancer risk among TZD users compared with nonusers was reduced by 33%, after adjustment for confounding interactions (Govindarajan et al, 2007). Although clinical data supporting the efficacy of TZDs in lung cancer is limited, in vitro studies as well as reports in experimental animals support this concept. In vitro studies available supporting the efficacy of TZDs in lung cancer are numerous (Tsubouchi et al, 2000; Satoh et al, 2002; Han et al, 2005). For example, the proliferation of A549 lung cancer cells was significantly inhibited by ciglitazone in a dose- and time-dependent manner both in vivo and in vitro, and PPAR-γ expression was markedly upregulated by ciglitazone treatment (Zhang et al, 2006). Troglitazone induced PPAR-γ expression and apoptosis in two human lung cancer cell lines, but not in normal cells (Li et al, 2005). These results suggest the potential for TZDs to target malignant cells without affecting normal cells (the goal of anti-cancer chemotherapy). In nude mice, direct injection of ciglitazone into A549-induced tumors suppressed the rate of tumor growth by 36% (Zhang et al, 2006).
While the modest anti-neoplastic effects of TZDs may preclude their use as monotherapy for treatment of lung cancer, combining TZDs with other anti-neoplastic agents could potentially enhance therapeutic efficacy. For example, the inhibitory effect of rosiglitazone on NSCLC cell growth was enhanced by the mammalian target of rapamycin (mTOR) inhibitor, rapamycin (Han and Roman, 2006). mTOR is a serine/threonine kinase that is activated by Akt and regulates protein synthesis, and rapamycin has been shown to inhibit cell growth by blocking the action of mTOR. In addition to the above study, rosiglitazone potentiated gefitinib's anti-proliferative effects by increasing the expression of the tumor suppressor, apoptotic gene, phosphatase and tensin homolog (PTEN) (Lee et al, 2006). The combination of the low-dose apoptosis inhibitor, MK886, ciglitazone, and 13-cis-retinoic acid, produced synergistic growth inhibition of lung cancer cells (A549 and H1299) suggesting that targeting PPAR-γ and retinoic acid action could be a promising approach to suppress lung cancer growth (Avis et al, 2005). In vitro synergistic anti-proliferative and apoptotic effects were also observed by combining a novel TZD with imatinib (Gleevec®) in various malignant cell lines (Zang et al, 2006). The opportunities to optimize anti-cancer efficacy with various chemotherapy permutations involving TZDs are numerous and suggest their potential benefit. Further, the experimental efficacy of low doses of these agents in these studies suggests potential clinical efficacy with reduced toxic side effects, a major concern in cancer chemotherapy. However, there are a few conflicting studies that suggest that TZD treatment may promote carcinogenesis or tumor development (Lefebvre et al, 1998; Saez et al, 2004). It may be the case that the specific tissue or type of cancer and its stage could contribute to the efficacy or failure of TZDs as anti-neoplastic agents. In support, rosiglitazone, ciglitazone, and prostaglandin J2 (15d-PGJ2) were all potent agonists of PPAR-γ transactivation in lung adenocarcinoma cell lines. However, these same ligands had no effect in squamous cell or large cell carcinomas of the lung (Allred and Kilgore, 2005). Prognostic indicators such as the ratio of PPAR-γ to RXR-α, for example, may be useful in predicting how cells may respond to specific combination treatments such as TZD and 9-cis-retinoic acid, an RXR-α ligand/agonist (Allred and Kilgore, 2005). In vivo and in vitro studies employing TZDs as anti-cancer agents (either alone or as combinatorial agents) for lung cancer are encouraging, however, clinical efficacy remains to be determined.
III. TZDs and breast cancer
Numerous studies have also examined the effects of TZDs in breast cancer. Breast cancer is the most frequent cancer in women and represents the second leading cause of cancer death in this population after lung cancer (Edwards et al, 2005; Al-Mansouri and Alokail, 2006). PPAR-γ is expressed in normal and malignant mammary epithelial cells, and TZDs suppress breast carcinoma proliferation in vitro and in experimental animal models (Suh et al, 1999; Nwankwo and Robbins, 2001; Jarrar and Baranova, 2007). Recently, PPAR-γ activation by conjugated linoleic acid was shown to have an anti-proliferative effect in MCF7 breast cancer cells (Bocca et al, 2007). Breast cancer cells, along with prostate cancer and melanoma cells were shown to undergo apoptosis with PPAR-γ ligands (Nunez et al, 2006). Additionally, PPAR-γ was shown to induce differentiation of malignant breast epithelial cells (Mueller et al, 1998). Interestingly, PPAR-γ acts as a tumor suppressor not only in breast, but also in skin and ovarian cancers (Nicol et al, 2004). However, PPAR-γ was also shown to act as a tumor promoter in breast carcinogenesis (Saez et al, 2004). This seemingly paradoxical outcome was proposed either to occur after a cancer initiation event had already progressed, or to possibly occur following PPAR-γ-mediated activation of a cytokine or growth factor such as TGF-β, which depending on the stage of breast cancer, can either be tumor suppressing or tumor promoting (Roberts and Wakefield, 2003). Nevertheless, the preponderance of evidence among studies examining TZDs in breast cancer cells suggests that PPAR-γ ligands inhibit proliferation and induce apoptosis both in vitro and in vivo. In addition, PPAR-γ ligands have been demonstrated to inhibit tumor angiogenesis and invasion in breast cancer (Fenner and Elstner, 2005).
Particularly relevant information is expected to come from human studies. In patient tissue samples, PPAR-γ immunoreactivity was significantly associated with improved clinical outcome in breast carcinoma patients by univariate analysis (Suzuki et al, 2006). In addition, a recent pilot trial examined short-term (2−6 weeks) treatment with rosiglitazone in 38 women with early-stage breast cancer. Rosiglitazone (8 mg/d), administered between the time of diagnostic biopsy and definitive surgery did not elicit significant effects on breast tumor cell proliferation analyzed as expression of Ki67, a marker of tumor growth and progression as well as proliferation (Yee et al 2007). In pretreatment tumors notable for nuclear expression of PPAR-γ as determined by immunohistochemistry, down-regulation of nuclear PPAR-γ expression occurred following rosiglitazone administration, contrary to what is expected. This TZD regimen was well tolerated and without serious adverse events (Yee et al, 2007). A prior trial reported in 2003 suggested that in patients with metastatic breast cancer, troglitizone failed to show any clinical benefits (Burstein et al, 2003). Overall, clinical studies are not encouraging. Nevertheless, more and longer term clinical studies are warranted to determine if the promising results obtained in pre-clinical and in-vitro studies can be extrapolated to humans.
IV. TZDs and colon cancer
In colon cancer, TZDs may also be of benefit (Kitamura et al, 1999). PPAR-γ mRNA and protein expression has been demonstrated in HT-29 colon cancer cells by RT-PCR and western blots, respectively (Sato et al, 2000; Chen et al, 2005), and PPAR-γ activation was associated with inhibition of cell growth through induction of apoptosis and suppression of the cell cycle (Takashima et al, 2001). Similarly both the synthetic TZD, pioglitazone, and the natural ligand, 15d-PGJ2, inhibited the proliferation of colon cancer cell lines in a dose-dependent manner that was reversed by the TZD antagonist GW9662 (Cerbone et al, 2007; Martinasso et al, 2007; Zhang et al, 2007).
In animal studies, a deficiency in intestinal PPAR-γ was associated with enhanced tumorigenicity in mouse small intestine and colon (McAlpine et al, 2006). Similarly, in mouse models of colon cancer, PPAR-γ agonists inhibited tumor growth or colon carcinogenesis (Yoshizumi et al, 2004; Chintharlapalli et al, 2006; Marin et al, 2006), although isolated studies suggest that PPAR-γ agonists induce colon tumors in mice (Yang et al, 2005c). Studies investigating PPAR-γ in human subjects with colon cancer are limited. In specimens from colon cancer patients, immunohistochemical analysis demonstrated a correlation between PPAR-γ and cell cycle-related molecules but no association was detected between PPAR-γ and patient survival (Theocharis et al, 2007). Furthermore, the risk reduction for colorectal cancers among diabetes patients taking TZDs did not reach statistical significance (Govindarajan et al, 2007), clearly indicating that more investigation will be required to establish a role for PPAR-γ in human colon carcinogenesis.
V. TZD mechanism of action in neoplasia
The molecular basis for the anti-tumor actions of TZDs remains incompletely elucidated. Recent studies employing gene expression analysis of TZD-treated tissues have provided new insights. For example, a recent study in ovarian cancer cells determined that ciglitazone upregulated genes predominantly associated with metabolic, differentiation and tumor suppressor pathways, whereas genes that were downregulated by ciglitazone were mainly involved in cell proliferation, cell cycle, cell organization, and steroid biosynthesis genes (Vignati et al, 2006). These findings are consistent with a genome-wide search for high-scoring PPREs in conserved regions of all human reference genes that found that genes with higher priority scores were those involved in cell cycle arrest, apoptosis and DNA damage response rather than those involved in fatty acid metabolism (Yu et al, 2005; Jarrar and Baranova, 2007). Thus, while distinct mechanisms for the anti-tumor actions of TZDs continue to be uncovered, a large proportion of the studies indicate that TZD-induced PPAR-γ activation promotes anti-tumor actions through apoptosis induction, differentiation, and growth (proliferation) arrest. The molecular evidence linking PPAR-γ activation and each of these mechanisms is discussed below.
A. Apoptosis
One of the main mechanisms of action by which TZDs act as anti-cancer agents involves the induction of apoptosis, an effect that complements the growth suppressive properties of TZDs. A schematic representation of these effects is shown in Figure 2. Numerous studies support the notion that PPAR-γ activation induces apoptosis and thus exerts anti-cancer effects (Clay et al, 1999). For instance, in lung cancer cells troglitazone induced apoptotic activity that was PPAR-γ-and ERK1/2-dependent (Li et al, 2006a). Troglitazone treatment reduced the anti-apoptotic protein, bcl-2, and caused nuclear accumulation and co-localization of PPAR-γ and ERK (Li et al, 2006a). Similarly, treating colon cancer cells with rosiglitazone caused apoptosis, detected with TUNEL staining and flow cytometry (Chen et al, 2005). Although the exact mechanisms by which apoptosis was induced in that study were not defined, bcl-2/bcl-x (anti-apoptotic proteins), p53, bad (pro-apoptotic proteins), and the transcription factor NF-κB were implicated in PPAR-γ ligand-induced apoptosis. Rosiglitazone treatment also increased PTEN protein levels which can lead to apoptosis through the negative regulation of Akt (Li et al, 1998). PTEN induction by PPAR ligands has also been reported to arrest tumor growth through PTEN's tumor suppressor effects. TZD treatment not only induced apoptosis and reduced the anti-apoptotic protein, bcl-2, in vitro, but also increased apoptotic bax expression in another study (Yang et al, 2005b). Further, TZDs reduced the anti-apoptotic protein, survivin (Figure 2), and caused dramatic sensitization of human breast cancer cells to apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and caspase activation (Lu et al, 2005b).
Figure 2. Mechanisms of TZD-induced apoptosis.
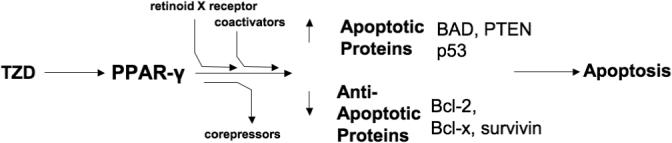
TZDs activate PPAR-γ, stimulating heterodimerization with the retinoid X receptor, recruitment of coactivators, and the dissociation of corepressors which ultimately causes apoptosis by decreasing anti-apoptotic proteins such as bcl-2/bcl-x and survivin, while increasing the levels of the pro-apoptotic proteins, p53, bad and phosphatase and tensin homolog (PTEN).
B. Growth/cell cycle arrest
In addition to apoptosis, PPAR-γ activation may reduce tumor development through the arrest of cancer cell proliferation, through effects on cell cycle checkpoints or growth factor inhibition. Several studies demonstrate that PPAR-γ activation suppresses proliferation rates in many cancer types (Weng et al, 2006). For instance, PPAR-γ activation not only caused apoptosis and reduced bcl-2 protein, but also inhibited the proliferation of lung cancer cells (Avis et al, 2005). The authors suggested that the observed apoptosis was mediated through the anti-proliferative effects of PPAR-γ-RXR interactions. In agreement, Elstner and colleagues showed that troglitazone combined with 9-cis-retinoic acid caused apoptosis in breast cancer cell lines, particularly in those in which bcl-2 expression levels were highest (Elstner et al, 2002).
One particularly well-known manner of suppressing proliferation rates involves cell cycle progression arrest. Cyclins are cell cycle regulators. Specifically, they are regulatory subunits of cell-cycle-specific kinases, and their activation is thought to regulate progress through the cell cycle. Cyclins are therefore potential oncogenes; and in fact, cyclin D1 overexpression and/or amplification are common features of several human cancers, thus promoting G1 phase progression (Musgrove et al, 1994; Chen and Harrison, 2005). Exposure to TZD for 24 h caused G0/G1 cell cycle arrest (Kawakami et al, 2002; Yang et al, 2005b). TZD treatment not only decreased protein levels of cyclin D1, but also reduced proliferating cell nuclear antigen, pRb, and Cdk4 and increased the cyclin-dependent kinase inhibitors p21 and p27, in a time-dependent manner (Yang et al, 2005b). Because the p21 and p27 kinase inhibitors inhibit CDK2/4 and CDK2 respectively, this can result in cell cycle arrest. In particular, increases in p21 expression levels have been attributed to Sp1 transcriptional activation (Han et al, 2004), as PPAR-γ has been shown to interact directly with transcription factors including Sp1 (Krey et al, 1995). In addition to p21, p18 was also demonstrated to be regulated by PPAR-γ (Morrison and Farmer, 1999). A schematic representation of some of these events is shown in Figure 3. These data are in agreement with other studies showing that levels of cyclins, specifically cyclin E, cyclin D2 and CDK 2, are decreased following TZD treatment (Zang et al, 2006). Lu and colleagues also demonstrated that TZD treatment profoundly reduced protein levels of cyclin D3 (Lu et al, 2005b). Conversely, p27 expression was increased and degradation was decreased in colon cancer cells following ciglitazone treatment (Chen and Harrison, 2005). Therefore, TZD downregulation or suppression of cell-cycle progression caused by cyclins represents an opportunity to halt uncontrolled cellular growth (Figure 3). Furthermore, ciglitazone inhibited A549 proliferation in mice, in a dose and time-dependent fashion and decreased the expression of cyclin D1. In the same study, it was further concluded that ciglitazone induced differentiation that was associated with cell cycle arrest (Zhang et al, 2006).
Figure 3. Mechanisms of TZD-induced growth arrest.
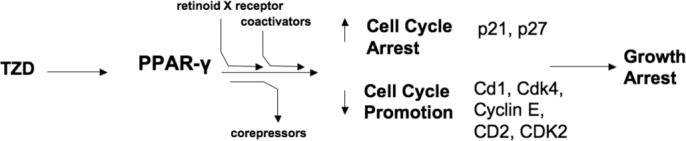
In addition to apoptosis, PPAR-γ activation may reduce tumor development through the arrest of cancer cell proliferation and effects on cell cycle checkpoints. TZD treatment activates PPAR-γ, stimulating heterodimerization with the retinoid X receptor, recruitment of coactivators, and the dissociation of corepressors resulting in decreased protein levels of activated cyclins that regulate progress through the cell cycle. These include: cyclin D1 (Cd1), as well as Cdk4, Cyclin E, CD2, and CDK2. Conversely, TZDs increase the cyclin-dependent kinase inhibitors p21 and p27 that can inhibit CDK2/4 and CDK2 respectively, ultimately causing cell cycle arrest.
C. Differentiation
Another mechanism by which PPAR-γ activation may exert anti-neoplastic effects is through the promotion of cellular differentiation, and early as well as recent evidence indicates that TZDs might have favorable effects in the treatment of a variety of tumors as differentiation-inducing agents (Tontonoz et al, 1997; Betz et al, 2005; Li et al, 2006b). PPAR-γ was demonstrated to induce differentiation in solid tumors both in vitro and in vivo (Kawamata et al, 2006). For example, TZD-induced differentiation of human cancer cells, defined as a shift toward a more steroidogenic phenotype, was mediated through activation of PPAR-γ-dependent pathways (Betz et al, 2005). TZD treatment in pancreatic cancer cells significantly inhibited growth through PPAR-dependent induction of pancreatic ductal differentiation without any increase in apoptosis (Ceni et al, 2005). In HT-29 colorectal cancer cells, TZD treatment inhibited growth and metastasis through differentiation-promoting effects (Yoshizumi et al, 2004). The ability of PPAR-γ activation to promote differentiation can be enhanced by the use of TZDs as combinatorial agents. For instance, the combination of the RXR agonist, bexarotene, with the PPAR-γ agonist, rosiglitazone, in colon cancer cells caused increased expression of the differentiation marker, CEA, while also decreasing cyclooxygenase-2 (COX-2) expression and prostaglandin-E2 (PGE2) synthesis (Cesario et al, 2006). These findings were later confirmed in vivo in a Moser xenograft tumor model, by the same group (Cesario et al, 2006). These reports and others suggest a potential role for combination regimens of RXR and PPAR-γ agonists in the treatment of colon and other cancers (Konopleva et al, 2004; Cesario et al, 2006). In cultured breast cancer cells, PPAR-γ ligands caused extensive lipid accumulation and changes in epithelial gene expression associated with a more differentiated, less malignant state (Mueller et al, 1998). In lung cancer cells, ciglitazone induced differentiation (Zhang et al, 2006), and PPAR-γ expression inhibited tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells and activating pathways that promoted a more differentiated epithelial phenotype (Bren-Mattison et al, 2005).
D. Additional considerations
The evaluation of other PPAR isoforms may also prove to be valuable in order to clarify the effect of PPARs on cellular differentiation. PPAR-β increased colonocyte differentiation as well as apoptosis in ligand-treated PPAR-β +/+ mice, whereas these effects were not found in PPAR-β −/− mice (Marin et al, 2006). Further, PPAR-β and PPAR-γ agonists altered mammary tumorigenesis and produced distinctive histopathologic patterns of tumor differentiation and development (Yin et al, 2005). Consistent with isoform-specific effects of PPARs, the specific TZD or PPAR ligand used may be of significant importance as differentiation effects seen with one ligand may not be observed in another. Clay et al, for example, reported that treating cells with 15dPGJ2 did not increase cellular differentiation, as had been seen in other neoplastic cells, but rather induced cellular events associated with programmed cell death or apoptosis (Clay et al, 1999).
VI. PPAR-γ-independent mechanisms of tumor suppression
In addition to PPAR-γ-dependent actions, TZDs demonstrate a number of important PPAR-γ-independent effects (Shiau et al, 2005; Han and Roman, 2006; Jarrar and Baranova, 2007). TZDs have been shown to stimulate the proteosomal degradation of cyclins D1 and D3 (Huang et al, 2005; Lu et al, 2005b), to block the G(1)-S transition through translation initiation inhibition (Palakurthi et al, 2001), and to scavenge toxic reactive oxygen species (ROS) (Inoue et al, 1997) through PPAR-γ-independent mechanisms. Additional PPAR-γ-independent actions of TZDs include the induction of cellular acidosis through inhibition of the Na+/H+ exchanger (de Dios et al, 2001; Turturro et al, 2004), calcium storage depletion (Palakurthi et al, 2001), and release of apoptotic factors from the mitochondria through the production of ROS (Pandhare et al, 2006). Other cited mechanisms by which TZDs exert anti-tumor effects in a PPAR-γ-independent manner include upregulation of PTEN/AMPK and down regulation of Akt/mTOR/p70S6 signaling cascades (Han and Roman, 2006). Because of these PPAR-γ-independent actions of TZDs, it must be emphasized that mechanistic investigations of individual PPAR-γ ligands in cancer therapy must account for both PPAR-γ-dependent and -independent pathways. The ultimate anti-cancer effect of a specific ligand may also depend on the target cell, the host's nutritional status, and the availability of co-activators or co-repressors in the cell of interest. Examples of ligand specific effects, independent of PPAR-γ activation, abound and further illustrate the need to thoroughly investigate PPAR-γ-independent effects. For example, many tumors are resistant to TRAIL, a promising agent that preferentially induces apoptosis in cancer cells, and Lu and colleagues suggest that both TRAIL sensitization and reduction in cyclin D3 protein levels induced by TZDs are likely PPAR-γ -independent because neither a dominant negative mutant of PPAR-γ nor PPAR-γ expression antagonized these effects (Lu et al, 2005). With the high frequency of dysfunctional apoptotic pathways in cancer cells, the degree to which a specific TZD can sensitize cancer cells to apoptosis-inducing agents, or the degree to which a TZD itself can induce apoptosis (independent of PPAR-γ status) will require further examination.
VII. Additional mechanisms of tumor suppression
In addition to apoptosis, differentiation induction, or growth arrest, other anti-tumor effects linked with PPAR-γ have been reported. Inhibition of invasion and metastasis is one such additional mechanism by which TZDs halt the cancer process. Metastasis was markedly promoted in diabetic and obese mice (Mori et al, 2006). Interestingly, obesity and diabetes have been associated with reduced PPAR-γ expression (Itoh et al, 1999; El Midaoui et al, 2006), suggesting that levels of PPAR-γ expression may be inversely related to metastatic potential. Angiogenesis is also an important mechanism in the progression and metastasis of tumors. Vascular endothelial growth factor (VEGF) is an important signaling protein involved in angiogenesis, and its overexpression has been associated with the process of metastasis. Pioglitazone not only normalized serum insulin and serum glucose but also serum VEGF levels in hyperinsulinemic, hyperglycemic rats (Yang et al, 2005a). Other studies have also confirmed the anti-angiogenic effects of PPAR-γ activation in tumors. These include studies reporting the inhibition of angiogenesis through suppression of chemokine production (Keshamouni et al, 2005), through PPAR-γ-mediated NO production and subsequent maxi-K channel opening (Kim and Cheon, 2006), as well as VEGF-mediated suppression of angiogenesis via VEGF promoter regulation (Peeters et al, 2005).
PPAR-γ activation has also been shown to counteract effects of the pro-inflammatory cytokines TNF-α and IL-1, and He and colleagues demonstrated that troglitazone inhibited insulin-like growth factor-I (IGF-I) through AMP-activated protein kinase (AMPK), an event which was thought to occur through a PPAR-γ-independent mechanism (He et al, 2006). In non-small cell lung carcinoma cells, PPAR-γ activation promoted ROS formation via proline oxidase induction, which ultimately caused cell death (Kim et al, 2007). And in breast cancer cells, where estrogen receptor status has a significant role in treatment outcome, Bonofiglio and colelagues showed that the estrogen receptor alpha binds to PPAR response elements and represses its transactivation. Physical interactions between the estrogen receptor and PPAR-γ that result in PPAR-γ signaling interference were also documented and thought to involve PI3K (Bonofiglio et al, 2005). These results are not entirely surprising, considering that it was recently shown that PPARs have a significant effect on many genes, including steroid biosynthesis genes (Vignati et al, 2006). The selected examples used to illustrate additional mechanisms of tumor suppression by TZD actions emphasize the diversity and complexity of the mechanisms which render TZDs anti-neoplastic
VIII. Future directions
The most convincing evidence demonstrating TZD efficacy in cancer therapy will undoubtedly come from clinical trials. Despite the numerous pre-clinical studies showing promise of TZDs as anti-cancer agents, currently available results of the few clinical trials that have already been performed are neither uniformly positive nor conclusive. For example, the effect of TZDs on the likelihood of colon, prostate or breast cancer development appears to be neutral, with the effect of TZDs being neither beneficial nor deleterious (Koro et al, 2007). A phase II trial examining rosiglitazone administration in patients with thyroid cancer found no relationship between PPAR-γ mRNA or protein in the neoplasms and radioiodine uptake status after rosiglitazone therapy (Kebebew et al, 2006). Furthermore, a diagnosis of cancer was significantly associated with TZD use, even after correcting for potential confounding variables (Ramos-Nino et al, 2007). However, the evaluation of the effectiveness of TZDs in cancer therapeutics must integrate potential differences among ligands within the same TZD class as identified in the pre-clinical studies discussed above. Additional clinical trials are currently ongoing (www.clinicaltrials.gov) that could clarify the role of TZDs in cancer, and results may not be available for some time. Cancer, as a complex, multifactorial disease will undoubtedly require the combination of agents in order to produce clinical efficacy. TZDs may provide more benefit when combined with other agents. The exploration of drug combinations at low doses may represent a promising strategy (Avis et al, 2005) as suggested by the potentiation of gefitinib by the co-administration of rosiglitazone (Yee et al, 2007).
It has also been suggested that in addition to cancer treatment, TZDs may have a role in cancer chemoprevention (Brown and Lippman, 2000; Badawi and Badr, 2002; Fan et al, 2003). However, other studies have provided evidence that PPAR-γ ligands may actually foster carcinogenesis. For example, the use of PPAR-γ ligands increased the development of colon tumors (Lefebvre et al, 1998). In another report, constitutive expression of PPAR-γ in the mammary glands accelerated tumor development (Saez et al, 2004), although it may be the case that expression of PPAR-γ without normal, physiological regulation may promote carcinogenesis. These conflicting results emphasize the need to increase the number and length of well-designed clinical studies that will carefully define the role of these promising agents.
IX. Conclusions
A rapidly expanding body of literature has examined the ability of PPAR-γ-activating TZD ligands to contribute to cancer therapy as evidenced by numerous in vitro and in vivo studies. The results of these studies, while mixed, have fostered sufficient interest to stimulate the investigation of TZDs in clinical trials. Additional studies of PPAR-γ ligands in combination with other agents or in chemopreventive strategies merit further consideration. Whether the results obtained from in vitro and preclinical studies investigating the anti-cancer potential of PPAR-γ ligands will extrapolate to efficacy in human trials, remains to be determined. Clinical trials of adequate power and duration are required to clarify the role that PPAR-γ activation may have in the treatment of cancer.
Acknowledgments
This work was supported by grants from the National Institutes of Health and the Veterans Affairs Research Service (AA16080, CMH; HL080543, JR).
Abbreviations
- AMPK
AMP-activated protein kinase
- COX-2
cyclooxygenase-2
- IGF-I
insulin-like growth factor-I
- mTOR
mammalian target of rapamycin
- PTEN
phosphatase and tensin homolog
- PGE2
prostaglandin-E2
- RXR
retinoid X receptor
- TZDs
thiazolidinediones
- VEGF
vascular endothelial growth factor
Biography
 From left to right: Jesse Roman, Carmelo Blanquicett, C. Michael Hart
From left to right: Jesse Roman, Carmelo Blanquicett, C. Michael Hart
References
- Allred CD, Kilgore MW. Selective activation of PPARgamma in breast, colon, lung cancer cell lines. Mol Cell Endocrinol. 2005;235:21–29. doi: 10.1016/j.mce.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Al-Mansouri LJ, Alokail MS. Molecular basis of breast cancer. Saudi Med J. 2006;27:9–16. [PubMed] [Google Scholar]
- Avis I, Martinez A, Tauler J, Zudaire E, Mayburd A, Abu-Ghazaleh R, Ondrey F, Mulshine JL. Inhibitors of the arachidonic acid pathway and peroxisome proliferator-activated receptor ligands have superadditive effects on lung cancer growth inhibition. Cancer Res. 2005;65:4181–4190. doi: 10.1158/0008-5472.CAN-04-3441. [DOI] [PubMed] [Google Scholar]
- Badawi AF, Badr MZ. Chemoprevention of breast cancer by targeting cyclooxygenase-2 and peroxisome proliferator-activated receptor-gamma (Review). Int J Oncol. 2002;20:1109–1122. [PubMed] [Google Scholar]
- Betz MJ, Shapiro I, Fassnacht M, Hahner S, Reincke M, Beuschlein F. Peroxisome proliferator-activated receptor-gamma agonists suppress adrenocortical tumor cell proliferation and induce differentiation. J Clin Endocrinol Metab. 2005;90:3886–3896. doi: 10.1210/jc.2004-1267. [DOI] [PubMed] [Google Scholar]
- Bocca C, Bozzo F, Francica S, Colombatto S, Miglietta A. Involvement of PPAR gamma and E-cadherin/beta-catenin pathway in the antiproliferative effect of conjugated linoleic acid in MCF-7 cells. Int J Cancer. 2007;121:248–256. doi: 10.1002/ijc.22646. [DOI] [PubMed] [Google Scholar]
- Bonofiglio D, Gabriele S, Aquila S, Catalano S, Gentile M, Middea E, Giordano F, Ando S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin Cancer Res. 2005;11:6139–6147. doi: 10.1158/1078-0432.CCR-04-2453. [DOI] [PubMed] [Google Scholar]
- Bren-Mattison Y, Van Putten V, Chan D, Winn R, Geraci MW, Nemenoff RA. Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC). Oncogene. 2005;24:1412–1422. doi: 10.1038/sj.onc.1208333. [DOI] [PubMed] [Google Scholar]
- Brown PH, Lippman SM. Chemoprevention of breast cancer. Breast Cancer Res Treat. 2000;62:1–17. doi: 10.1023/a:1006484604454. [DOI] [PubMed] [Google Scholar]
- Burstein HJ, Demetri GD, Mueller E, Sarraf P, Spiegelman BM, Winer EP. Use of the peroxisome proliferator-activated receptor (PPAR) gamma ligand troglitazone as treatment for refractory breast cancer: a phase II study. Breast Cancer Res Treat. 2003;79:391–397. doi: 10.1023/a:1024038127156. [DOI] [PubMed] [Google Scholar]
- Ceni E, Mello T, Tarocchi M, Crabb DW, Caldini A, Invernizzi P, Surrenti C, Milani S, Galli A. Antidiabetic thiazolidinediones induce ductal differentiation but not apoptosis in pancreatic cancer cells. World J Gastroenterol. 2005;11:1122–1130. doi: 10.3748/wjg.v11.i8.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerbone A, Toaldo C, Laurora S, Briatore F, Pizzimenti S, Dianzani MU, Ferretti C, Barrera G. 4-Hydroxynonenal and PPARgamma ligands affect proliferation, differentiation, apoptosis in colon cancer cells. Free Radic Biol Med. 2007;42:1661–1670. doi: 10.1016/j.freeradbiomed.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Cesario RM, Stone J, Yen WC, Bissonnette RP, Lamph WW. Differentiation and growth inhibition mediated via the RXR:PPARgamma heterodimer in colon cancer. Cancer Lett. 2006;240:225–233. doi: 10.1016/j.canlet.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Chen F, Harrison LE. Ciglitazone-induced cellular anti-proliferation increases p27kip1 protein levels through both increased transcriptional activity and inhibition of proteasome degradation. Cell Signal. 2005;17:809–816. doi: 10.1016/j.cellsig.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Chen WC, Lin MS, Bai X. Induction of apoptosis in colorectal cancer cells by peroxisome proliferators-activated receptor gamma activation up-regulating PTEN and inhibiting PI3K activity. Zhonghua Yi Xue Za Zhi. 2005;118:1477–1481. [PubMed] [Google Scholar]
- Chintharlapalli S, Papineni S, Safe S. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit colon cancer cell and tumor growth through PPARgamma-dependent and PPARgamma-independent pathways. Mol Cancer Ther. 2006;5:1362–1370. doi: 10.1158/1535-7163.MCT-06-0002. [DOI] [PubMed] [Google Scholar]
- Clay CE, Namen AM, Atsumi G, Willingham MC, High KP, Kute TE, Trimboli AJ, Fonteh AN, Dawson PA, Chilton FH. Influence of J series prostaglandins on apoptosis and tumorigenesis of breast cancer cells. Carcinogenesis. 1999;20:1905–1911. doi: 10.1093/carcin/20.10.1905. [DOI] [PubMed] [Google Scholar]
- de Dios ST, Hannan KM, Dilley RJ, Hill MA, Little PJ. Troglitazone, but not rosiglitazone, inhibits Na/H exchange activity and proliferation of macrovascular endothelial cells. J Diabetes Complications. 2001;15:120–127. doi: 10.1016/s1056-8727(01)00141-6. [DOI] [PubMed] [Google Scholar]
- Edwards BK, Brown ML, Wingo PA, Howe HL, Ward E, Ries LA, Schrag D, Jamison PM, Jemal A, Wu XC, Friedman C, Harlan L, Warren J, erson RN, Pickle LW. Annual report to the nation on the status of cancer, 1975−2002, featuring population-based trends in cancer treatment. J Natl Cancer Inst. 2005;97:1407–1427. doi: 10.1093/jnci/dji289. [DOI] [PubMed] [Google Scholar]
- El Midaoui A, Wu L, Wang R, de Champlain J. Modulation of cardiac and aortic peroxisome proliferator-activated receptor-gamma expression by oxidative stress in chronically glucose-fed rats. Am J Hypertens. 2006;19:407–412. doi: 10.1016/j.amjhyper.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Elstner E, Williamson EA, Zang C, Fritz J, Heber D, Fenner M, Possinger K, Koeffler HP. Novel therapeutic approach: ligands for PPARgamma and retinoid receptors induce apoptosis in bcl-2-positive human breast cancer cells. Breast Cancer Res Treat. 2002;74:155–165. doi: 10.1023/a:1016114026769. [DOI] [PubMed] [Google Scholar]
- Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM, Saladin R, Najib J, Laville M, Fruchart JC, Deeb S, Vidal-Puig A, Flier J, Briggs MR, Staels B, Vidal H, Auwerx J. The organization, promoter analysis, expression of the human PPARgamma gene. J Biol Chem. 1997;272:18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- Fan YY, Spencer TE, Wang N, Moyer MP, Chapkin RS. Chemopreventive n-3 fatty acids activate RXRalpha in colonocytes. Carcinogenesis. 2003;24:1541–1548. doi: 10.1093/carcin/bgg110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner MH, Elstner E. Peroxisome proliferator-activated receptor-gamma ligands for the treatment of breast cancer. Expert Opin Investig Drugs. 2005;14:557–568. doi: 10.1517/13543784.14.6.557. [DOI] [PubMed] [Google Scholar]
- Gervois P, Fruchart JC, Staels B. Drug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nutr Clin Pract. 2007;3:145–156. doi: 10.1038/ncpendmet0397. [DOI] [PubMed] [Google Scholar]
- Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER, Midathada MV, Kim L, Kim PJ, Owens RJ, Lang NP. Thiazolidinediones and the risk of lung, prostate, colon cancer in patients with diabetes. J Clin Oncol. 2007;25:1476–1481. doi: 10.1200/JCO.2006.07.2777. [DOI] [PubMed] [Google Scholar]
- Han S, Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther. 2006;5:430–437. doi: 10.1158/1535-7163.MCT-05-0347. [DOI] [PubMed] [Google Scholar]
- Han S, Rivera HN, Roman J. Peroxisome proliferator-activated receptor-gamma ligands inhibit alpha5 integrin gene transcription in non-small cell lung carcinoma cells. Am J Respir Cell Mol Biol. 2005;32:350–359. doi: 10.1165/rcmb.2004-0345OC. [DOI] [PubMed] [Google Scholar]
- Han S, Sidell N, Fisher PB, Roman J. Up-regulation of p21 gene expression by peroxisome proliferator-activated receptor gamma in human lung carcinoma cells. Clin Cancer Res. 2004;10:1911–1919. doi: 10.1158/1078-0432.ccr-03-0985. [DOI] [PubMed] [Google Scholar]
- He G, Sung YM, Digiovanni J, Fischer SM. Thiazolidinediones inhibit insulin-like growth factor-i-induced activation of p70S6 kinase and suppress insulin-like growth factor-I tumor-promoting activity. Cancer Res. 2006;66:1873–1878. doi: 10.1158/0008-5472.CAN-05-3111. [DOI] [PubMed] [Google Scholar]
- Huang JW, Shiau CW, Yang YT, Kulp SK, Chen KF, Brueggemeier RW, Shapiro CL, Chen CS. Peroxisome proliferator-activated receptor gamma-independent ablation of cyclin D1 by thiazolidinediones and their derivatives in breast cancer cells. Mol Pharmacol. 2005;67:1342–1348. doi: 10.1124/mol.104.007732. [DOI] [PubMed] [Google Scholar]
- Inoue I, Katayama S, Takahashi K, Negishi K, Miyazaki T, Sonoda M, Komoda T. Troglitazone has a scavenging effect on reactive oxygen species. Biochem Biophys Res Commun. 1997;235:113–116. doi: 10.1006/bbrc.1997.6512. [DOI] [PubMed] [Google Scholar]
- Itoh H, Doi K, Tanaka T, Fukunaga Y, Hosoda K, Inoue G, Nishimura H, Yoshimasa Y, Yamori Y, Nakao K. Hypertension and insulin resistance: role of peroxisome proliferator-activated receptor gamma. Clin Exp Pharmacol Physiol. 1999;26:558–560. doi: 10.1046/j.1440-1681.1999.03082.x. [DOI] [PubMed] [Google Scholar]
- Jarrar MH, Baranova A. PPARgamma activation by thiazolidinediones (TZDs) may modulate breast carcinoma outcome: the importance of interplay with TGFbeta signalling. J Cell Mol Med. 2007;11:71–87. doi: 10.1111/j.1582-4934.2007.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami S, Arai G, Hayashi T, Fujii Y, Xia G, Kageyama Y, Kihara K. PPARgamma ligands suppress proliferation of human urothelial basal cells in vitro. J Cell Physiol. 2002;191:310–319. doi: 10.1002/jcp.10099. [DOI] [PubMed] [Google Scholar]
- Kawamata H, Tachibana M, Fujimori T, Imai Y. Differentiation-inducing therapy for solid tumors. Curr Pharm Des. 2006;12:379–385. doi: 10.2174/138161206775201947. [DOI] [PubMed] [Google Scholar]
- Kebebew E, Peng M, Reiff E, Treseler P, Woeber KA, Clark OH, Greenspan FS, Lindsay S, Duh QY, Morita E. A phase II trial of rosiglitazone in patients with thyroglobulin-positive and radioiodine-negative differentiated thyroid cancer. Surgery. 2006;140:960–966. doi: 10.1016/j.surg.2006.07.038. discussion 966−967. [DOI] [PubMed] [Google Scholar]
- Keshamouni VG, Arenberg DA, Reddy RC, Newstead MJ, Anthwal S, Standiford TJ. PPAR-gamma activation inhibits angiogenesis by blocking ELR+CXC chemokine production in non-small cell lung cancer. Neoplasia (New York, NY) 2005;7:294–301. doi: 10.1593/neo.04601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Ahn JH, Cheon HG. Apoptotic action of PPAR(gamma) activation in human non-small cell lung cancer is mediated via proline oxidase-induced ROS formation. Mol Pharmacol. 2007;72:674–685. doi: 10.1124/mol.107.035584. [DOI] [PubMed] [Google Scholar]
- Kim KY, Cheon HG. Antiangiogenic effect of rosiglitazone is mediated via peroxisome proliferator-activated receptor gamma-activated maxi-K channel opening in human umbilical vein endothelial cells. J Biol Chem. 2006;281:13503–13512. doi: 10.1074/jbc.M510357200. [DOI] [PubMed] [Google Scholar]
- Kitamura S, Miyazaki Y, Shinomura Y, Kondo S, Kanayama S, Matsuzawa Y. Peroxisome proliferator-activated receptor gamma induces growth arrest and differentiation markers of human colon cancer cells. Jpn J Cancer Res. 1999;90:75–80. doi: 10.1111/j.1349-7006.1999.tb00668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Elstner E, McQueen TJ, Tsao T, Sudarikov A, Hu W, Schober WD, Wang RY, Chism D, Kornblau SM, Younes A, Collins SJ, Koeffler HP, Andreeff M. Peroxisome proliferator-activated receptor gamma and retinoid X receptor ligands are potent inducers of differentiation and apoptosis in leukemias. Mol Cancer Ther. 2004;3:1249–1262. [PubMed] [Google Scholar]
- Koro C, Barrett S, Qizilbash N. Cancer risks in thiazolidinedione users compared to other anti-diabetic agents. Pharmacoepidemiol Drug Saf. 2007;16:485–492. doi: 10.1002/pds.1352. [DOI] [PubMed] [Google Scholar]
- Krey G, Mahfoudi A, Wahli W. Functional interactions of peroxisome proliferator-activated receptor, retinoid-X receptor, Sp1 in the transcriptional regulation of the acylcoenzyme-A oxidase promoter. Mol Endocrinol. 1995;9:219–231. doi: 10.1210/mend.9.2.7776972. [DOI] [PubMed] [Google Scholar]
- Lee SY, Hur GY, Jung KH, Jung HC, Lee SY, Kim JH, Shin C, Shim JJ, In KH, Kang KH, Yoo SH. PPAR-gamma agonist increase gefitinib's antitumor activity through PTEN expression. Lung Cancer. 2006;51:297–301. doi: 10.1016/j.lungcan.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Lefebvre AM, Chen I, Desreumaux P, Najib J, Fruchart JC, Geboes K, Briggs M, Heyman R, Auwerx J. Activation of the peroxisome proliferator-activated receptor gamma promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nature medicine. 1998;4:1053–1057. doi: 10.1038/2036. [DOI] [PubMed] [Google Scholar]
- Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, Parsons R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- Li M, Lee TW, Mok TS, Warner TD, Yim AP, Chen GG. Activation of peroxisome proliferator-activated receptor-gamma by troglitazone (TGZ) inhibits human lung cell growth. J Cell Biochem. 2005;96:760–774. doi: 10.1002/jcb.20474. [DOI] [PubMed] [Google Scholar]
- Li M, Lee TW, Yim AP, Mok TS, Chen GG. Apoptosis induced by troglitazone is both peroxisome proliferator-activated receptor-gamma- and ERK-dependent in human non-small lung cancer cells. J Cell Physiol. 2006a;209:428–438. doi: 10.1002/jcp.20738. [DOI] [PubMed] [Google Scholar]
- Li MY, Lee TW, Yim AP, Chen GG. Function of PPARgamma and its ligands in lung cancer. Crit Rev Clin Lab Sci. 2006b;43:183–202. doi: 10.1080/10408360600552587. [DOI] [PubMed] [Google Scholar]
- Lu J, Imamura K, Nomura S, Mafune K, Nakajima A, Kadowaki T, Kubota N, Terauchi Y, Ishii G, Ochiai A, Esumi H, Kaminishi M. Chemopreventive effect of peroxisome proliferator-activated receptor gamma on gastric carcinogenesis in mice. Cancer Res. 2005a;65:4769–4774. doi: 10.1158/0008-5472.CAN-04-2293. [DOI] [PubMed] [Google Scholar]
- Lu M, Kwan T, Yu C, Chen F, Freedman B, Schafer JM, Lee EJ, Jameson JL, Jordan VC, Cryns VL. Peroxisome proliferator-activated receptor gamma agonists promote TRAIL-induced apoptosis by reducing survivin levels via cyclin D3 repression and cell cycle arrest. J Biol Chem. 2005b;280:6742–6751. doi: 10.1074/jbc.M411519200. [DOI] [PubMed] [Google Scholar]
- Marin HE, Peraza MA, Billin AN, Willson TM, Ward JM, Kennett MJ, Gonzalez FJ, Peters JM. Ligand activation of peroxisome proliferator-activated receptor beta inhibits colon carcinogenesis. Cancer Res. 2006;66:4394–4401. doi: 10.1158/0008-5472.CAN-05-4277. [DOI] [PubMed] [Google Scholar]
- Martinasso G, Oraldi M, Trombetta A, Maggiora M, Bertetto O, Canuto RA, Muzio G. Involvement of PPARs in Cell Proliferation and Apoptosis in Human Colon Cancer Specimens and in Normal and Cancer Cell Lines. PPAR Res. 2007;2007:93416. doi: 10.1155/2007/93416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlpine CA, Barak Y, Matise I, Cormier RT. Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ mice. Int J Cancer. 2006;119:2339–2346. doi: 10.1002/ijc.22115. [DOI] [PubMed] [Google Scholar]
- Mori A, Sakurai H, Choo MK, Obi R, Koizumi K, Yoshida C, Shimada Y, Saiki I. Severe pulmonary metastasis in obese and diabetic mice. Int J Cancer. 2006;119:2760–2767. doi: 10.1002/ijc.22248. [DOI] [PubMed] [Google Scholar]
- Morrison RF, Farmer SR. Role of PPARgamma in regulating a cascade expression of cyclin-dependent kinase inhibitors, p18(INK4c) and p21(Waf1/Cip1), during adipogenesis. J Biol Chem. 1999;274:17088–17097. doi: 10.1074/jbc.274.24.17088. [DOI] [PubMed] [Google Scholar]
- Mueller E, Drori S, Aiyer A, Yie J, Sarraf P, Chen H, Hauser S, Rosen ED, Ge K, Roeder RG, Spiegelman BM. Genetic analysis of adipogenesis through peroxisome proliferator-activated receptor gamma isoforms. J Biol Chem. 2002;277:41925–41930. doi: 10.1074/jbc.M206950200. [DOI] [PubMed] [Google Scholar]
- Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPAR gamma. Mol Cell. 1998;1:465–470. doi: 10.1016/s1097-2765(00)80047-7. [DOI] [PubMed] [Google Scholar]
- Musgrove EA, Lee CS, Buckley MF, Sutherland RL. Cyclin D1 induction in breast cancer cells shortens G1 and is sufficient for cells arrested in G1 to complete the cell cycle. Proc Natl Acad Sci U S A. 1994;91:8022–8026. doi: 10.1073/pnas.91.17.8022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol CJ, Yoon M, Ward JM, Yamashita M, Fukamachi K, Peters JM, Gonzalez FJ. PPARgamma influences susceptibility to DMBA-induced mammary, ovarian and skin carcinogenesis. Carcinogenesis. 2004;25:1747–1755. doi: 10.1093/carcin/bgh160. [DOI] [PubMed] [Google Scholar]
- Nunez NP, Liu H, Meadows GG. PPAR-gamma ligands and amino acid deprivation promote apoptosis of melanoma, prostate, breast cancer cells. Cancer Lett. 2006;236:133–141. doi: 10.1016/j.canlet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Nwankwo JO, Robbins ME. Peroxisome proliferator-activated receptor- gamma expression in human malignant and normal brain, breast and prostate-derived cells. Prostaglandins Leukot Essent Fatty Acids. 2001;64:241–245. doi: 10.1054/plef.2001.0266. [DOI] [PubMed] [Google Scholar]
- Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM, Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001;61:6213–6218. [PubMed] [Google Scholar]
- Pandhare J, Cooper SK, Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J Biol Chem. 2006;281:2044–2052. doi: 10.1074/jbc.M507867200. [DOI] [PubMed] [Google Scholar]
- Peeters LL, Vigne JL, Tee MK, Zhao D, Waite LL, Taylor RN. PPARgamma represses VEGF expression in human endometrial cells: implications for uterine angiogenesis. Angiogenesis. 2005;8:373–379. doi: 10.1007/s10456-005-9027-4. [DOI] [PubMed] [Google Scholar]
- Ramos-Nino ME, Maclean CD, Littenberg B. Association between cancer prevalence and use of thiazolidinediones (TZDs): results from the Vermont Diabetes Information System. BMC Med. 2007;5:17. doi: 10.1186/1741-7015-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez E, Rosenfeld J, Livolsi A, Olson P, Lombardo E, Nelson M, Banayo E, Cardiff RD, Izpisua-Belmonte JC, Evans RM. PPAR gamma signaling exacerbates mammary gland tumor development. Genes & development. 2004;18:528–540. doi: 10.1101/gad.1167804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Ishihara S, Kawashima K, Moriyama N, Suetsugu H, Kazumori H, Okuyama T, Rumi MA, Fukuda R, Nagasue N, Kinoshita Y. Expression of peroxisome proliferator-activated receptor (PPAR)gamma in gastric cancer and inhibitory effects of PPARgamma agonists. British journal of cancer. 2000;83:1394–1400. doi: 10.1054/bjoc.2000.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Toyoda M, Hoshino H, Monden T, Yamada M, Shimizu H, Miyamoto K, Mori M. Activation of peroxisome proliferator-activated receptor-gamma stimulates the growth arrest and DNA-damage inducible 153 gene in non-small cell lung carcinoma cells. Oncogene. 2002;21:2171–2180. doi: 10.1038/sj.onc.1205279. [DOI] [PubMed] [Google Scholar]
- Shiau CW, Yang CC, Kulp SK, Chen KF, Chen CS, Huang JW, Chen CS. Thiazolidenediones mediate apoptosis in prostate cancer cells in part through inhibition of Bcl-xL/Bcl-2 functions independently of PPARgamma. Cancer Res. 2005;65:1561–1569. doi: 10.1158/0008-5472.CAN-04-1677. [DOI] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, Merval R, Lebret M, Torra IP, Delerive P, Fadel A, Chinetti G, Fruchart JC, Najib J, Maclouf J, Tedgui A. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- Standiford TJ, Keshamouni VG, Reddy RC. Peroxisome proliferator-activated receptor-(gamma) as a regulator of lung inflammation and repair. Proc Am Thorac Soc. 2005;2:226–231. doi: 10.1513/pats.200501-010AC. [DOI] [PubMed] [Google Scholar]
- Suh N, Wang Y, Williams CR, Risingsong R, Gilmer T, Willson TM, Sporn MB. A new ligand for the peroxisome proliferator-activated receptor-gamma (PPAR-gamma), GW7845, inhibits rat mammary carcinogenesis. Cancer Res. 1999;59:5671–5673. [PubMed] [Google Scholar]
- Suzuki T, Hayashi S, Miki Y, Nakamura Y, Moriya T, Sugawara A, Ishida T, Ohuchi N, Sasano H. Peroxisome proliferator-activated receptor gamma in human breast carcinoma: a modulator of estrogenic actions. Endocr Relat Cancer. 2006;13:233–250. doi: 10.1677/erc.1.01075. [DOI] [PubMed] [Google Scholar]
- Takashima T, Fujiwara Y, Higuchi K, Arakawa T, Yano Y, Hasuma T, Otani S. PPAR-gamma ligands inhibit growth of human esophageal adenocarcinoma cells through induction of apoptosis, cell cycle arrest and reduction of ornithine decarboxylase activity. Int J Oncol. 2001;19:465–471. [PubMed] [Google Scholar]
- Theocharis S, Giaginis C, Parasi A, Margeli A, Kakisis J, Agapitos E, Kouraklis G. Expression of Peroxisome Proliferator-Activated Receptor-gamma in Colon Cancer: Correlation with Histopathological Parameters, Cell Cycle-Related Molecules, Patients' Survival. Dig Dis Sci. 2007;52:2305–2311. doi: 10.1007/s10620-007-9794-4. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, Brun RP, Mueller E, Altiok S, Oppenheim H, Evans RM, Spiegelman BM. Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor. Proc Natl Acad Sci U S A. 1997;94:237–241. doi: 10.1073/pnas.94.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubouchi Y, Sano H, Kawahito Y, Mukai S, Yamada R, Kohno M, Inoue K, Hla T, Kondo M. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-gamma agonists through induction of apoptosis. Biochem Biophys Res Commun. 2000;270:400–405. doi: 10.1006/bbrc.2000.2436. [DOI] [PubMed] [Google Scholar]
- Turturro F, Friday E, Fowler R, Surie D, Welbourne T. Troglitazone acts on cellular pH and DNA synthesis through a peroxisome proliferator-activated receptor gamma-independent mechanism in breast cancer-derived cell lines. Clin Cancer Res. 2004;10:7022–7030. doi: 10.1158/1078-0432.CCR-04-0879. [DOI] [PubMed] [Google Scholar]
- Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet. 1999;354:141–148. doi: 10.1016/S0140-6736(98)10364-1. [DOI] [PubMed] [Google Scholar]
- Vignati S, Albertini V, Rinaldi A, Kwee I, Riva C, Oldrini R, Capella C, Bertoni F, Carbone GM, Catapano CV. Cellular and molecular consequences of peroxisome proliferator-activated receptor-gamma activation in ovarian cancer cells. Neoplasia (New York, NY) 2006;8:851–861. doi: 10.1593/neo.06433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng JR, Chen CY, Pinzone JJ, Ringel MD, Chen CS. Beyond peroxisome proliferator-activated receptor gamma signaling: the multi-facets of the antitumor effect of thiazolidinediones. Endocr Relat Cancer. 2006;13:401–413. doi: 10.1677/erc.1.01182. [DOI] [PubMed] [Google Scholar]
- Yang B, Lin P, Carrick KM, McNulty JA, Clifton LG, Winegar DA, Strum JC, Stimpson SA, Pahel GL. PPARgamma agonists diminish serum VEGF elevation in diet-induced insulin resistant SD rats and ZDF rats. Biochem Biophys Res Commun. 2005a;334:176–182. doi: 10.1016/j.bbrc.2005.06.078. [DOI] [PubMed] [Google Scholar]
- Yang FG, Zhang ZW, Xin DQ, Shi CJ, Wu JP, Guo YL, Guan YF. Peroxisome proliferator-activated receptor gamma ligands induce cell cycle arrest and apoptosis in human renal carcinoma cell lines. Acta Pharmacol Sin. 2005b;26:753–761. doi: 10.1111/j.1745-7254.2005.00753.x. [DOI] [PubMed] [Google Scholar]
- Yang K, Fan KH, Lamprecht SA, Edelmann W, Kopelovich L, Kucherlapati R, Lipkin M. Peroxisome proliferator-activated receptor gamma agonist troglitazone induces colon tumors in normal C57BL/6J mice and enhances colonic carcinogenesis in Apc1638 N/+ Mlh1+/− double mutant mice. Int J Cancer. 2005c;116:495–499. doi: 10.1002/ijc.21018. [DOI] [PubMed] [Google Scholar]
- Yee LD, Williams N, Wen P, Young DC, Lester J, Johnson MV, Farrar WB, Walker MJ, Povoski SP, Suster S, Eng C. Pilot study of rosiglitazone therapy in women with breast cancer: effects of short-term therapy on tumor tissue and serum markers. Clin Cancer Res. 2007;13:246–252. doi: 10.1158/1078-0432.CCR-06-1947. [DOI] [PubMed] [Google Scholar]
- Yin Y, Russell RG, Dettin LE, Bai R, Wei ZL, Kozikowski AP, Kopelovich L, Glazer RI. Peroxisome proliferator-activated receptor delta and gamma agonists differentially alter tumor differentiation and progression during mammary carcinogenesis. Cancer Res. 2005;65:3950–3957. doi: 10.1158/0008-5472.CAN-04-3990. [DOI] [PubMed] [Google Scholar]
- Yoshizumi T, Ohta T, Ninomiya I, Terada I, Fushida S, Fujimura T, Nishimura G, Shimizu K, Yi S, Miwa K. Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, inhibits growth and metastasis of HT-29 human colon cancer cells through differentiation-promoting effects. Int J Oncol. 2004;25:631–639. [PubMed] [Google Scholar]
- Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J Biol Chem. 2005;280:13600–13605. doi: 10.1074/jbc.M409468200. [DOI] [PubMed] [Google Scholar]
- Zang C, Liu H, Waechter M, Eucker J, Bertz J, Possinger K, Koeffler HP, Elstner E. Dual PPARalpha/gamma ligand TZD18 either alone or in combination with imatinib inhibits proliferation and induces apoptosis of human CML cell lines. Cell Cycle. 2006;5:2237–2243. doi: 10.4161/cc.5.19.3259. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhang H, Xing L. Influence of ciglitazone on A549 cells growth in vitro and in vivo and mechanism. J Huazhong Univ Sci Technolog Med Sci. 2006;26:36–39. doi: 10.1007/BF02828033. [DOI] [PubMed] [Google Scholar]
- Zhang YQ, Tang XQ, Sun L, Dong L, Qin Y, Liu HQ, Xia H, Cao JG. Rosiglitazone enhances fluorouracil-induced apoptosis of HT-29 cells by activating peroxisome proliferator-activated receptor gamma. World J Gastroenterol. 2007;13:1534–1540. doi: 10.3748/wjg.v13.i10.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]