

Abstract
We report the synthesis of diinsininone (33), the aglycone of (±)-diinsinin (2). Thereby, we complete the first construction of a proanthocyanidin (PA) type-A compound incorporating a [3.3.1]-bicyclic ketal as its characteristic core. Our strategy utilizes a coupling between a benzopyrilium salt and a flavanone that proves applicable to other PA type-A compounds. During this undertaking, treatment of naringenin (9) with 2-iodoxybenzoic acid (IBX) followed by reductive work-up affords eriodictyol (10). This reactivity mirrors that of catechol hydroxylase (F3H) found in the flavonoid pathway. Other interesting transformations include the formation of flavonoids through an ortho-quinone methide (o-QM) cycloaddition–oxidation sequence and regioselective β-glycosidations of several unprotected flavanones suggesting a likely synthesis of 2 from the aglycone 33.
Keywords: Proanthocyanidin, [3.3.1]-Bicyclic ketal, Benzopyrilium, o-Quinone methide, Benzopyran, IBX, Catechol, Flavonoid
1. Introduction
Chemists should ponder probable biosynthetic pathways leading to their target, because few things are more humbling to one’s best-laid plans than nature’s simpler solution. Nature has been conducting and optimizing chemical experiments for millions of years; therefore, matching or exceeding nature’s accumulated wisdom is a daunting challenge. However, the precise sequence used by nature to assemble its products is rarely known. In these instances, synthetic chemists are empowered with the ability to illuminate nature’s hidden pathways by determining the reactivity inherent to its intermediates.
Consider the [3.3.1]-bicyclic compounds diinsininol (1) and diinsinin (2) shown in Figure 1. In nature’s quest to build these compounds in an efficient manner, it might fashion a single flavan component using genomics, and then cause this entity to dimerize, forming bonds i and ii, whereupon oxidases and lipases would complete its creation. By mimicking this purported pathway, we hoped to reveal some of nature’s more closely guarded secrets regarding chemoselectivity, diastereoselectivity and enantioselectivity during the assembly process leading to 1 and 2.
Figure 1.
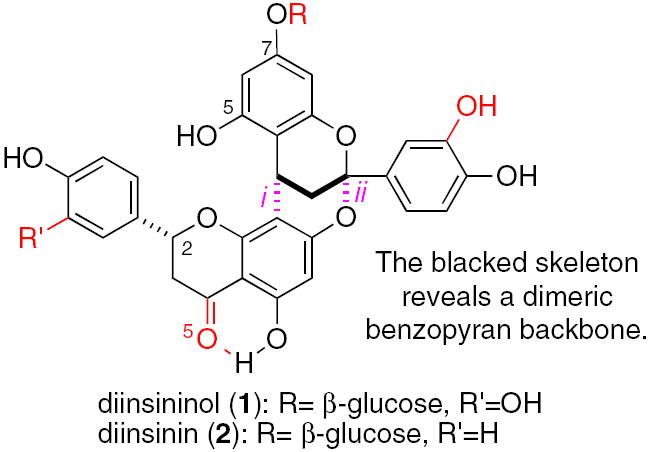
PA type-A structures, diinsininol (1) and diinsinin (2).
In 1996, the compounds 1 and 2 were discovered in the rhizome of Sarcophyte piriei Hutch, a parasitic plant that grows on the roots of the Acacia species of tropical shrubs and trees.1 A decoction of its tuber is used in Somalian folk medicine; an activity that led to their eventual isolation and evaluation.2 Diinsininol (1) and diinsinin (2) inhibit prostaglandin synthesis (IC50 values of 9.20 μM and 13.14 μM, respectively) and platelet activating factor-induced exocytosis (IC50 values of 49 μM and 39 μM, respectively). These biological activities are common among drugs that inhibit cyclooxygenase-2 (COX-2) and protect cells from tumor growth; tumors with high COX-2 levels are usually larger and metastasize faster than those without.3
2. Biological relevance
The flavonoid carbon skeleton encompasses a range of structural and biological diversity and is widely distributed throughout the plant kingdom.4 Because of its abundance, it often proves useful as a starting material for synthesis. Its structural diversity on the other hand enables evaluation of an assortment of pharmacological targets. More than 5000 flavonoids have been isolated varying in the hydroxylation, methoxylation, glycosylation, and acylation patterns of the carbon backbone.5 This arsenal of structures evolved as a defense to oxidative stress and hungry herbivores.6
Flavonoids protect organisms from oxidative stress by destroying reactive oxygen species that would have otherwise caused cell damage. It has long been known that foods high in antioxidants lower the risk of many diseases, including cancer, cataract, heart disease, and rheumatoid arthritis.7 The so-called ‘French Paradox’ stems from the observation that French people consuming as much as 1200 mg of flavonoids per liter of red wine with structures similar to 1 and 2 display lower incidences of coronary disease despite a higher intake of saturated fat.8-12
In addition to these suppositious medical benefits, compounds 1 and 2 resemble antihyperglycemic components found within cinnamon, which have been studied for their ability to potentiate insulin, maintain normal blood sugar levels, and assist in fat metabolism.13 Currently insulin resistance, which is referred to as metabolic syndrome X, affects 50 million people. Syndrome X is linked to an unhealthy lifestyle, genetic factors, and the progression to type-II diabetes.14 In 2004, several powerful antihyperglycemic compounds were identified in cinnamon and determined to be trimer and tetramers of PA type-A compounds displaying the characteristic [3.3.1]-bicyclic ketal core.15
3. Relevance of the speculative biosynthetic pathways
The biosyntheses of 1 and 2 initially follows the flavonoid pathway, which proceeds from phenylalanine through trans-cinnamate, chalcones, flavanones, anthocyanidins, to flavans. It is among the best studied of biosynthetic pathways in nature.16-18 The optically active compounds 1 and 2 suggest a double union between two products emerging from the flavonoid pathway. It is reasonable to speculate that an organism would utilize an identical path for these structurally related natural products. Paths A and B have been proposed (Scheme 1). In our minds, these proposed routes are profoundly different and suggest different consequences for reactivity and stereochemistry leading to the [3.3.1]-bicyclic ketal. Path B, which has been shown to afford the singly linked type-B PA structures, was proposed by Nonaka to yield the [3.3.1]-bicyclic ketal structure found in type-A PA compounds by an oxidative cyclization. A catechin type-B dimer has been transformed into the corresponding type-A structure with picrylhydrazyl radical and hydrogen peroxide.19 Path B appears to entail the chemoselective addition of a flavanone or flavan nucleophile to an electrophile arising from the para-quinone methide or its corresponding cation. This initial coupling should be inherently diastereoselective. However, if a Path B-oxidation sequence does result in the biosyntheses of 1 and 2, it is remarkable because the original stereocenters ultimately become compromised when carried to the optically active natural products. Path A, on the other hand, suggests that a benzopyrylium salt can undergo a net [3+3]-cycloaddition in an enantioselective fashion chaperoned by an unknown enzyme,20 or perhaps proceeds in a diastereoselective fashion with the directing stereochemistry again compromised when forming the final natural products.
Scheme 1.
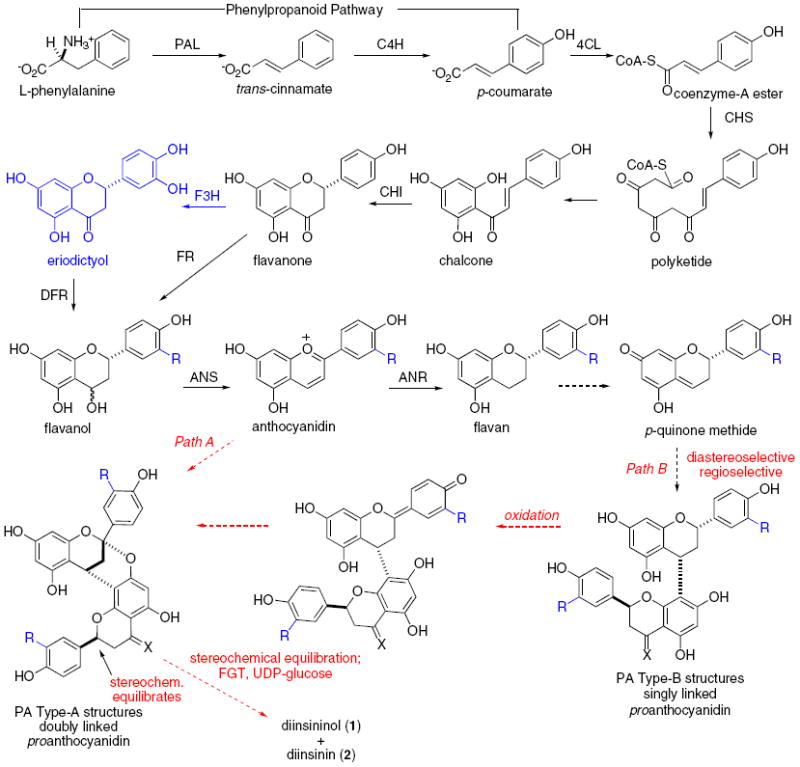
Known (solid arrows) and speculative (dashed arrows) biosynthetic pathways leading to 1 and 2. PAL: phenylalanine ammonia lyase; C4H: cinnamate 4-hydroxylase; 4CL: 4-coumarate: CoA ligase; CHS: chalcone synthase; CHI: chalcone flavanone isomerase; F3H: flavanone 3-hydroxylase; FR: flavanone reductase; DFR: dihydroxyflavanone reductase; ANS: anthocyanidin synthase; ANR: anthocyanidin reductase.
The oxidation state of the components at the time of coupling remains unclear as does the timing of the oxidation that distinguishes between the two natural products. Flavanone 3-hydroxylase (F3H) may catalyze hydroxylation of (2S)-flavanone to eriodictyol. A combination of di-hydroxylated and mono-hydroxylated materials leads to diinsininone (33), while a combination of di-hydroxylated systems provides diinsininolone (34). Alternatively, F3H or some derivative of this enzyme may establish divergence in oxidation states (catechol vs phenol) in the final or penultimate step of the biosynthesis. Similarly, glycosylation may occur as the final or penultimate step of the biosynthesis. Divergence among the pathways for similar components seems energetically wasteful for an organism.
4. Synthetic relevance
To the best of our knowledge, there are no previous synthetic endeavors addressing diinsininol (1), diinsinin (2), or similar type-A PA compounds in the literature. While there are numerous methods for the construction of chromans, the enantioselective construction of flavonoids is a surprisingly difficult task and it is difficult to improve upon their biosynthetic pathway.21 Recently, our group devised a strategy for the enantioselective assembly of non-natural flavonoids by a [4+2]-cycloaddition between an o-QM and a chiral enol ether. We utilized this process in the first total synthesis of the flavonoid (+)-mimosifoliol.22 We initially envisioned extending this strategy to the chroman functionality embedded within diinsininol (1) and diinsinin (2). The additional hydroxyl residue that distinguishes 1 from 2 could be introduced using our IBX oxidation–reduction sequence for the construction of o-quinols from phenols.28
5. Related path A and B synthetic endeavors
Previous syntheses of the singly linked PA type-B structures have followed the putative biosynthetic route, Path B.23-26 However, unless other stereochemical substituents adjoin the site of reactivity and direct the process Path B couplings appear to result in a complex diastereomeric mixture of products. Moreover, regioselectivity and chemoselectivity prove problematic for most systems. Far less information is available regarding the likelihood of a successful Path A coupling. However, Jurd reports a simple benzopyrylium salt undergoes coupling with phloroglucinol to result in a similar [3.3.1]-bicyclic ketal in 25% yield.27
6. Our synthetic studies
We began surveying these problems by constructing the flavans, flavanones, and dihydroxyflavanones, which might arise in the putative biosynthetic pathway, and then proceeded to investigate the inherent ability of these structures to form type-A and B PA adducts (Scheme 2). Recently, our group had shown that o-QMs generated from o-Boc salicyl-alcohols would undergo cycloadditions with electron-rich olefins forming chroman adducts that resemble compound 5.22 However, our original two-pot process, involving reduction and coupling proved unfruitful with aldehyde 3. This substrate mandates the use of an in situ reduction–cycloaddition protocol. The tris-o-Boc salicylaldehyde 3 (0.10 M diethyl ether, −78 °C), styrene 4 (40 equiv), and magnesium bromide dietherate (1.0 equiv) are subjected to lithium aluminum hydride (1.0 equiv) and thereby produce the flavan 5 in a 45% isolated yield.
Scheme 2.
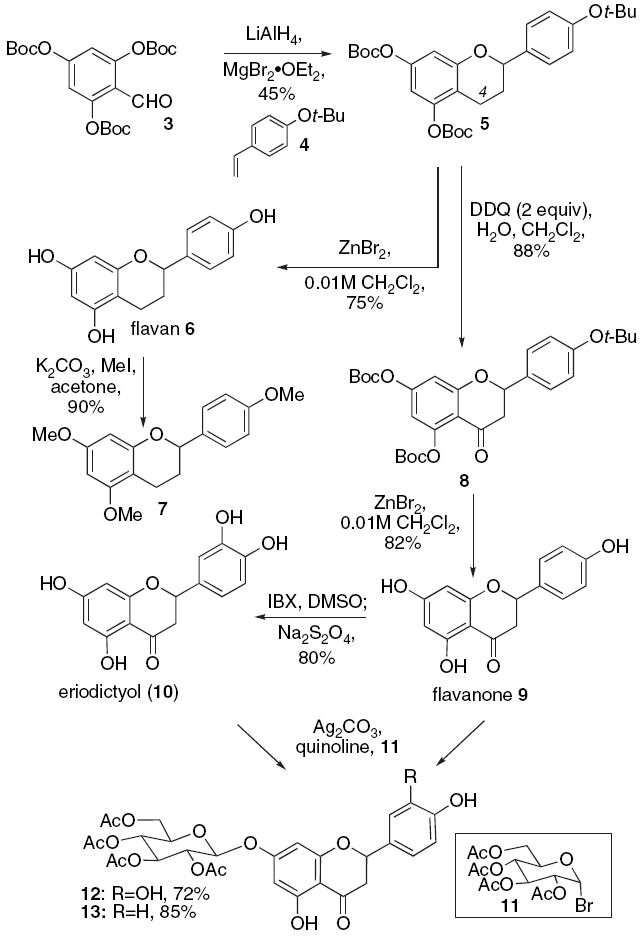
Synthesis of flavan, flavanone, and catechol.
C(4)-oxidation of flavan 5 (0.10 M methylene chloride) occurs with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (2 equiv) in the presence of water (1.0 equiv) to afford the flavanone 8 in 88% yield. Cleavage of the t-butyl residues found in compounds 5 and 8 (0.01 M methylene chloride) is accomplished with zinc bromide (10 equiv) to afford the flavan 6 and the flavanone 9, respectively. Per-methylation of 6 (0.10 M acetone) using methyl iodide (5.0 equiv) and potassium carbonate (4.0 equiv) affords tris-methylated flavan ether 7 and facilitates its structural identification.
Next, we used our one-pot two-step chemical process that mimics the enzyme F3H found in the flavan biosynthesis.28 The unprotected flavanone 9 (0.03 M dimethyl sulfoxide) is subjected to 2-iodoxybenzoic acid (2.0 equiv) and forms a highly reactive o-quinone intermediate, which is reduced during work-up with sodium sulfite (3.0 equiv) to provide eriodictyol (10) in an isolated 80% yield. As previously observed, phenols bearing electron-withdrawing groups such as −C(O)R, −CHO, and −NO2 fail to undergo oxidation with 2-iodoxybenzoic acid.28 This reaction can be viewed as electrophilic aromatic substitution.29 The −C(O)R substituent of flavanone 9 deactivates the A-ring and effectively prevents its oxidation. With the phenol 9 and catechol 10 in hand, methods for their regioselective glycosidation were explored. The insolubility of substrates 9 and 10 severely limited the conditions that could be employed. After some experimentation, we found that phenol 9 and catechol 10 (0.10 M quinoline) undergo regioselective β-glycosidation with acetobromo-α-d-glucose 11 (1.50 equiv) and silver carbonate (1.50 equiv) to produce compounds 13 and 12 in good yield.30 The following order of reactivity was observed for flavanone 9 and appears to follow the relative acidity of each phenol: C(5)−OH<c(4′)−OH<c(7)−OH. Given this result, we expect the glycosidation of the aglycones of 1 and 2 to be fairly straightforward.
Next, we turned our attention to the formation of the C(4)–C(8) bond following a putative Path B biosynthesis. Given our experiences with diastereoselective additions to o-QMs, we decided to explore this reaction within the context of a flavanone (Scheme 3). The glycosidation reaction revealed that the hydroxyl residues of flavanones would also undergo regioselective alkylation. We decided to exploit this inherent reactivity for selective protection. Sequential treatment of flavanone 9 with methyl iodide, tert-butyldimethylsilyl triflate, and di-tert-butyl dicarbonate and the appropriate bases affords compound 15 in good yield (75%).
Scheme 3.
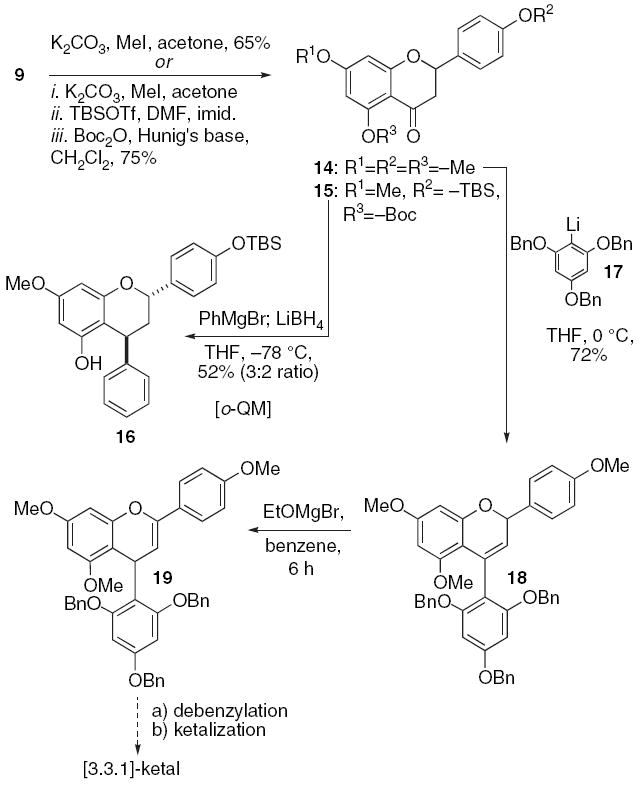
o-QM reaction with flavanone backbone.
Reaction of the o-OBoc flavanone 15 (0.05 M tetrahydrofuran) with phenylmagnesium bromide (1.0 equiv) followed by lithium borohydride (1.0 equiv) results in flavan 16 formation in a 52% yield and a 3:2 ratio of diastereomers. We presume that the reaction proceeds through an o-QM intermediate. The lack of stereoselectivity was somewhat troubling and did not bode well for applying any of these Path B strategies to the syntheses of 1 and 2. Therefore, we sought another method for carbon–carbon bond formation. The flavanone 9 was per-methylated to afford compound 14. Treatment of flavanone 14 (0.2 M tetrahydrofuran, −78 °C) with the phloroglucinol lithium 17 (3.0 equiv) affords the chromene 18 by spontaneous elimination of the tertiary alcohol intermediate. Isomerization of the double bond within compound 18 (0.07 M benzene, 65 °C) occurs upon treatment with ethoxymagnesium bromide (2.0 equiv) to form the enol ether 19.31 We envisioned that debenzylation of compound 19 and subsequent ketalization would afford the desired [3.3.1]-bicyclic system. However, treatment of the debenzylated adduct with strong acid results in loss of the phloroglucinol fragment from adduct 19.
With compound (±)−7 in hand, we considered other strategies mimicking Path B (Scheme 4).32 Treatment of the flavan (±)−7 (0.10 M methylene chloride) with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (0.5 equiv) affords a mixture of more than four stereoisomers of the dimer 22 in a combined 52% yield. Given this result, we expected optically pure 7 to give more than two diastereomers; there must be atropisomerism among the trans and/or cis adducts. To partially circumvent this stereochemical problem, we examined the oxidation of 7 (0.1 M methylene chloride) with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (1.0 equiv) and in the presence of tribenzylated phloroglucinol (20) (3 equiv). The reaction proceeds in a 60% yield and affords the type-B PA structure 21 in a 3:2 ratio favoring the trans diastereomer. In addition to the stereochemistry issues raised by this strategy, there are numerous protecting group issues that would have to be resolved as well. Therefore, we began to investigate a Path A inspired synthesis.
Scheme 4.
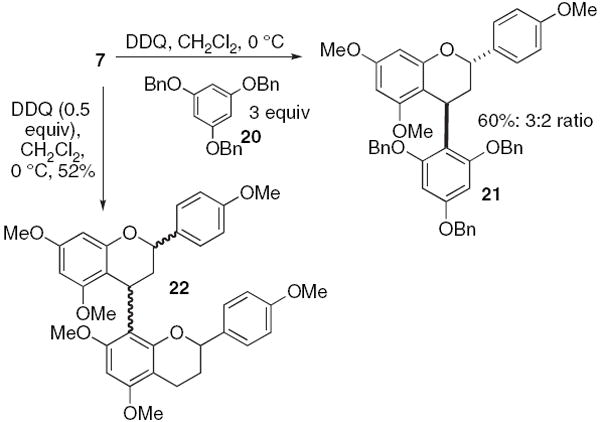
DDQ dimerization and coupling.
A Path A strategy requires rapid and easy access to the corresponding benzopyrylium salts. We initially considered methods to construct these systems from 7, 9, and 10. However, careful scrutiny of the literature reveals oxidative methods are plagued by low yields, crude mixtures.33 Instead, we opted to explore a new protocol recently reported by Mas (Scheme 5).34 2,4,6-Triacetoxybenzaldehyde (24) undergoes condensation with hydroxyacetophenones 25 and 26 in a saturated hydrochloric acid solution in methanol to result in the benzopyrylium salts 27 and 28 in good yields.
Scheme 5.
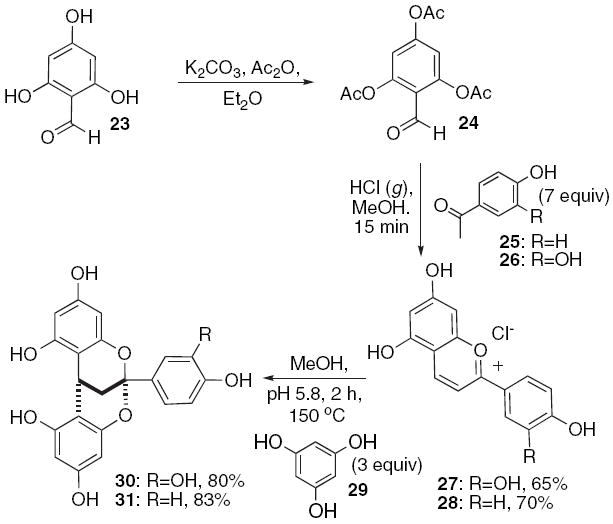
Mas’s procedure provides benzopyrylium salts that readily undergo addition with phloroglucinol.
We find that addition of an aqueous buffered solution of phloroglucinol 29 (3 equiv) to the respective benzopyryliums 27 and 28 (0.20 M methanol) affords the ketals 30 and 31, respectively, in outstanding yields. Optimal conditions employ a Emrys microwave reactor with heating to 150 °C for 120 min. Isolation of non-ketalized products from reactions conducted at lower temperatures reveals that the carbon–carbon bond formation precedes the oxygen–carbon bond formation. While we entertained the possibility of desymmetrizing the meso compounds 30 and 31, we first examined the addition of flavans and flavanones to the benzopyrilium salts 27 and 28.
The benzopyrilium salt 27 (0.10 M methanol) combines with flavan 6 (6 equiv) using the previously described microwave equipment and results in the regioselective formation of the type-A PA compound 32 in 42% yield (Scheme 6). However, a 1:1 mixture of diastereomers is observed. Upon lowering the temperature to 120 °C and lengthening the reaction time to 20 h for the subsequent combination of the benzopyrilium 27 with the flavanone 9, diinsininone (33) forms. To our delight, compound 33 emerges from this extended reaction as a single diastereomer. However, the similar conditions proved entirely unproductive for the combination of the benzopyrilium 27 with the hydroxylated flavanone 10.
Scheme 6.
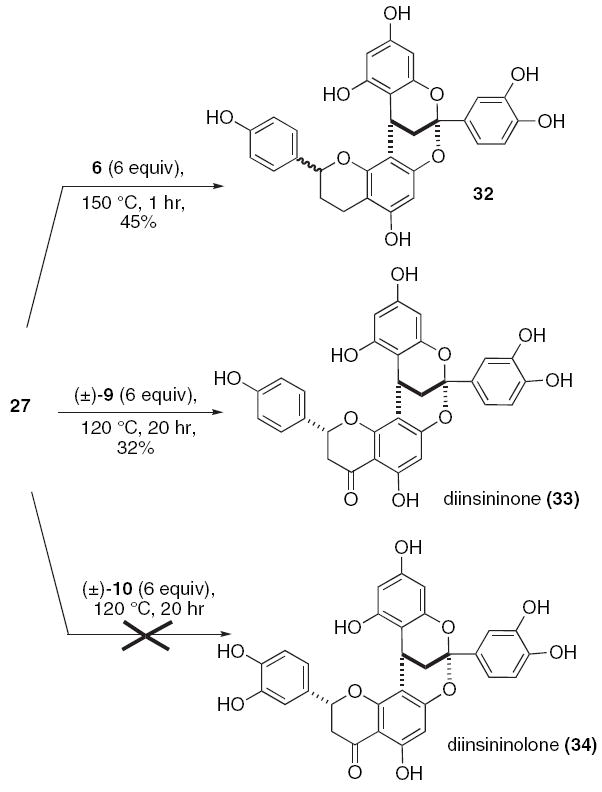
Reactions of the benzopyrilium salt 27 with flavan and flavanone.
The formation of a single diastereomer of diinsininone (33) is explained by the equilibria shown in Scheme 7. It should be noted that we had originally imagined degrading naringin (35) to afford optically active (S)-C(2) flavanone 9. However, in our hands, hydrolysis of the sugar results in formation of racemic 9 as determined by a Chiralcel OD-H column.35
Scheme 7.
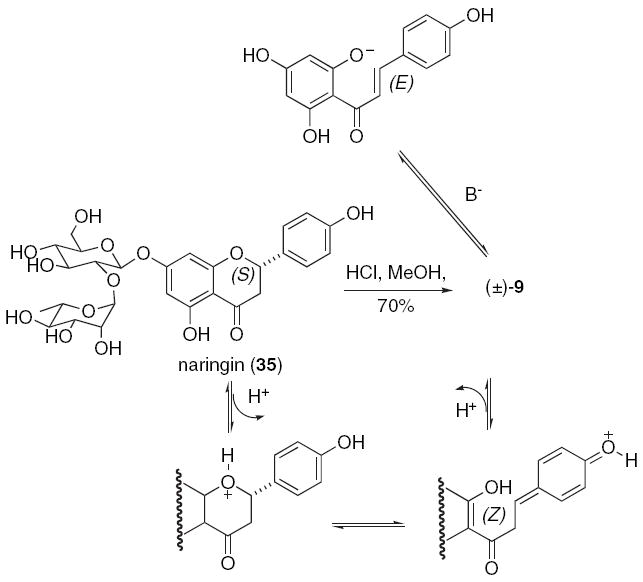
Acid and base-catalyzed racemization of a C(2)-stereocenter.
7. Conclusions
One day, a better understanding of the PA biosynthetic pathway may allow the genetic engineering of plants for the production of compounds coveted for their antioxidant and antihyperglycemic activities. Until that day arrives, chemical synthesis remains the best method for the construction of these compounds and enables the exploration of their biological activities. The preceding synthetic studies have illuminated inherent reactivity of several intermediates belonging to the flavonoid pathway. These studies suggest that compounds 1 and 2 most likely arise from an identical biosynthetic pathway proceeding through a Path A benzopyrilium coupling. Whether the oxidative divergence among these structures arise as the ultimate or penultimate step remains to be determined. Related natural products belonging to the Dragon’s Blood family of flavanones, a name that derives from the red resin plant exudates, can also be accessed by appropriately adapting the above strategy (Fig. 2).32b
Figure 2.
Natural products of the Dragon’s Blood family.
8. Experimental
8.1. Aldehyde 3
A flame-dried flask equipped with a stir-bar and a nitrogen line containing 2,4,6-trihydroxybenzaldehyde (2 g, dehydrated with P2O5, 0.05 M CH2Cl2), Hunig’s Base (1.36 mL, 0.6 equiv), DMAP (four crystals), and Boc2O (7.74 g, 3.2 equiv) was stirred overnight. The reaction was quenched with 1 M NH4Cl and extracted twice with CH2Cl2. The organic layer was washed with water, brine, dried over MgSO4, and concentrated in vacuo. The crude mixture was then chromatographed on silica gel, eluting with petroleum ether/EtOAc (95:5) to afford the desired product in 75% yield. 1H NMR (400 MHz, CDCl3): δ 10.24 (s, 1H), 6.88 (s, 2H), 1.46 (s, 18H), 1.42 (s, 9H).
8.2. Tris-protected flavan 5
A flame-dried flask equipped with stir-bar and nitrogen line, was charged with aldehyde 3 (50 mg, 0.1 M Et2O), 1-tert-butoxy-4-vinylbenzene (0.19 mL, 40 equiv), and MgBr2·OEt2 (28.4 mg, 1.00 equiv). The solution was cooled to −78 °C and LiAlH4 (4 mg, 1.00 equiv) was added. The reaction was warmed to rt over 3 h then quenched with 1 M NaHCO3 and extracted with Et2O. The ether layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (95:5) to afford the desired product in 45% yield. 1H NMR (400 MHz, CDCl3): δ 7.08 (d, 2H, J=9 Hz), 6.70 (d, 2H, J=9 Hz), 6.39 (s, 1H), 6.34 (s, 1H), 5.20–4.90 (m, 1H), 2.60–2.20 (m, 4H), 1.45 (s, 9H), 1.40 (s, 18H).
8.3. Flavan 6
A flame-dried flask equipped with stir-bar and nitrogen line, was charged with flavan 5 (20.0 mg, 0.01 M CH2Cl2) and ZnBr2 (87.5 mg, 10 equiv). The solution was stirred at rt for 6 h. The reaction was quenched with 1 M HCl and extracted twice with EtOAc. The ether layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (50:50) to afford the desired product in 75% yield. 1H NMR (400 MHz, CD6CO): δ 7.27 (d, 2H, J=9 Hz), 6.85 (d, 2H, J=9 Hz), 6.00 (s, 1H), 5.87 (s, 1H), 4.89–4.86 (m, 1H), 2.72–2.66 (m, 2H), 2.11–1.88 (m, 2H).
8.4. Tris-methylated chroman 7
A round-bottom flask equipped with a stir-bar was charged with flavan 6 (1.67 g, 0.2 M acetone), K2CO3 (340 mg, 4 equiv), and MeI (0.19 mL, 5 equiv). The reaction was stirred overnight and filtered. The reaction was diluted with H2O and extracted twice with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (80:20) to afford the desired product in 90% yield. 1H NMR (400 MHz, CDCl3): δ 7.36 (d, 2H, J=9 Hz), 6.93 (d, 2H, J=9 Hz), 6.13 (s, 1H), 6.09 (s, 1H), 4.93–4.92 (m, 1H), 3.83 (s, 3H), 3.80 (s, 3H), 3.77 (s, 3H), 2.80–2.60 (m, 4H), 2.20–1.97 (m, 4H).
8.5. Flavanone 8
Flavan 5 (30 mg, 1 equiv, 0.01 M CH2Cl2), H2O (1 μL, 1 equiv), and DDQ (recrystallized from hot CHCl3, 26.5 mg, 2 equiv) were added to a round-bottom flask equipped with a stir-bar, a septum and stirred under N2 (g) at rt overnight. The reaction was diluted with H2O and extracted twice with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (70:30) to afford the desired product in 88% yield. 1H NMR (400 MHz, CDCl3): δ 7.08 (d, 2H, J=9 Hz), 6.70 (d, 2H, J=9 Hz), 6.54 (s, 1H), 6.48 (s, 1H), 5.55–5.50 (m, 1H), 3.38–3.13 (m, 2H), 1.56 (s, 18H), 1.40 (s, 9H).
8.6. 4′,5,7-Trihydroxyflavanone (9)
A flame-dried flask equipped with stir-bar and nitrogen line, was charged with flavanone 8 (20.0 mg, 0.01 M CH2Cl2) and ZnBr2 (85.2 mg, 10 equiv). The solution was stirred at rt for 6 h. The reaction was quenched with 1 M HCl and extracted twice with EtOAc. The ether layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (50:50) to afford the desired product in 82% yield. 1H NMR (400 MHz, CD6CO): δ 12.19 (s, 1H), 9.59 (s, 1H), 8.55 (s, 1H), 7.39 (d, 2H, J=9 Hz), 6.90 (d, 2H, J=9 Hz), 5.95 (s, 1H), 5.94 (s, 1H), 5.48–5.44 (m, 1H), 3.22–3.15 (m, 2H), 2.75–2.70 (m, 2H); 13C NMR (100 MHz, CD6CO): δ 197.4, 167.3, 165.4, 164.5, 158.8, 130.9, 129.1, 116.3, 103.3, 96.8, 95.9, 80.0, 43.6, 30.5; HRMS (ESI-TOF) (M++H+) m/z calcd for C15H12O5 273.0685, found 273.0765.
8.7. Eriodictyol (10)28
To a solution of flavanone 9 (1.011 g, 0.03 M DMSO) was added IBX36 (2.135 g, 2 equiv) and stirred at rt for 5 h. As the reaction progressed, the color changed from a translucent yellow solution to an opaque brown solution. Na2S2O4 (1.94 g, 3 equiv) was added to the reaction and stirred for an additional 4 h. The reaction was quenched with water (300 mL) and extracted with EtOAc (3×100 mL). The combined organic extracts stirred with pH 8.5 buffer solution to remove o-iodobenzoic acid. The organic layer was washed with brine, dried with MgSO4, filtered, and concentrated. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (50:50) to afford the desired product in 80% yield. 1H NMR (400 MHz, CD6CO): δ 7.03 (s, 1H), 6.87 (s, 2H), 5.96–5.94 (m, 2H), 5.41–5.38 (m, 1H), 3.18 (m, 1H), 2.75 (m, 1H); 13C NMR (100 MHz, CD6CO): δ 167.7, 137.8, 135.7, 134.8, 116.8, 116.5, 102.0, 89.7, 86.5, 85.2, 73.6, 67.2, 66.3, 50.4, 14.0; HRMS (ESI-TOF) (M++Na+) m/z calcd for C15H12O6 311.0633, found 311.0525.
8.8. Acetobromo-α-d-glucose (11)
d-Glucose (2.05 g, 0.40 M pyridine) and Ac2O (12 mL, 0.44 M) were stirred at rt overnight. The reaction was quenched with H2O, extracted with CDCl3, washed with NaHCO3, dried with Na2SO4, and afforded pentaacetyl-glucose. 1H NMR (400 MHz, CDCl3): δ 5.72–5.71 (m, 1H), 5.28–5.24 (m, 1H), 5.16–5.11 (m, 2H), 4.32–4.28 (m, 1H), 4.13–4.10 (m, 1H), 3.86 (m, 1H), 2.12 (s, 3H), 2.09 (s, 3H), 2.04 (s, 3H), 2.03 (s, 3H), 2.02 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 170.8, 170.3, 169.6, 169.5, 169.2, 91.9, 73.0, 72.9, 70.4, 67.9, 61.6, 21.0, 20.9, 20.8, 20.7; HRMS (ESI-TOF) (M++Na+) m/z calcd for C16H22O11 413.1162, found 413.1057. When the pentaacetoglucose (2 g, 0.57 M acetic acid) was dissolved, HBr (0.64 mL, 48% in acetic acid) was added drop-wise. The reaction was stirred overnight, poured over an ice/H2O slurry, extracted with CHCl3, neutralized with NaHCO3, washed with H2O, dried with Na2SO4, and afforded acetobromo-α-d-glucose (11). 1H NMR (400 MHz, CDCl3): δ 6.62–6.61 (m, 1H), 5.59–5.55 (m, 1H), 5.19–5.15 (m, 1H), 4.86–4.83 (m, 1H), 4.36–4.30 (m, 2H), 4.15–4.12 (m, 1H), 2.11 (s, 3H), 2.10 (s, 3H), 2.06 (s, 3H), 2.05 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 170.7, 170.1, 170.0, 169.7, 86.8, 72.3, 70.8, 70.4, 67.4, 61.1, 20.9, 20.9, 20.8, 20.7; HRMS (ESI-TOF) (M++Na+) m/z calcd for C14H19O9Br 433.0212, found 433.0126.
8.9. Naringenin glucoside 1330
A solution of flavanone 9 (9.4 mg, 0.14 M quinoline), bromide 11 (21 mg, 1.50 equiv), and Ag2CO3 (13.8 mg, 1.5 equiv) was stirred for 6 h at rt. The reaction was poured into methanol, filtered through a short pad of Celite, and evaporated in vacuo. The residue was dissolved into EtOAc, washed with 1 N HCl, washed with brine, and dried over MgSO4. After evaporation, the resulting crude product was purified by flash chromatography eluting with hexane/EtOAc (1:1) to afford the desired product in 85% yield. The product was an inseparable mixture of diastereomers. 1H NMR (400 MHz, DMSO): δ 12.03 (s, 1H), 9.65 (s, 1H), 7.31 (d, 2H, J=9 Hz), 6.79 (d, 2H, J=9 Hz), 6.16–6.09 (m, 2H), 5.67–5.63 (m, 1H), 5.53–5.50 (m, 1H), 5.37–5.34 (m, 1H), 5.06 (m, 2H), 4.30–4.26 (m, 1H), 4.15–4.05 (m, 2H), 3.40–3.33 (m, 1H), 2.76–2.70 (m, 1H), 2.00–1.95 (m, 12H); (ESI-TOF) (M++Na) m/z calcd for C29H30O14 625.1636, found 625.1528.
8.10. Trimethylated naringenin 14
Naringenin (9, 1 g, 0.2 M acetone), K2CO3 (3.05 g, 6 equiv), and MeI (1.83 mL, 8 equiv) were stirred overnight. The reaction was filtered and concentrated in vacuo. The resulting crude product was purified by flash chromatography eluting with petroleum ether/EtOAc (70:30) to afford the desired product in 65% yield. 1H NMR (400 MHz, CDCl3): δ 7.47 (d, 2H, J=9 Hz), 6.98 (d, 2H, 9 Hz), 6.18 (s, 1H), 6.16 (s, 1H), 5.45–5.40 (m, 1H), 3.85 (s, 3H), 3.82 (s, 3H), 3.81 (s, 3H), 3.03–2.85 (m, 2H), 2.64–2.58 (m, 2H).
8.11. Tris-protected naringenin 15
Naringenin (9, 162 mg, 0.1 M DMF), Na2CO3 (69 mg, 1.10 equiv), and MeI (0.4 mL, 1.10 equiv) were stirred at rt overnight. The reaction was quenched with H2O and extracted with EtOAc. The organic layer was washed repeatedly with H2O, washed with brine, and dried over MgSO4. The resulting crude product was purified by flash chromatography (petroleum ether/EtOAc 50:50) to afford mono-protected naringenin. 1H NMR (400 MHz, CD6CO): δ 12.15 (s, 1H), 8.57 (s, 1H), 7.40 (d, 2H, J=8 Hz), 6.89 (d, 2H, J=8 Hz), 6.04 (s, 1H), 6.03 (s, 1H), 5.39–5.35 (m, 1H), 3.82 (s, 3H), 3.10–3.03 (m, 2H), 2.78–2.70 (m, 2H). mono-Protected narigenin (66 mg, 0.1 M THF), 2,6-lutidine (0.028 mL, 1.05 equiv), and TBSOTf (0.056 mL, 1.05 equiv) were stirred at rt for 4 h. The reaction was quenched with H2O, extracted with EtOAc, and dried with Na2SO4. The resulting crude product was purified by flash chromatography (petroleum ether/EtOAc 80:20) to afford bis-protected naringenin. 1H NMR (400 MHz, CD6CO): δ 12.15 (s, 1H), 7.47 (d, 2H, J=8 Hz), 6.96 (d, 2H, J=8 Hz), 6.07 (s, 1H), 6.05 (s, 1H), 5.55–5.51 (m, 1H), 3.86 (s, 3H), 3.27–3.19 (m, 2H), 2.81–2.77 (m, 2H), 1.00 (s, 9H), 0.33 (s, 6H). The bis-protected compound (0.1 M CH2Cl2), DMAP (one crystal), Hunig’s Base (0.037 mL, 1.0 equiv), and Boc2O (100 mg, 1.0 equiv) were stirred at rt for 1 h. The reaction was quenched with H2O, extracted with EtOAc, and dried with Na2SO4. The resulting crude product was purified by flash chromatography (petroleum ether/EtOAc 90:10) to afford tris-protected naringenin 15 in 75% yield. 1H NMR (400 MHz, CD6CO): δ 7.50–7.48 (d, 2H, J=8 Hz), 6.98–6.95 (d, 2H, J=8 Hz), 6.50 (s, 1H), 6.37 (s, 1H), 5.52–5.49 (s, 1H), 3.90 (s, 3H), 3.08–3.05 (m, 2H), 2.85 (m, 2H), 1.54 (s, 9H), 1.01 (s, 9H), 0.25 (s, 6H).
8.12. Compound 16
A flame-dried flask equipped with stir-bar and nitrogen line, was charged with flavanone 15 (8.0 mg, 1 equiv, 0.05 M THF) and cooled to −78 °C. Phenylmagnesium bromide (8.0 μL, 1.50 equiv, 3 M THF) was added drop-wise and stirred for 1 h. LiBH4 (1.0 mg, 1.00 equiv) was added to the solution, warmed to rt, quenched with H2O, and extracted with EtOAc. The ether layer was washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (90:10) to afford the desired product in 52% yield.
8.13. Chromene 18
Bromotribenzylated phloroglucinol (65 mg, 0.3 M THF, 3 equiv) was cooled to −78 °C, n-BuLi (0.052 mL, 2.5 M THF, 3.00 equiv) was added drop-wise, and the reaction stirred until complete by TLC (eluting with petroleum ether/EtOAc 90:10). This solution was added drop-wise to a stirring solution of flavan 15 (13.7 mg, 0.2 M THF) at −78 °C. After 1 h, the reaction was quenched with H2O, extracted EtOAc, washed with brine, and dried with MgSO4. The reaction was purified by flash chromatographed with EtOAc/hexane (70:30) to afford the desired product in 72% yield. 1H NMR (400 MHz, CDCl3): δ 7.53–7.22 (m, 17H), 6.61–6.59 (m, 2H), 6.35–6.30 (m, 2H), 6.14–6.12 (m, 1H), 5.92–5.91 (m, 1H), 4.91 (d, 1H, J=5 Hz), 4.76 (d, 1H, J=5 Hz), 5.05–4.96 (m, 6H), 3.73 (s, 3H), 3.72 (s, 3H), 3.30 (s, 3H); HRMS (ESI-TOF) (M–H)+ m/z calcd for C45H40O7 691.7900, found 691.2711.
8.14. Enol ether 1931
A flame-dried flask equipped with a stir-bar and a nitrogen line containing EtOH (2 equiv, 0.5 M THF) was cooled to 0 °C. Phenylmagnesium bromide (2 equiv) was added to the solution warmed to rt, then evaporated to dryness. The chromene 18 (1 equiv, 0.7 M benzene) was added to the EtOMgBr, equipped with a reflux condenser, stir-bar, nitrogen line, and refluxed for 6 h. The reaction was quenched with H2O, extracted EtOAc, washed with brine, and dried with MgSO4. The reaction was purified by flash chromatography eluting with EtOAc/hexane (70:30) to afford the desired product in 65% yield. 1H NMR (400 MHz, CDCl3): δ 7.53–7.05 (m, 15H), 7.03 (d, 2H, J=8 Hz), 6.66 (d, 2H, J=8 Hz), 6.47 (s, 1H), 6.36 (d, 1H, J=10 Hz), 6.28–6.27 (m, 1H), 6.00 (s, 1H), 4.94 (s, 1H), 4.98 (s, 2H), 4.88–4.83 (m, 4H), 4.66 (d, 1H, J=10 Hz), 3.74 (s, 3H), 3.72 (s, 3H), 3.33 (s, 3H).
8.15. Chroman 21
To a flame-dried flask equipped with a stir-bar and a nitrogen line containing flavan 7 (20 mg, 0.10 M CH2Cl2) and tribenzylphloroglucinol (33.6 mg, 3 equiv) was added DDQ (recrystallized from hot CHCl3, 15.8 mg, 1 equiv) at rt. The reaction was stirred for 3 h, quenched with H2O, extracted with EtOAc, the organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (70:30) to afford the desired product in 60% yield.
8.16. Dimer 22
To a flame-dried flask equipped with a stir-bar and a nitrogen line containing flavan 7 (30 mg, 0.10 M CH2Cl2) stirring at rt was added DDQ (recrystallized from hot CHCl3, 11.2 mg, 0.5 equiv). The reaction was stirred for 3 h, quenched with H2O, extracted with EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude mixture was then chromatographed through silica gel, eluting with petroleum ether/EtOAc (40:60) to afford the desired product in 52% yield.
8.17. 2,4,6-Triacetoxybenzaldehyde (24)
A flame-dried flask equipped with a stir-bar and a nitrogen line containing phenol 23, dehydrated with P2O5, (2.58 g, 1 equiv, 0.15 M Et2O), K2CO3 (8.41 g, 3.6 equiv), and Ac2O (16.8 mL, 11 equiv) was stirred overnight. The reaction was filtered and concentrated to afford the desired product in 80% yield. 1H NMR (400 MHz, CDCl3): δ 10.13 (s, 1H), 6.97 (s, 2H), 2.38 (s, 6H), 2.31 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 185.6, 168.7, 167.9, 155.2, 1530.0, 118.3, 114.9, 21.4, 21.0; δ IR (CH2Cl2, υmax cm−1) 1778, 1697, 1616, 1580, 1433, 1370, 1298, 1180, 1122, 1051, 1022; HRMS (ESI-TOF) (M++Na) m/z calcd for C13H12O7 303.2302, found 303.0484.
8.18. General method for benzopyrilium salt formation34
A stream of anhydrous gaseous HCl was first passed through a solution of hydroxyacetophenone (1.6 g, 7 equiv, 0.12 M MeOH) at 0 °C, until the concentration of HCl rose to 15% by mass. A solution of aldehyde 24 (0.43 g, 1 equiv, 0.06 M MeOH) was then added drop-wise at 0 °C. At the end of the addition, the reaction mixture was kept for a few minutes at rt and evaporated to dryness. The resulting residue was vigorously stirred in Et2O, which afforded the crude product after filtration and washing with the same solvent. The pure product was obtained by recrystallization in MeOH–HCl (95:5) in 65 and 70% yields for compounds 27 and 28, respectively. Luteolinidin Chloride (27). 1H NMR (400 MHz, CD3OD–TFA 99:1): δ 8.95 (d, 1H, J=9 Hz), 7.94 (d, 1H, J=9 Hz), 7.78 (dd, 1H, J=9, 2 Hz), 7.65 (d, 1H, J=2 Hz), 6.98 (d, 1H, J=9 Hz), 6.87 (d, 1H, J=2 Hz), 6.63 (d, 1H, J=2 Hz); 13C NMR (100 MHz, CD3OD–TFA 99:1): δ 172.4, 171.9, 160.2, 159.6, 156.1, 149.0, 148.0, 125.3, 121.6, 117.9, 115.9, 113.5, 110.6, 103.3, 96.1. Apigeninidin Chloride (28). 1H NMR (400 MHz, CD3OD–TFA 99:1): δ 9.06 (dd, 1H, J=9, 1 Hz), 8.27 (d, 2H, J=9 Hz), 8.01 (d, 1H, J=9 Hz), 7.06 (d, 2H, J=9 Hz), 6.92 (dd, 1H, J=2, 1 Hz), 6.64 (d, 1H, J=2 Hz); 13C NMR (100 MHz, CD3OD–TFA 99:1): δ 172.7, 172.4, 167.3, 160.6, 160.0, 149.5, 133.1, 121.3, 118.4, 113.8, 110.3, 103.2, 96.1.
8.19. General method for the condensation of benzopyrilium salt with phloroglucinol27
Benzopyrilium salt 27 (20 mg, 0.6 M MeOH/pH 5.8 buffer (1:1) and phloroglucinol (26.2 mg, 3 equiv) were heated at 150 °C for 2 h in an Emrys microwave reactor. The reaction was concentrated to dryness on silica gel and purified by flash chromatographed with EtOAc/hexane (80:20) to afford the desired product in 80% yield. Compound 30. 1H NMR (400 MHz, CD6CO): δ 7.19 (d, 1H, 2 Hz), 7.05 (dd, 1H, J=2, 8 Hz), 6.88 (d, 1H, J=2 Hz), 6.06 (s, 4H), 4.33 (t, 1H, J=3 Hz), 2.22 (d, 3H, J=3 Hz); 13C NMR (100 MHz, CD6CO): δ 158.1, 154.7, 154.3, 146.4, 145.6, 134.9, 118.4, 115.8, 114.2, 107.5, 99.4, 97.3, 96.7, 34.5, 21.4; HRMS (ESI-TOF) (M++Na) m/z calcd for C21H16O8 419.0843, found 419.0758. An alternative method for easier purification is as follows. The reaction is concentrated to dryness and submitted to pyridine (0.3 mL) and Ac2O (0.5 mL) overnight. The reaction is quenched with 1 M HCl, repeatedly extracted with EtOAc, and dried with MgSO4. The reaction was purified with flash chromatography eluting with EtOAc/hexane (60:40) and the ketal and non-ketal compounds were isolated. Acetylated compound 30. 1H NMR (400 MHz, CDCl3): δ 7.56–7.27 (m, 3H), 6.74 (d, 2H, J=2 Hz), 6.54 (d, 2H, J=2 Hz), 4.28 (t, 1H, J=3 Hz), 2.42 (s, 6H), 2.33 (s, 3H), 2.32 (s, 3H), 2.29 (s, 3H, J=3 Hz), 2.26 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 169.2, 169.0, 168.3, 153.7, 150.2, 147.8, 142.9, 142.1, 139.0, 124.3, 123.7, 121.6, 115.7, 109.8, 108.5, 98.4, 33.0, 22.9, 21.7, 21.3, 20.9, 20.8; IR (CH2Cl2, υmax cm−1) 1772, 1618, 1369, 1198, 1222, 1024; HRMS (ESI-TOF) (M++Na) m/z calcd for C33H28O14 671.1479, found 671.1378. Enol ether 30. 1H NMR (400 MHz, CDCl3): δ 7.53–7.22 (m, 3H), 6.90–6.82 (m, 4H), 5.32 (d, 1H, J=4 Hz), 4.90 (d, 1H, J=4 Hz), 2.48–2.25 (m, 21H); HRMS (ESI-TOF) (M++Na) m/z calcd for C35H30O15 713.1585, found 713.1511.
8.20. Acetylated compound of 31
Benzopyrilium salt 28 (30 mg, 0.6 M MeOH/pH 5.8 buffer (1:1) and phloroglucinol (39.0 mg, 3 equiv) were heated at 150 °C for 2 h in an Emrys microwave reactor. The reaction was concentrated to dryness and submitted to pyridine (0.3 mL) and Ac2O (0.5 mL) overnight. The reaction is quenched with 1 M HCl, repeatedly extracted with EtOAc, and dried with MgSO4. The crude product was purified by flash chromatography with EtOAc/hexane (60:40) and the ketal and non-ketal product were isolated in 83% yield. 1H NMR (400 MHz, CDCl3): δ 7.68 (d, 2H, J=9 Hz), 7.18 (d, 2H, J=9 Hz), 6.75 (d, 2H, J=2 Hz), 6.55 (d, 2H, J=2 Hz), 4.27 (t, 1H, J=3 Hz), 2.43 (s, 6H), 2.29 (d, 3H, J=3 Hz), 2.33 (s, 3H), 2.27 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 169.5, 169.2, 169.0, 153.8, 151.4, 150.2, 147.8, 137.8, 127.3, 121.8, 115.8, 109.7, 108.5, 98.9, 33.1, 23.0, 21.7, 21.4, 21.3; δ IR (CH2Cl2, υmax cm−1) 1768, 1620, 1434, 1369, 1201, 1184, 1130, 1116, 1072, 1022, 904; HRMS (ESI-TOF) (M++Na) m/z calcd for C31H26O12 613.1424, found 613.1337. Enol ether 31. 1H NMR (400 MHz, CDCl3): δ 7.63 (d, 2H, J=9 Hz), 7.10 (d, 2H, J=9 Hz), 6.90–6.60 (m, 4H), 5.30 (d, 1H, J=4 Hz), 4.91 (d, 1H, J=4 Hz), 2.38–2.13 (m, 18H); HRMS (ESI-TOF) (M++Na) m/z calcd for C33H28O13 655.1530, found 655.1423.
8.21. Compound 32
Benzopyrilium salt 27 (8.5 mg, 0.6 M MeOH/pH 5.8 buffer (1:1) and flavan 6 (43 mg, 6 equiv) were heated at 150 °C for 2 h in an Emrys microwave reactor. The reaction was concentrated to dryness and submitted to pyridine and Ac2O overnight. The reaction was quenched with 1 M HCl, repeatedly extracted with EtOAc, and dried with MgSO4. The desired product was purified by gradient chromatography with EtOAc/hexane (60:40–80:20) and the desired product was isolated in 45% yield. 1H NMR (400 MHz, CDCl3): δ 7.58–7.50 (m, 4H), 7.46–7.38 (m, 4H), 7.16–7.08 (m, 4H), 6.75–6.68 (m, 2H), 6.53–6.51 (m, 2H), 6.29–6.27 (m, 2H), 5.17–4.92 (m, 2H), 4.14 (br s, 2H), 2.97 (m, 8H), 2.44–2.31 (m, 40H); HRMS (ESI-TOF) (M++Na) m/z calcd for C42H36O15 803.2054, found 803.1935.
8.22. Diinsininone (33)
Benzopyrilium salt 27 (8.9 mg, 0.6 M MeOH/pH 5.8 buffer (1:1) and flavanone 9 (47 mg, 6 equiv) were heated at 120 °C for 20 h in an Emrys microwave reactor. The reaction was concentrated to dryness and submitted to pyridine and Ac2O overnight. The reaction was quenched with 1 M HCl, repeatedly extracted with EtOAc, and dried with MgSO4. The desired product was purified by gradient chromatography with EtOAc/hexane (60:40–80:20) and the desired product was isolated in 32% yield. 1H NMR (400 MHz, CDCl3): δ 7.57–7.48 (m, 8H), 7.24–7.19 (m, 6H), 6.74–6.72 (m, 2H), 6.51–6.50 (m, 4H), 5.60–5.51 (m, 2H), 4.47 (br s, 1H), 4.44 (br s, 1H), 3.09–3.00 (m, 2H), 2.78–2.27 (m, 2H), 2.39–2.27 (m, 40H); HRMS (ESI-TOF) (M++Na) m/z calcd for C42H34O16 817.1847, found 817.1593.
Acknowledgments
Research support from the University of California Coordinating Committee on Cancer Research in the form of two awards (19990641) and (SB010064) are greatly appreciated along with additional support of NSF (CHE-9971211), PRF (PRF34986-G1), NIH (GM-64831), the UC-AIDS Initiative (K00-SB-039) and Research Corporation (R10296). We are also thankful for the support of our M.S. facility funded in part by DAAD19-00-1-0026.
References and notes
- 1.Ogundaini A, Farah M, Perera P, Samuelsson G, Lars B. J Nat Prod. 1996;59:587–590. doi: 10.1021/np960386k. [DOI] [PubMed] [Google Scholar]
- 2.Samuelsson G, Farah MH, Claeson P, Hagos M, Thulin M, Hedberg O, Warfa AM, Hassan AO, Elmi AH, Abdulrahman AD, Elmi AS, Adbi YA, Alin MJ. J Ethnopharmacol. 1991;35:25–63. doi: 10.1016/0378-8741(91)90132-w. [DOI] [PubMed] [Google Scholar]
- 3.Sheehan KM, Sheahan K, O’Donoghue DP, Mac-Sweeney F, Conroy RM, Fitzgerald DJ, Murray FE. JAMA. 1999;282:1254–1257. doi: 10.1001/jama.282.13.1254. [DOI] [PubMed] [Google Scholar]
- 4.Haborne JB. Phytochemistry. 1966;5:589–600. [Google Scholar]
- 5.Pietta P, Gardana C, Pietta A. Flavanoids in Herbs. In: Rice-Evans CA, Packer L, editors. Flavonoids in Health and Disease. 2. Marcel Dekker; New York, NY: 2003. pp. 43–69. [Google Scholar]
- 6.Winkel-Shirley B. Curr Opin Plant Biol. 2002;5:218–223. doi: 10.1016/s1369-5266(02)00256-x. [DOI] [PubMed] [Google Scholar]
- 7.Rice-Evans CA, Packer L, editors. Flavonoids in Health and Disease. Marcel Dekker; New York, NY: 2003. [Google Scholar]
- 8.Renaud S, De Lorgeril M. Lancet. 1992;339:1523–1526. doi: 10.1016/0140-6736(92)91277-f. [DOI] [PubMed] [Google Scholar]
- 9.Simonetti P, Gardana C, Pietta P. Caffeic Acid as Biomarker of Red Wine Intake. In: Packer L, editor. Methods in Enzymology. Vol. 335. Academic; San Diego, CA: 2001. pp. 122–130. [DOI] [PubMed] [Google Scholar]
- 10.(a) Frankel EN, Waterhouse AL, Kinsella JE. Lancet. 1993;341:454–457. doi: 10.1016/0140-6736(93)90206-v. [DOI] [PubMed] [Google Scholar]; (b) Soleas GI, Diamandis EP, Goldberg DM. J Clin Lab Anal. 1997;11:287–313. doi: 10.1002/(SICI)1098-2825(1997)11:5<287::AID-JCLA6>3.0.CO;2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frei B. Crit Rev Food Sci Nutr. 1995;35:83–98. doi: 10.1080/10408399509527689. [DOI] [PubMed] [Google Scholar]
- 12.Ursini F, Rapuzzi I, Toniolo R, Tubaro F, Bontempelli G. Characterization of Antioxidant Effect of Procyanidins. In: Packer L, editor. Methods in Enzymology. Vol. 335. Academic; San Diego, CA: 2001. pp. 338–360. [DOI] [PubMed] [Google Scholar]
- 13.(a) McCarty MF. Med Hypotheses. 2005;64:151–158. doi: 10.1016/j.mehy.2004.03.036. [DOI] [PubMed] [Google Scholar]; (b) Imparl-Radosevich J, Deas S, Polansky MM, Baedke DB, Ingebritsen TS, Anderson RA, Graves D. Horm Res. 1998;50:177–182. doi: 10.1159/000023270. [DOI] [PubMed] [Google Scholar]; (c) Khan A, Safdar M, Ali Kahn MM, Kattak KN, Anderson RA. Diabetes Care. 2003;26:3215–3218. doi: 10.2337/diacare.26.12.3215. [DOI] [PubMed] [Google Scholar]
- 14.Kraja AT, Rao DC, Weder AB, Mosley TH, Turner ST, Hsiung CA, Quertermous T, Cooper R, Curb JD, Province MA. Nutr Metab. 2005;2:29–42. doi: 10.1186/1743-7075-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson RA, Broadhurst CL, Polansky MM, Schmidt WF, Khan A, Flanagan VP, Schoene NW, Graves DJ. J Agric Food Chem. 2004;52:65–70. doi: 10.1021/jf034916b. [DOI] [PubMed] [Google Scholar]
- 16.Marles MAS, Ray H, Gruber MY. Phytochemistry. 2003;64:367–383. doi: 10.1016/s0031-9422(03)00377-7. [DOI] [PubMed] [Google Scholar]
- 17.Winefield CS, Lewis DH, Swinny EE, Zhang H, Arathoon HS, Fischer TC, Halbwirth H, Stich K, Gosch C, Forkmann G, Davies KM. Physiol Plant. 2005;124:419–430. [Google Scholar]
- 18.Lo S-ZC, Nicholson RL. Plant Physiol. 1998;116:979–989. doi: 10.1104/pp.116.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Kondo K, Kurihara M, Fukuhara K, Tanaka T, Suzuki T, Miyata N, Toyoda M. Tetrahedron Lett. 2000;41:485–488. [Google Scholar]; (b) Nonaka G-I, Morimoto S, Kinjo J-E, Nohara T, Nishioka I. Chem Pharm Bull. 1987;35:149–155. [Google Scholar]
- 20.Harborne JB, Baxter H, editors. The Handbook of Natural Flavonoids. Vol. 1. Wiley; New York, NY: 1999. pp. vii–xvi. [Google Scholar]
- 21.Marais JPJ, Ferreira D, Slade D. Phytochemistry. 2005;66:2145–2176. doi: 10.1016/j.phytochem.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 22.(a) Selenski C, Pettus TRR. J Org Chem. 2004;69:9196–9203. doi: 10.1021/jo048703c. [DOI] [PubMed] [Google Scholar]; (b) Selenski C, Mejorado LH, Pettus TRR. Synlett. 2004:1101–1104. [Google Scholar]; (c) Jones RM, Selenski C, Pettus TRR. J Org Chem. 2002;67:6911–6915. doi: 10.1021/jo020224v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Selenski C, Pettus TRR. o-Quinone Methides. In: Griesbeck AG, editor. Science of Synthesis. Georg Thieme; Stuttgart: in press. [Google Scholar]; (e) Van De Water R, Magdziak D, Chau J, Pettus TRR. J Am Chem Soc. 2000;122:6502–6503. [Google Scholar]
- 23.(a) Steynberg PJ, Nel RJJ, van Rensburg H, Bezuiden-houdt BCB, Ferreira D. Tetrahedron. 1998;54:8153–8158. [Google Scholar]; (b) Botha JJ, Ferreira D, Roux DG. J Chem Soc Perkin Trans. 1981;1:1235–1245. [Google Scholar]; (c) Steenkamp JA, Ferreira D, Roux DG. Tetrahedron Lett. 1985;26:3045–3048. [Google Scholar]; (d) Delcour JA, Ferreira D, Roux DG. J Chem Soc Perkin Trans. 1983;1:1711–1717. [Google Scholar]; (e) Botha JJ, Ferreira D, Roux DG. J Chem Soc Perkin Trans. 1981;1:1235–1245. [Google Scholar]
- 24.(a) Arnaudinaud V, Nay B, Nuhrich A, Deffieux G, Mérillon J-M, Monti J-P, Vercauteren J. Tetrahedron Lett. 2001;42:1279–1281. [Google Scholar]; (b) Arnaudinaud V, Nay B, Sarah V, Nuhrich A, Deffieux G, Mérillon J-M, Monti J-P, Vercauteren J. Tetrahedron Lett. 2001;42:5669–5671. [Google Scholar]
- 25.(a) Saito A, Nakajima N, Tanaka A, Ubukata M. Tetrahedron. 2002;58:7829–7837. [Google Scholar]; (b) Saito A, Nakajima N, Tanaka A, Ubukata M. Tetrahedron Lett. 2003;58:5449–5452. [Google Scholar]; (c) Saito A, Emoto M, Tanaka A, Doi Y, Shoji K, Mizushina Y, Ikawa H, Yoshida H, Matsuura N, Nakajima N. Tetrahedron. 2004;60:12043–12049. [Google Scholar]; (d) Saito A, Doi Y, Tanaka A, Matsuura N, Ubukata M, Nakajima N. Bioorg Med Chem. 2004;12:4783–4790. doi: 10.1016/j.bmc.2004.07.024. [DOI] [PubMed] [Google Scholar]; (e) Saito A, Tanaka A, Ubukata M, Nakajima N. Synlett. 2004:1069–1073. [Google Scholar]
- 26.(a) Tückmantel W, Kozikowski AP, Romanczyk LJ., Jr J Am Chem Soc. 1999;121:12073–12081. [Google Scholar]; (b) Kozikowski AP, Tückmantel W, Hu Y. J Org Chem. 2001;66:1287–1296. doi: 10.1021/jo001462y. [DOI] [PubMed] [Google Scholar]; (c) Kozikowski AP, Tückmantel W, Böttcher G, Romanczyk LJ., Jr J Org Chem. 2003;68:1641–1658. doi: 10.1021/jo020393f. [DOI] [PubMed] [Google Scholar]
- 27.(a) Jurd L, Waiss AC., Jr Tetrahedron. 1965;21:1471–1483. [Google Scholar]; (b) Jurd L. J Heterocycl Chem. 1981;18:429–430. [Google Scholar]; (c) Jurd L, Lundin R. Tetrahedron. 1968;24:2653–2661. [Google Scholar]
- 28.Magdziak D, Rodriguez AA, van de Water RW, Pettus TRR. Org Lett. 2002;4:285–288. doi: 10.1021/ol017068j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Y, Zhang J, Pettus TRR. Org Lett. 2005;7:5841–5844. doi: 10.1021/o1023749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oyama K-I, Kondo T. Tetrahedron. 2004;60:2025–2034. [Google Scholar]
- 31.(a) Caisraghi G, Casnati G. Chem Commun. 1970:321. [Google Scholar]; (b) Caisraghi G, Casnati G, Salerno G. J Chem Soc C. 1971:2546–2548. [Google Scholar]
- 32.(a) Camarda L, Merlini L, Nasini G. Flavonoids from Natural Red Resins. In: Farkas L, Kallay F, Gabor M, Wagner H, editors. Flavonoids and Bioflavonoids, 1981. Akadémiai Kiado: Budapest; 1982. pp. 311–320. [Google Scholar]; (b) Arnone A, Nasini G, de Pava OV. J Nat Prod. 1997;60:971–975. doi: 10.1021/np9702188. [DOI] [PubMed] [Google Scholar]
- 33.(a) Sweeney JG, Iacobucci GA. Tetrahedron. 1981;37:1481–1483. [Google Scholar]; (b) Sweeney JG, Iacobucci GA. Tetrahedron. 1977;33:2927–2932. [Google Scholar]; (c) Robinson GM, Robinson R, Todd AR. J Chem Soc. 1934:809–812. [Google Scholar]; (d) Leon A, Robinson R. J Chem Soc. 1931:2732–2737. [Google Scholar]; (e) Pratt DD, Robinson R. J Chem Soc. 1925:1128–1138. [Google Scholar]
- 34.Mas T. Synthesis. 2003;12:1878–1880. [Google Scholar]
- 35.Caccamese S, Caruso C, Parrinello N, Savarino A. J Chromatogr. 2005;1076:155–162. doi: 10.1016/j.chroma.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 36.Frigerio M, Santagostino M, Sputore S. J Org Chem. 1999;64:4537–4538. [Google Scholar]