

Abstract
We examined the effect of a metallic silver particle on Förster resonance energy transfer (FRET) between a nearby donor-acceptor pair. A donor- labeled oligonucleotide was chemically bound to a single silver particle and then an acceptor- labeled complementary oligonucleotide was conjugated by hybridization. The photophysical behavior of FRET between the donor-acceptor pair on the metal particle was investigated using both ensemble emission spectra and single- molecule fluorescence detections. Both the emission intensities and lifetimes indicated an enhanced FRET efficiency due to the metal particle. This interaction led to an increase in the Förster distance for energy transfer from 8.3 to 13 nm. The rate constant of FRET near the silver particle was 21-fold faster than that of unbound donor-acceptor pair. These results suggest the use of metal-enhanced FRET for measuring proximity of large biomolecules or for energy transfer based assays.
Keywords: silver nanoparticle, Förster resonance energy transfer (FRET), metal plasmon resonance, metal-enhanced FRET, single molecule fluorescence detection
Introduction
The classic work of Drehage showed that the lifetime of a fluorophore is altered by a nearby metal mirror and that the lifetimes depend on the distance from the metal. Since these publications in the 1970’s, there has been theoretical2 and experimental3 interest in the behavior of fluorophores near metals. This interest has now extended to thin metal films and nanoparticles which display collective oscillations of electrons known as surface plasmons.4–5 Fluorophores are known to display the optical properties of oscillating point dipoles.6 The rates of radiation and the spatial distribution of the radiation can be dramatically altered due to near field interactions of the fluorophore with the metal.7 These interactions can increase the rates of fluorophore excitation and/or emission,7–10 a phenomenon which is now being used to develop sensitive fluorescence detection methods.11–12
Fluorescence resonance energy transfer (FRET), which is commonly used for biological research and sensing, occurs by the dipole-dipole interactions between an excited donor (D) molecule and an acceptor (A). This interaction is strongly dependent on the donor-acceptor distance, so FRET can be used to measure molecular distances or donor-to-acceptor proximity.13 The strength of the donor-acceptor interactions are usually described in terms of the Förster distance R0 at which FRET is 50% efficient. Förster distance is in the range of 2 to 6 nm, which is acceptable for measuring distances on modest size biomolecules but is often too short on some larger proteins, protein complexes, or moderate lengths of DNA. Hence it is of interest to identify methods to increase the distances over which FRET occurs.
Recent studies of metal plasmon resonances are revealing unexpected electrodynamic properties. One example is the energy transfer along chains of non-contacting metal particles, which occurs due to coherent energy transfer from particles-to-particle through dipole or multiple interactions.14 Furthermore, theory published in the 1980’s suggested that metal particles could increase the efficiency of FRET between donor and acceptor close to a metal particle.15 This theory suggests that metal particles can increase the strength of the donor-acceptor interactions. At present, there exists only a modest amount of experimental data on the modification of FRET by metal particles. Several reports from this laboratory using ensemble spectroscopy have shown increases in FRET.16 To our knowledge there have been no reports on the single- molecule FRET near metallic structures. Single molecule spectroscopy (SMS) is advantageous because of its ability to bypass the ensemble averaging and allow resolution of actual distributions of the spectral parameters.17 Such details of the underlying distribution become crucially important when the system under study is heterogeneous. In this study, we designed a single donor-acceptor pair separated by a rigid hybridized DNA duplex chain and bound this donor-acceptor pair to a single silver nanoparticle. FRET between the donor and acceptor was investigated using both the ensemble and single molecule measurements. Using single molecule spectroscopy (SMS), it is possible to select and study complexes which contain both a donor and an acceptor. Additionally, we can use the well known single-step photobleaching of fluorophores to select those particles which have single donor-acceptor pairs.
While the single molecule method allows us to identify single donor-acceptor pairs, our present approach does not allow sele ction based on metal particle size. Fortunately, the synthetic methods for forming particles are highly evolved and yield good homogeneity of particle size.18 The metal particles, which are protected by the organic monolayers on the metal cores, are used in this experiment. These monolayers protected particle s provide versatile and quantitative functionalization of the organic coating layers accomplished via ligand exchange, as well as the size controlling by the preparation conditions. In this work, N-(2-mercapto-propionyl)glycine (abbreviated as tiopronin) were used to prepare the tiopronin-coated silver particles by a modified Brust method. 19,20 These silver particles displayed a good chemical stability and solubility in water. The average diameter of metal core was 20 nm. 20 The metal particles were succinimidylated with (2-mercapto-propionylamino) acetic acid 2,5-dioxo-pyrrolidin-1-ylester via ligand exchange (Scheme 1). A donor-labeled single stranded oligonucleotide was chemically bound to the single silver particle via condensation between the terminated succinimidyl ester on the silver particle and the amino group on the oligonucleotide, and then an acceptor-labeled oligonucleotide was conjugated to the metal particle by the hybridization with the bound donor- labeled oligonucleotide. The donor-acceptor pair was separated by a rigid DNA duplex chain. In order to reduce the quenching by the absorbance from the metal particle at excitation and emission wavelengths, 21 two near infrared fluorophores cyanine (cy) 5 and 5.5 were employed as the donor and acceptor in FRET model, respectively.
Scheme 1.

Tiopronin-coated silver particle was succinimidylated via ligand exchange, followed by binding the aminated donor-labeled oligonucleotide and hybridization with the complementary acceptor- labeled oligonucleotide.
Experimental Section
All reagents and spectroscopic grade solvents were used as received from Fisher or Aldrich. RC dialysis membrane (MWCO 50,000) was obtained from Spectrum Laboratories, Inc. Nanopure water (>18.0 MΩ.cm) purified using Millipore Milli-Q gradient system, was used in all experiments. (2-mercapto-propionylamino) acetic acid 2,5-dioxo-pyrrolidin-1-ylester was synthesized as previous reported.22 Four oligonucleotides (Scheme 2) were synthesized by the Biopolymer Laboratory in the University of Maryland at Baltimore, in which one is labeled by the donor Cy5 and the complementary one is labeled by the acceptor Cy5.5. Oligos 1 and 2 were synthesized with one amino group substituted on the pyrimidine ring of thymine base.
Scheme 2.

Oligonucleotide sequences used in the experiments.
Preparing tiopronin-coated metal nanoparticles and binding aminated single-stranded oligonucleotides
Tiopronin-coated silver nanoparticles were prepared using a modified Brust reaction with a mole ratio of tiopronin/silver nitrate = 1/6 in methanol using reduction by an excess amount of sodium borohydride. 19,23 After filtration, the residual precipitated particles were washed thoroughly with methanol and acetone. Particle (1 mg/mL) and tiopronin (10 mM) were then co-dissolved in water and the solution was stirred for 24 h for annealing of the particles. 24 The water was removed under vacuum and the residual was washed thoroughly with methanol and acetone. The residual solid tiopronin-coated particles were further purified by dialysis against water.
The tiopronin-coated silver particles were succinimidylated by a ligand exchange reaction (Scheme 1). (2- mercapto-propionylamino) acetic acid 2,5-dioxo-pyrrolidin-1-ylester (2 × 10−8 M) and tiopronin-coated silver particle (1mg/mL, 4 × 10−8 M) were co-dissolved in a mixing solve nt of water/methanol (v/v = 1/1) and stirred for 24 h. Methanol was removed under vacuum. Free organic components were removed thoroughly by dialysis against water (MWCO 50,000). The animated oligonucleotides (oligo 1 or 2 in Scheme 2) were chemically bound onto the silver particles by adding oligonucleotides (1 μM) to the succinimidylated silver particles (1 mg/mL) in water with continuous stirring for 24 h. Unbound oligonucleotides were removed by extensive dialysis against water (MWCO 50,000).
Hybridization of oligonucleotides on tiopronin metal nanoparticles
The acceptor- labeled complementary oligonucleotides (oligo 3 or 4) were conjugated by hybridization with the bound oligo 1 or 2 on the metal particles in 10 mM NaCl buffer solution at pH = 7.4. 20 Unbound oligonucleotides were removed by dialysis against the buffer solution (MWCO 50,000). Using the same method, the unlabeled complementary oligonucleotide (oligo 4) was also hybridized to the bound oligo 1 or 2 on the silver particle to serve as controls. Unbound oligonucleotides were removed by dialysis against buffer solution (MWCO 50,000).
Spectra, lifetime, FRET, and TEM measurements
Absorption spectra were monitored with a Hewlett Packard 8453 spectrophotometer. Ensemble fluorescence spectra were recorded in solution with a Cary Eclipse Fluorescence Spectrophotometer. Ensemble lifetimes were measured by PicoQuant Modular Fluorescence Lifetime Spectrometer (Fluo Time 100) using PicoQuant 635 – 690 nm LED (LDH 8-1-568) as the light source with a resolution of 34 ps. A 650 nm filter was used to isolate the donor emission. Data were analyzed using single or dual-exponential model. For SMS-FRET measurements, a dilute solution was dispersed on a pre-cleaned glass coverslip. The coverslips (18×18 μm, Corning) used in the experiments were first soaked in a 10:1 (v:v) mixture of concentrated H2SO4 and 30% H2O2 overnight, extensively rinsed with water, sonicated in absolute ethanol for 2 min and dried with air stream. The sample solution was adjusted to a nanomolar concentration to give an appropriate surface density for single molecule studies. All single molecule measurements were performed using a time-resolved confocal microscopy (MicroTime 200, PicoQuant). Briefly, it consists of an inverted microscope coupled to a high-sensitivity detection setup. A single- mode pulsed laser diode (635 nm, 100ps, 40 MHz) (PDL800, PicoQuant) was used as the excitation source. An oil immersion objective (Olymp us, 100×, 1.3NA) was employed both for focusing laser light onto sample and collecting fluorescence emission from the sample. The fluorescence that passed a dichroic mirror (Q655LP, Chroma) was focused onto a 75 μm pinhole for spatial filtering to reject out-of- focus signals. The donor and acceptor emission were separated using a 50/50 nonpolarizing beam-splitter plate, which were then focused onto two single photon avalanche diodes (SPAD) (SPCM-AQR-14, Perkin Elmer Inc). Emission filters were used to eliminate the residual excitation and to minimize spectral crosstalk. Time-dependent fluorescence data were collected with a dwell time of 50 ms. The data was stored in the time-tagged-time-resolved (TTTR) mode, which allows recording every detected photon with its individual timing information. In combination with a pulsed diode laser, Instrument Response Function (IRF) widths of about 300 ps FWHM can be obtained, which permits the recording of sub- nanosecond fluorescence lifetimes, extendable to less than 100 ps with reconvolution. Lifetimes were estimated by fitting to a ?2 value of less than 1.2 and with a residuals trace that was fully symmetrical about the zero axis.
Transmission electron micrographs (TEM) were taken with a side-entry Philips electron microscope at 120 keV. Samples were cast from water solutions onto standard carbon-coated (200–300 Å) Formvar films on copper grids (200 mesh) by placing a droplet of a 1 mg/mL aqueous sample solution on grids. The size distribution of metal core was analyzed with Scion Image Beta Release 2 counting at least 200 particles.
Results and Discussion
The tiopronin-coated silver particles synthesized under the current conditions display good solubility in water. They are stable for months when dry or dispersed in water. The TEM images show that the metal particles are approximately homogeneous in size distribution and have an average diameter of 20 nm for the metal cores (Figure 1). Hence, the average chemical composition of the metal particle is estimated to be ca. (Ag)2.5×105(Tio)5.0×103. (2- mercapto-propio nylamino) acetic acid 2,5-dioxo-pyrrolidin-1-ylester (2 × 10−8 M) and tiopronin-coated silver particle (1mg/mL, 4 × 10−8 M) were co-dissolved in water with a mole ratio of 1/2. According to the report from Murray et al, the ligand exchange reaction occurs at a 1:1 mole ratio, 18 so the average number of succinimidyl ligands per particle is less than 0.5 even when all succinimidyl ligands are bound to the metal particles. This indicates that only half of metal particles are succinimidylated by a single functionalized ligand via the ligand exchange reaction. The low molar ratio of succinimidylation on each metal particle guarantees only a single oligonucleotide is bound to a silver particle.
Figure 1.
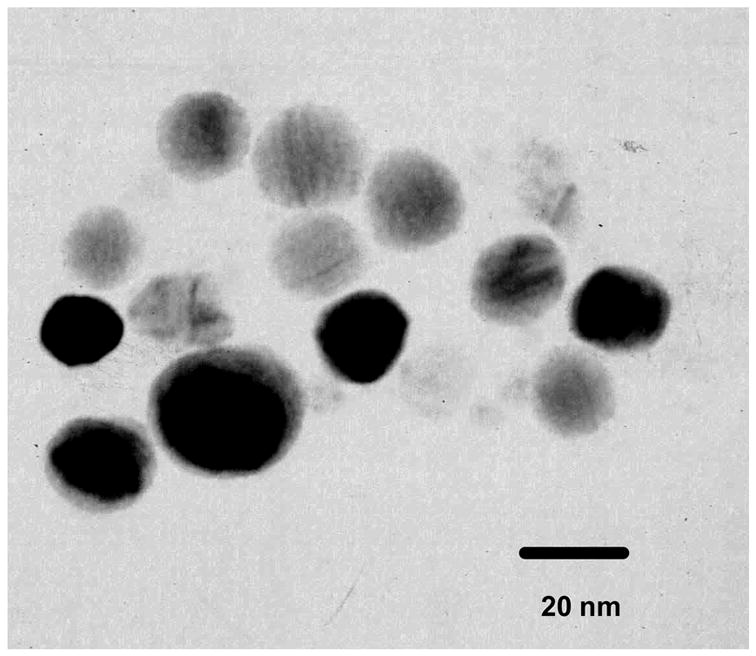
Transmission electron micrographs (TEM) of silver particles were taken with a side-entry Philips electron microscope at 120 keV.
The animated oligonucleotides (oligo 1 or 2 in Scheme 2) were chemically bound to the metal particle via condensation between the succinimidyl ester on the metal particle and the amine moiety on the oligonucleotide. Acceptor- labeled oligonucleotides (oligo 3) were conjugated to the metal particles by the hybridization with the oligo 1 or 2 bound on the metal particles, respectively, in 10 mM NaCl buffer solution at pH = 7.4. 20 As a control the unlabeled complementary oligonucleotide (oligo 4) was also conjugated to the metal particle. Tiopronin-coated silver particles displayed a typical plasmon absorbance near 400 nm (Figure 2). 20 The absorbance spectrum was not altered significantly with the succinimidylation, oligonucleotide binding, or hybridization of complementary oligonucleotide due to the low quantity of reaction on the surface of silver particle. Because the carboxylic ligands on the metal particle were depronated at the neutral pH of this buffer solutio n, the oligonucleotide ligands could not be physically absorbed onto the surface of particle. Moreover, the shortest distance from the oligonucleotide to the surface of silver particle was estimated to be 2.5 nm. Hence, the negative-charged oligonucleotides are expected to stay away from the negative-charged metal particle. This repulsion may alter the degree of stretching of oligonucleotide, but we feel that the relatively short oligonucleotide in this study can be simply regarded as a rigid rod. However, in the unlikely event the oligonucleotide is stretched when bound to the particle then the amount of energy transfer should decrease, which is opposite to the effect we observed (see below). Since the labeled oligonucleotide is distant from the particle it will be mobile in solution. This mobility should minimize the effects of donor-to-acceptor orientation or fluorophore orientation relative to the metal surface.
Figure 2.

Absorbance spectra of tiopronin-coated silver particles, succinimidylated silver particles, aminated oligo 1 bound silver particles, and oligo 3 conjugated silver particles by hybridization in buffer solution.
First we performed ensemble fluorescence measurement of the labeled oligonucleotide. Upon excitation at 610 nm, the free donor- labeled oligo 1 displayed an emission maximum at 661 nm (Figure 3), while acceptor-labeled oligo 3 displayed a maximum at 702 nm upon excitation at 650 nm. Oligo 1 and 3 were then hybridized at a molar ratio of 1:1 in buffer solution, and the hybridized oligo 1–3 showed an emission band at 661 nm and a rising shoulder at near 700 nm upon excitation at 610 nm. The emission maximum at 661 nm was ascribed to the donor and the shoulder at 700 nm to the acceptor. The emission intensity at 661 nm of hybridized oligo 1–3 decreased in comparison with that of free oligo 1 at the same concentration, which was due to FRET between the donor and acceptor pair. According to the change in the donor emission intensity, the efficiency of energy transfer was estimated to be 0.15. 6
Figure 3.

Emission spectra of oligo 1, 3, and hybridized oligo 1–3 in 10 mM NaCl buffer solution at pH = 7.4. Excitation was at 610 and 650 nm as shown.
Next we examined the ensemble spectra of the labeled oligonucleotides bound to silver particles. Oligo 1 bound to the silver particle displayed an emission at 663 nm upon excitation at 610 nm (Figure 4). Conjugated oligo 3 on the metal particle displayed an emission maximum at 705 nm upon excitation at 650 nm. These results showed that binding to the metal particle did not significantly alter the emission wavelengths. However, the bound oligo 1–3, which contains both donor and acceptor, displayed a very different emission spectrum from the free oligo 1–3. In the presence of metal particle the donor intensity was dramatically reduced while the acceptor emission was enhanced. This result indicates that the efficiency of energy transfer was increased by the metal particle. The transfer efficiency on the silver particle was estimated to be 0.70, 6 times higher than that of free donor – acceptor pair in solution. Oligo 2–4 bound to the metal particle displayed only a weak scattering background from the particle without a significant contribution to the emission from oligo 1–3. To verify that the spectral change was attributed to the presence of metal particles the silver was dissolved by adding a few drops of aqueous 0.1N NaCN and incubating for 15 min. In that process, oligo 1–3 was released completely from the metal particles. The released oligonucleotide pairs displayed an emission spectrum nearly identical to that of the free oligonucleotide pair, indicating that the change of FRET was primarily brought by the nearby metal particle.
Figure 4.

Emission spectra of oligo 1, 3, hybridized oligo 1–3, and hybridized 2–4 bound to the silver particle in 10 mM NaCl buffer solution at pH = 7.4. Excitation was at 610 and 650 nm as shown.
We believe our synthetic procedure resulted in a mostly homogeneous preparation with regard to particle size and the limit of one donor-acceptor pair per silver particle. However, the ensemble emission spectra cannot reveal any unexpected heterogeneity. The presence of such heterogeneity could compromise our interpretation of the data. Hence, we used single molecule spectroscopy to identify single donor-acceptor pairs in the sample complexed with silver particle s. An accepted indication of observing a single molecule is single-step photobleaching.
In a typical single- molecule experiment we recorded the intensity images of the donors and the acceptors simultaneously. This was accomplished using the two detection channels of our single molecule instrument and the appropriate emission filters. Upon excitation with the 635 nm laser, a portion of fluorescent spots in the donor channel were accompanied by relatively weak spots in the acceptor channel. There was a considerable amount of cross-talk between the channels due to spectra overlap of the two fluorophores. The intensities of the individual spots provided rough estimation of the degree of FRET. Representative example s of the time traces collected in the donor and acceptor channels in the absence of a metal particle are shown in Figure 5, showing that the intensity level from donor was brighter than that from the acceptor. This result was analogous to that observed from the ensemble emission spectra (Figure 3) where the acceptor was found to be weak relative to the donor emission. The traces shown in Figure 5 are representative of the single donor-acceptor pairs we examined. The acceptor emission decreased simultaneously with decrease photobleaching of the donor, indicating that most of the acceptor emission is the result of FRET and not direct excitation. The small fraction of molecules which did not display single step photobleaching was not included in the analysis.
Figure 5.

Respective time-trace intensities of a free donor-acceptor pair as seen in (a) the donor channel or (b) the acceptor channel. The insets represent the respective fluorescence images. The images are 150 × 150 pixels, with in integration time of 0.6 ms per pixel. Both images were recorded simultaneously using two SPAD detectors.
Next we examined single donor-acceptor pairs bound to silver particles. After binding of oligo 1–3 to a single silver particle, the emission intensity in the donor channel decreased while the intensity in the acceptor increased (Figure 6). While proximity of the donor and acceptor may have affected their total int ensities we believe the relative intensities of the donor and acceptor represent the efficiency of energy transfer.
Figure 6.

Respective time-trace intensities of a donor-acceptor pair bound on the silver particle as seen in (a) the donor channel or (b) the acceptor channel. The insets represent the respective fluorescence images. The images are 150 × 150 pixels, with in integration time of 0.6 ms per pixel. Both images were recorded simultaneously using two SPAD detectors.
Since most complexes contain some underlying heterogeneity the results observed for a single molecule may not be representative of the entire sample. Hence we examined a large number of individual molecules and used the resulting data to understand the overall properties of the sample. We selected individual donor-acceptor pairs and recorded the intensity traces for each molecule. The efficiency of energy transfer is estimated from the total photons in the donor and acceptor channels for each individual fluorescence spot. A correction factor was used for the detection efficiencies of the detectors 16,25 and for cross-talk between the detection channels. Histograms of the efficiencies for both free and particle bound donor-acceptor pairs were constructed using data from more than 50 single- molecule time traces (Figure 7a). Only spots displaying single-step photobleaching were included in the analysis. We found that the single molecule FRET efficiencies displayed a dramatic increase from 0.22 for the free donor-acceptor pair to 0.87 for the particle bound oligo pair. Hence the transfer efficiency was, on average, 4-fold larger for the donor-acceptor pair on the silver particles.
Figure 7.

Histograms of FRET on the free donor-acceptor pairs and that of the donor-acceptor pairs bound to the silver particle s: (a) the efficiency and (b) the relative distance counting from more than 50 single time traces.
One way to visualize the effect of the metal particle is to use the transfer efficiency to calculate the apparent donor-acceptor distance. The apparent distance r between the donor and acceptor can be calculated using6
| (1) |
where E is the efficiency of energy transfer and R0 is the Förster distance at which 50% energy is transferred and is characteristic of the donor-acceptor pair. We use the term “apparent” because the oligonucleotide is rigid and the silver particle is not expected to alter the donor-acceptor distance. If the silver particle increases the strength of the donor-acceptor interaction, the transfer efficiency increases and the donor and acceptor appear to be close together. For the present study it is not necessary to calculate the exact distances, but instead can consider the apparent as donor-to-acceptor distance r relative to R0 (r/R0). In the absence of silver particles the energy transfer efficiency near 22% corresponds to an apparent distance of 1.1 R0. When bound to the silver particles the transfer efficiency of 87% corresponds to an apparent distance of 0.75 R0. Hence, the presence of the silver particle results in an increase in the apparent donor-to-acceptor distance of nearly 40%.
It is also possible to interpret the data in terms of a change in R0 near the metal particles. This is a reasonable approach because the DNA is rigid and the donor-to-acceptor distance is not expected to change upon binding to the metal particle. From this perspective a change in transfer efficiency from 0.22 to 0.87 corresponds to an increase in the apparent Förster distance from 8.3 nm to 13 nm, an increase of 60% (Figure 7b). This suggests that the metal particle increases the strength of the donor-to-acceptor interaction. It is interesting to note that the largest reported Förster distances are all less than 10 nm.
It is of interest to compare the transfer efficiencies from the ensemble and single molecule experiments. Although the calculated efficiencies from the ensemble spectra and single-molecule measurements are different, the increase of Förster distances are approximately the same, indicating that FRET is enhanced similarly for the complexes dissolved in solution or cast on a solid substrate. Given the estimated length of 9.2 nm between the donor and the acceptor, the theoretical value of transfer efficiency is about 0.20, which is good agreement with the single-molecule results from the free donor-acceptor pair.
In addition to a change in the energy transfer, the metal particles can affect the intrinsic decay rates of fluorophores. This effect can be observed by measurements of the intensity decay.13 We measured the ensemble donor intensity decay for the samples in solution and analyzed the data in terms of a single exponential model. It was found that the donor lifetime of the oligo 1 (0.94 ns) in solution became shorter when hybridized with the oligo 3 (0.83 ns, Figure S1 in supporting information), so that the change in donor lifetime appears to be mostly due to FRET. The FRET efficiency was estimated to be 0.12 from the changes in lifetime. This value is close to that calculated from the intensity change. The lifetime of oligo 1 was shortened to 0.40 ns when bound to the silver particle, which was ascribed to the coupling between the fluorophore and metal particle. The lifetime of oligo 1 on the metal particle was not altered significantly when hybridized by oligo 4 (0.41 ns), but shortened to 0.15 ns when hybridized by the oligo 3, verifying the FRET was enhanced by the metal particle. The efficiency of energy transfer was estimated to be 0.63 from the donor lifetimes. It was noticed that the efficiencies calculated from the lifetimes were close to the corresponding values from the intensities (0.15 and 0.70).
The changes of FRET near the metal particle can also be studied from the single molecule lifetime measurements. The lifetimes of the free oligo 1–3 was 2.33 ns (Figure S2 in supporting information), but decreased to 0.85 ns when the donor-acceptor pair bound to the single silver particle. We found that the lifetimes obtained from the ensemble spectra or single- molecule measurements are different, probably due to the different environments for the fluorophores: in solution for the ensemble measurements and on solid substrate from the single molecule measurements. But both decreased with binding the donor-acceptor pair to the silver particle.
The single molecule donor lifetimes can be used to obtain information about the rates of energy transfer kT. The relationship of kT to the transfer efficiency is given by Equation 2,6
| (2) |
where, kT is the rate of energy transfer and τD−1 is the total lifetime of donor. When bound onto the metal particle, the rate of energy transfer due to coupling with the metal particle (km) has to be considered and hence eq. 2 becomes7
| (3) |
Because the transfer efficiency and lifetime are available from experiments, kT and km can be calculated from Equations 2 and 3. The rate of energy transfer kT is estimated to be 1.6 × 107 s−1 for the unbound oligo 1–3 when using the data from the ensemble spectra in solution. Similarly, the rate of transfer due to the metal particle was estimated to be 3.4 × 108 s−1. This value is 20-fold greater than the constant rate of unbound donor-acceptor pair, and demonstrated the important role of metal particles on energy transfer.
Conclusion
In this study, we investigated the effect of nano-size silver particles on the energy transfer between the donor and acceptor bound to an oligonucleotide. Because DNA is a rigid biomolecule the donor-to-acceptor distance is not expected to change upon binding to the metal particle. Hence, any changes in FRET will be due to changes in the donor-acceptor interaction caused by the metal. The silver particles had an average 20 nm diameter of metal core. In order to reduce the quenching by the metal particle at the excitation and emission wavelengths we used two near- infrared fluorophores Cy5 and Cy5.5 as the donor and acceptor, respectively. The donor and acceptor were separated by a rigid hybridized oligonucleotide and bound to a single metal particle. The emission properties were monitored using both the ensemble emission spectra in solution and single molecule measurements on a solid substrate. The efficiency of FRET was shown to be greatly enhanced and the Förster distance was obviously lengthened from 8.3 to 13 nm when binding the donor-acceptor pair to the single silver particle. Time-resolved donor intensity decays also showed an increase in transfer efficiency, and further allowed estimation of the rates of energy transfer. The rate constant for energy transfer was found to increase 21- fold for the donor-acceptor pair bound to the silver particle as compared to the unbound donor-acceptor pair. This result indicates that compared with the regular FRET, the metal enhanced FRET provides an opportunity for detecting donor and acceptor proximity over a longer distance than the standard Förster distance.
Supplementary Material
Acknowledgments
This research was supported by a grant from NIH, HG-02655, NCRR, and RR-08119.
References
- 1.Drexhage KH. J Lumin. 1970;12:693. [Google Scholar]
- Drexhage KH. In: Progress in Optics. Wolf E, editor. XII. North Holland: Amsterdam; 1974. [Google Scholar]
- 2.(a) Sokolov K, Chumanov G, Cotton TM. Anal Chem. 1998;70:3898. doi: 10.1021/ac9712310. [DOI] [PubMed] [Google Scholar]; (b) Tarcha PJ, De Saja-Gonzalez J, Rodriguez-Llorente S, Aroca R. Appl Spectrosc. 1999;53:43. [Google Scholar]; (c) De Saja-Gonzalez J, Aroca R, Nagao Y, De Saja JA. Spectrochim Acta A. 1997;53:173. [Google Scholar]
- 3.(a) Tews KH. J Lumin. 1974;9:223. [Google Scholar]; (b) Chance RR, Prock A, Silbey R. J Chem Phys. 1974;60:2744. [Google Scholar]; (c) Persson BNJ. J Phys C Solid State Phys. 1978;11:4251. [Google Scholar]; (d) Gersten J, Nitzan A. J Chem Phys. 1981;75:1139. [Google Scholar]; (e) Ruppin R. J Chem Phys. 1982;76:1681. [Google Scholar]; (f) Barnes WL. J Modern Optics. 1998;45:661. [Google Scholar]
- 4.(a) Kummerlen J, Leitner A, Brunner H, Aussenegg FR, Wokaun A. Mol Phys. 1993;80:1031. [Google Scholar]; (b) Antunes PA, Constantino CJL, Aroca RF, Duff J. Langmuir. 2001;17:2958. [Google Scholar]; (c) Kamat PV. J Phys Chem B. 2002;106:7729. [Google Scholar]; (c) Kelly KL, Coronado E, Zhao LL, Schatz GC. J Phys Chem B. 2003;107:668. [Google Scholar]; (d) Hao E, Li S, Bailey RC, Zou S, Schatz GC, Hupp JT. J Phys Chem B. 2004;108:1224. [Google Scholar]
- 5.(a) Kumbhar AS, Kinnan MK, Chumanov G. J Am Chem Soc. 2005;127:12444. doi: 10.1021/ja053242d. [DOI] [PubMed] [Google Scholar]; (b) Bruzzone S, Malvaldi M, Arrighini GP, Guidotti C. J Phys Chem B. 2005;109:3807. doi: 10.1021/jp045451a. [DOI] [PubMed] [Google Scholar]; (c) Hubert C, Rumyantseva A, Lerondel G, Grand J, Kostcheev S, Billot L, Vial A, Bachelot R, Royer P, Chang S-h, Gray SK, Wiederrecht GP, Schatz GC. Nano Lett. 2005;5:615. doi: 10.1021/nl047956i. [DOI] [PubMed] [Google Scholar]; (d) Millstone JE, Park S, Shuford KL, Qin L, Schatz GC, Mirkin CA. J Am Chem Soc. 2005;127:5312. doi: 10.1021/ja043245a. [DOI] [PubMed] [Google Scholar]
- 6.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum Published; New York: 1999. [Google Scholar]
- 7.(a) Lakowicz JR. Anal Biochem. 2005;337:171. doi: 10.1016/j.ab.2004.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lakowicz JR. Anal Biochem. 2001;298:1. doi: 10.1006/abio.2001.5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gryczynski I, Malicka J, Shen YB, Gryczynski Z, Lakowicz JR. J Phys Chem B. 2002;106:2191. doi: 10.1021/jp013013n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Malicka J, Gryczynski I, Gryczynski Z, Lakowicz JR. Anal Biochem. 2003;315:57. doi: 10.1016/S0003-2697(02)00702-9. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Aslan K, Huang J, Wilson GM, Geddes CD. J Am Chem Soc. 2006;128:4206. doi: 10.1021/ja0601179. [DOI] [PubMed] [Google Scholar]
- 9.(a) Shen Y, Swiatkiewicz J, Lin TC, Markowicz P, Prasad PN. J Phys Chem B. 2002;106:4040. [Google Scholar]; (b) Yonzon CR, Jeoung E, Zou S, Schatz GC, Mrksich M, Van Duyne RP. J Am Chem Soc. 2004;126:12669. doi: 10.1021/ja047118q. [DOI] [PubMed] [Google Scholar]; (c) Yu F, Persson B, Lofas S, Knoll W. J Am Chem Soc. 2004;126:8902. doi: 10.1021/ja048583q. [DOI] [PubMed] [Google Scholar]; (d) Lee IYS, Suzuki H, Ito K, Yasuda Y. J Phys Chem B. 2004;108:19368. [Google Scholar]
- 10.(a) Ekgasit S, Thammacharoen C, Yu F, Knoll W. Anal Chem. 2004;76:2210. doi: 10.1021/ac035326f. [DOI] [PubMed] [Google Scholar]; (b) Song JH, Atay T, Shi S, Urabe H, Nurmikko AV. Nano Lett. 2005;5:1557. doi: 10.1021/nl050813r. [DOI] [PubMed] [Google Scholar]; (c) Baluschev S, Yu F, Miteva T, Ahl S, Yasuda A, Nelles G, Knoll W, Wegner G. Nano Lett. 2005;5:2482. doi: 10.1021/nl0517969. [DOI] [PubMed] [Google Scholar]; (d) Kawasaki M, Mine S. J Phys Chem B. 2005;109:17254. doi: 10.1021/jp053167t. [DOI] [PubMed] [Google Scholar]
- 11.(a) Rosi NL, Mirkin CA. Chem Rev. 2005;105:1547. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]; (b) Wabuyele MB, Vo-Dinh T. Anal Biochem. 2003;77:7810. doi: 10.1021/ac0514671. [DOI] [PubMed] [Google Scholar]
- 12.Lakowicz JR. Emerging biomedical application of time-resolved florescence spectroscopy, Topic in Fluorescence spectroscopy. In: Lakowicz JR, editor. Probe Design and Chemical Sensing. Vol. 4. Plenum Press; New York: 1994. [Google Scholar]
- 13.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Kluwer Academic/Plenum Publishers; New York: 1999. [Google Scholar]
- 14.(a) Citrin DS. Nano lett. 2004;4:1561. [Google Scholar]; (b) Citrin DS. Nano lett. 2005;5:985. doi: 10.1021/nl050513+. [DOI] [PubMed] [Google Scholar]
- 15.(a) Hua XM, Gersten JI, Nitzan A. J Chem Phys. 1985;83:3650. [Google Scholar]; (b) Gersten JI, Nitzan A. Chem Phys Letts. 1984;104:31. [Google Scholar]
- 16.(a) Malicka J, Gryczynski I, Fang J, Kusba J, Lakowicz JR. Anal Biochem. 2003;315:160. doi: 10.1016/S0003-2697(02)00710-8. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lakowicz JR, Kusba J, Shen Y, Malicka J, D’Auria S, Gryczynski Z, Gryczynski I. J Fluoro. 2003;13:69. doi: 10.1023/A:1022306630924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Deniz AA, Dahan M, Grunwell JR, Ha TJ, Faulhaber AE, Chemla DS, Weiss S, Schultz PG. Proc Natl Acad Sci USA. 1999;96:3670. doi: 10.1073/pnas.96.7.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ha TJ.Methods 2001257811558999 [Google Scholar]; (c) Ebenstein Y, Mokari T, Banin U. J Phys Chem B. 2004;108:93. [Google Scholar]; (d) Tinnefeld P, Sauer M. Angew Chem Int Ed. 2005;44:2642. doi: 10.1002/anie.200300647. [DOI] [PubMed] [Google Scholar]
- 18.(a) Hayat MA, editor. Colloidal Gold: Principles, Methods, and Applications. Academic Press; San Diego: 1991. [Google Scholar]; (b) Kreibig U, Vollmer M. Optical Properties of Metal Clusters. Springer-Verlag; Berlin and Heidelberg, Germany: 1995. [Google Scholar]; (c) Feldheim DL, Foss CA. Metal Nanoparticles. Synthesis, Characterization and Applications. Marcel Dekker, Inc; New York: 2002. [Google Scholar]
- 19.(a) Templeton AC, Wuelfing WP, Murray RW. Acc Chem Res. 2000;33:27. doi: 10.1021/ar9602664. [DOI] [PubMed] [Google Scholar]; (b) Hostetler MJ, Wingate JE, Zhong CJ, Harris JE, Vachet RW, Clark MR, Londono JD, Green SJ, Stokes JJ, Wignall GD, Glish GL, Porter MD, Evans ND, Murray RW. Langmuir. 1998;14:17–30. [Google Scholar]; (c) Ingram RS, Hostetler MJ, Murray RW. J Am Chem Soc. 1997;119:9175. [Google Scholar]; (d) Huang T, Murray RW. Langmuir. 2002;18:7077. [Google Scholar]; (b) Huang T, Murray RW. J Phys Chem B. 2001;105:12498. [Google Scholar]
- 20.Zhang J, Malicka J, Gryczynski I, Lakowicz JR. J Phys Chem B. 2005;109:7643. doi: 10.1021/jp0490103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aguila A, Murray RW. Langmuir. 2000;16:5949. [Google Scholar]
- 22.Zhang J, Roll D, Geddes CD, Lakowicz JR. J Phys Chem B. 2004;108:12210. doi: 10.1021/jp037772c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brust M, Walker M, Bethell D, Schiffrin DJ, Whyman R. J Chem Soc, Chem Commun. 1994:801. [Google Scholar]
- 24.(a) Hicks JF, Miles DT, Murray RW. J Am Chem Soc. 2002;124:13322. doi: 10.1021/ja027724q. [DOI] [PubMed] [Google Scholar]; (b) Schaaff TG, Shafigullin MN, Khoury JT, Vezmar I, Whetten RI. J Phys Chem. 2001;105:8785. [Google Scholar]
- 25.(a) Bemey C, Danuser G. Biophys J. 2003;84:3992. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ha T, Enderle Th, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Proc Natl Acad Sci USA. 1996;12:6264. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Koberling F. FRET Analysis of Free Diffusing Molecules using the MicroTime 200. PicoQuant GmbH. 2004;(6) [Google Scholar]
- 26.Hohng S, Joo C, Ha T. Biophys J. 2004;87:1328. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Karymov M, Daniel D, Sankey OF, Lyubchenko YL. Biophys J. 2005;102:8186. doi: 10.1073/pnas.0407210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.