

Abstract
Efficient photopolymerization of a potentially expandable monomer is of practical importance for a variety of polymeric applications demanding dimensional stability, particularly if the polymerization process is well controlled based on a detailed investigation of the reaction. In the current study, photoinitiated polymerization kinetics of 2-methylene-7-phenyl-1,4,6,9-tetraoxaspiro[4.4]nonane (MPN) either with cationic initiation alone or with combined cationic/free radical initiation was examined using real-time FT-IR. A proposed mechanism based on the simplified propagation steps of the cationic double ring-opening polymerization of MPN was confirmed by both computer modeling and NMR spectroscopic analysis of resulting polymers as well as the experimentally observed apparent activation energy. According to this mechanism, α-position attack is the predominant mode for the second ring opening during cationic polymerization of MPN. Further, cationic photopolymerization was performed along with a free radical co-initiator or with exposure to moisture to get an improved understanding of the complex cationic double ring-opening polymerization. As a result, free radical-promoted cationic polymerization helps increase the polymerization rate of MPN while even a trace amount of moisture was found to significantly impact both the reaction kinetics and the polymerization course.
INTRODUCTION
Volumetric shrinkage during polymerization results from the transformation of highly mobile monomer molecules to the relatively immobile, compact polymer chains. Polymerization shrinkage and consequent shrinkage stress cause a variety of problems in practical applications, such as dental restorative materials1, adhesives2, stereolithography3 and coatings4. Various efforts have been directed to reduce shrinkage and shrinkage stress, such as the use of high molecular weight monomers5, 6 or reactive prepolymers/oligomers with low functional group concentrations7, modified polymerization conditions8 and thiol-ene systems7. Nevertheless, these methods do not have the potential to achieve zero volumetric shrinkage or even expansion during polymerization.
In the 1970s, spiro orthocarbonate (SOC) monomers were initially developed9 and several examples of this class of monomers were found to produce a volumetric expansion during double ring-opening polymerization due to the compact nature of the bicyclic monomer and the relatively more flexible open-chain structure associated with the polymer. Since then, numerous investigations into the synthesis and polymerization of SOCs have been reported10–12. However, the majority of these double ring-opening polymerization studies of SOCs have been performed under solution polymerization conditions and typically with cationic initiators such as boron trifluoride etherate over a wide range of temperatures, usually at elevated temperatures11, 12. These polymerization conditions limit the practical utility of these potentially expandable monomers and complicate any interpretation of volume change, whether positive or negative. As a consequence, incommensurate to the extensive studies on SOCs, there are relatively few evaluations of these monomers under conditions that might favor their practical applications.
Photopolymerization offers a number of advantages over thermal polymerization, such as spatial and temporal control as well as no volatile solvent13. A few studies on photopolymerization of SOC monomers at ambient conditions have been reported14, 15 with most conducted as free radically or cationically initiated polymerizations in combination with methacrylate or epoxide comonomers. In addition, SOCs are known to be susceptible to acid-catalyzed hydrolysis16, which would result in the formation of small molecules and/or low molecular weight oligomers. These decomposition processes will certainly affect dimensional change and polymer properties obtained. In order to better understand the effects of SOC monomers in copolymerization systems, it is important to investigate the homopolymerization mechanism and kinetics of SOCs.
As initially demonstrated by Sakai et al., the ring-opening polymerization mode is related to ring size for unsubstituted spiro orthocarbonates17. SOCs with six-membered cyclic structures generally undergo double ring opening polymerization to form polyethercarbonate, although multiple competing mechanisms also result in the liberation of small molecules. In contrast, SOCs composed of five-membered or seven-membered rings exhibit relatively higher cationic polymerizability due to greater ring strain but mainly undergo single ring-opening polymerization with the elimination of ethylene carbonate or tetrahydrofuran, respectively. These undesired side reactions would significantly reduce the mechanical properties of the final polymer that are already expected to be low due to the flexible linear nature of the repeat unit in these addition polymers and the relatively low molecular weights typically obtained. However, subsequent studies11, 12 demonstrated that the cationic polymerization mechanisms of SOCs are also directed by substitution patterns. SOCs with aromatic groups substituted adjacent to the spiro ether, unlike analogs with aliphatic ring substitution, undergo clean double ring-opening polymerization without elimination, although the polymer molecular weight is still relatively low. The formation of a stable benzyl cation during ring-opened polymer propagation appears to help direct and drive this process. This is one of the reasons why the phenyl-substituted MPN monomer was chosen in the current study.
Known as a liquid SOC monomer with an exocyclic methylene group, MPN (Figure 1) was reported as a cationically photopolymerizable SOC under ambient conditions18. However, despite the lack of any kinetic study, such an unsaturated SOC monomer has already been utilized in the development of new photocurable materials for potential practical applications, such as low shrinkage dental restoratives15, 19. Several SOCs synthesized in our lab, including 2,8-dimethyl-1,5,7,11-tetraoxaspiro[5.5]undecane, 3,9-dimethyl-1,5,7,11-tetraoxaspiro[5.5]undecane, 2-methyl-7-chloromethyl-1,4,6,9-tetraoxaspiro[4.4]nonane and unsaturated 2,7-dimethylene-1,4,6,9-tetraoxaspiro[4.4]nonane, were found to be reasonably active toward initiators such as boron trifluoride etherate but to have only minimal reactivity in photopolymerizations with iodonium salt initiators at room temperature20. Thus, it is important to understand the polymerization mechanism of MPN to help design efficiently photopolymerizable SOC monomers in the future.
Figure 1.
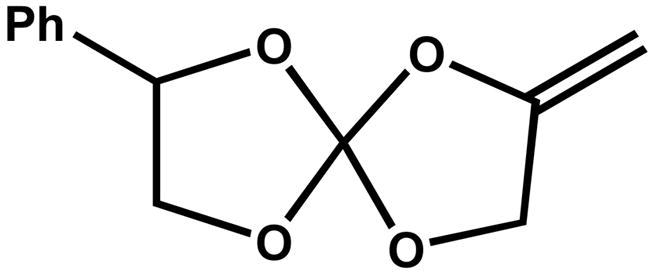
Structure of MPN.
The main purpose of this investigation is to study the photoinitiated cationic polymerization of MPN in detail to gain a better understanding of the kinetics as well as the ring-opening polymerization mechanism and mechanisms of competing reactions. This information on MPN may eventually help identify new SOC monomers that allow the development of novel low shrinkage or expanding materials.
EXPERIMENTAL SECTION
Materials
The free radical photoinitiator used was 2,2-dimethoxy-2-phenyl acetophenone (DMPA). The cationic photoinitiators used were [4-[(2-hydroxytetradecyl)oxy]-phenyl]phenyliodonium hexafluoroantimonate (ISb), ferrocenium hexafluorophosphate (FeP) and (tert-butoxycarbonylmethoxynaphthyl)-diphenylsulfonium triflate (SS). The initiators and all other chemicals utilized for monomer synthesis and polymer processing were obtained from Aldrich Chemical Co. (Milwaukee, WI) and were used as received. The MPN monomer was synthesized mainly according to the four-step procedure of Bolln18.
Methods
1H- and 13C-NMR spectra (Varian Inova-400) were recorded using TMS as internal standard in CDCl3 at ambient temperature. The polymerization reaction was monitored in real-time with a FT-IR spectrometer (Thermo-Nicolet Magna 560). To initiate photopolymerization, a UV light source (Acticure, EXFO, Mississaugua, Ontario, Canada) with 320–500 nm filter was used. Thick films for near-IR experiments were prepared by sandwiching samples in rectangular molds made from glass slides at the thickness of about 1 mm. Thin films (thickness: 10 – 15 μm) for mid-IR experiments were prepared by laminating samples between NaCl crystals. A horizontal accessory21 was used to position the sample horizontally while perpendicular to the IR beam. For photopolymerizations conducted at various temperatures, a temperature cell, which was designed to fit in the horizontal accessory, was used.
Initiation condition selection
Photopolymerization with 0.5 mol% of the three cationic photoinitiators showed that FeP could initiate MPN polymerization at room temperature although difficulties with spontaneous polymerizations even in amber vials were encountered. MPN with SS produced very low polymerization rates, though the solubility of SS in MPN is much better than that of ISb. Hence, the ISb photoinitiator with relatively fast but controlled polymerization kinetics was chosen in this study. Due to the limited solubility of ISb in MPN, a 0.3 mol% concentration was used throughout the subsequent cationic photopolymerization study. The compositions used in the study of free radical-promoted cationic polymerization were ISb at 0.3 mol% together with DMPA at 0.0 mol%, 0.1 mol%, 0.3 mol% and 0.6 mol%. All initiator molar percentages are relative to the monomer. Unless otherwise noted, all photopolymerizations were conducted at room temperature with an incident UV irradiation of 20 mW/cm2 for 720 s followed by continued monitoring of the dark cure for an additional 180 s.
RESULTS AND DISCUSSION
Measurement of double ring-opening polymerization conversion
Mid-infrared (MIR) spectra of MPN upon cationic photopolymerization were evaluated to identify absorbance bands that potentially could be used to monitor the ring-opening polymerization process during subsequent real-time studies of polymerization kinetics. The characteristic tetraoxaspiro C-O absorbance at 1212 cm−1 (Figure 2a) is in a spectral region of complex overlapping absorbance bands that would make it difficult to follow the peak area evolution accurately during polymerization. To study the kinetics of MPN polymerization using MIR, an alternative well-resolved peak that can indicate double ring-opening polymerization is required. As shown in Figure 2b, the exocyclic carbon-carbon double bond peak at 1697 cm−1 appears to be a good choice if the double bond conversion is directly related to the double ring-opening polymerization conversion. In other words, there should be no 1,2-addition to leave the SOC moiety as a pendent group in the polymer. To validate this method, the resulting polymer was first dissolved in chloroform and then precipitated in methanol. The precipitated polymer, which has no double bond peak, also has no characteristic tetraoxaspiro C-O absorbance in FT-IR spectrum. This result along with the consistent ratio between carbonate carbonyl and keto carbonyl peaks in the IR as shown in Figure 2b, and the agreement with the NMR spectral assignments indicate that the desired double ring opening is the primary reaction pathway. Therefore, double bond conversion during polymerization is useful to represent the kinetics of the cationic double ring-opening polymerization of MPN. However, the double bond peak (C=C) in the MIR range at 1697 cm−1 still has a slight overlap with the keto carbonyl peak formed during polymerization, which complicates the measurement of the double bond peak area and compromises the accuracy, especially at the later stages of the polymerization with very substantial carbonyl absorbance. To avoid this problem, most of the kinetic data in the current study are based on the disappearance of the well-resolved double bond peak (=CH2) at 6202 cm−1 in near-infrared (NIR; Figure 2c), which is widely used to monitor (meth)acrylate and other vinyl monomer polymerization kinetics for thick films22. Since MPN has very limited reactivity (well less than 5% conversion) in ambient temperature photopolymerization with only DMPA as the free radical initiator, the NIR double bond peak area was used to monitor the double ring-opening conversion in the subsequent free radical-promoted cationic polymerization study as well.
Figure 2.

FT-IR spectra during photopolymerization focusing on (a) tetraoxaspiro C-O absorption at 1212 cm−1, (b) exocyclic methylene absorption at 1697 cm−1 in mid-IR and (c) exocyclic methylene absorption at 6202 cm−1 in near-IR. Arrows indicate time progression.
Mechanism of MPN cationic double ring-opening polymerization
Bolln18 reported that MPN has about 4.1 % volumetric shrinkage during polymerization, which is fairly consistent with the 6.1 % shrinkage value at complete conversion obtained in this study based on the monomer and polymer densities, both measured with gas pycnometer23. Therefore, even with an efficient double ring-opening polymerization, MPN has significant volumetric shrinkage. In addition, as mentioned before, not all SOCs can undergo efficient double ring-opening polymerization at ambient photopolymerization conditions. Thus, it is important to understand the polymerization mechanism of MPN in order to design novel potentially expandable SOC monomers that can undergo efficient double ring-opening polymerization.
According to Bolln18, there are two possible major pathways for cationic double ring-opening polymerization of MPN (Figure 3a). The first is to polymerize through the stable benzyl cation 3a, and the other is through the rearranged benzyl cation 3c that would result in slightly different polymer structures 4a, 4c. However, due to the relatively high stability of the single-ring opened intermediate trioxocarbonium 224, which is stabilized by three alkoxy groups, cation 3a–3c may not exist as intermediate carbocations in any appreciable concentration during photopolymerization. Thermodynamic calculation (MOPAC 2002, AM1 semi-empirical quantum mechanical method, CAChe®, Fujitsu America) results confirmed that the single ring-opening intermediate cation 2 with a heat of formation of 40.5 kcal/mol is the predominant cation species over cation 3a–3c with heats of formation of 54.6, 46.2 and 49.3 kcal/mol, respectively. Therefore, two possible mechanistic pathways involving bi-molecular reaction17 for the cationic homopolymerization of MPN are proposed here as shown in Figure 3b. The mechanism of the first ring opening to cation 2 is the same as suggested by Bolln18 with the formation of energetically favored carbonyl group and stabilized trioxocarbonium. However, the proposed pathways for the second ring-opening process, involving nucleophilic attack of the monomer on α and β carbons in carbocation 2, lead to products 3α and 3β, respectively. To determine the probability of these two possible polymerization pathways, molecular modeling calculations were also performed. Firstly, the geometries of the reactant (1 + 2) and the resulting products (3α and 3β) were optimized and heats of formation as well as free energies were calculated. Then, the heat of formation and free energy of transition states of the proposed simplified propagation steps involving one monomer molecule and intermediate 2 (Figure 3b) were also calculated. The calculated results for both α and β nuclephilic attacks are shown in Figure 4. As clearly demonstrated, both α and β attacks resulted in products with similar free energies at around −15 kcal/mol, which means negligible selectivity based on the thermodynamic stability of products 3α and 3β. However, the free energy of the transition state for α attack is much lower than that for β attack. Based on this, the calculated propagation rate of α attack is 14 orders of magnitude faster than that of β attack at room temperature. From the calculated results, it is concluded that α attack, which results in product 3α is the predominant pathway for the MPN cationic double ring-opening polymerization at ambient conditions and the resonance stabilization is much more important than the steric energy in determining the mode of attack during polymerization.
Figure 3.

(a) Bolln’s and (b) proposed bi-molecular mechanism of the cationic double ring-opening polymerization of MPN.
Figure 4.
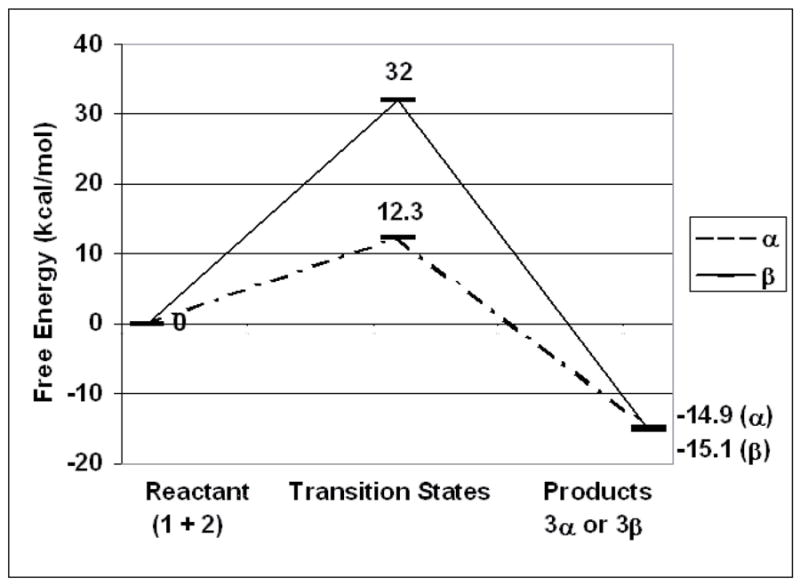
Calculated simplified propagation reaction coordinates for α and β attack.
This mechanism based on the calculation of activation energy was also substantiated by NMR spectra of the resulting polymer. As shown in the 1H spectrum (Figure 5), the peak at about 3.6 ppm represents the benzylic proton on C7 (denoted with a star; Figure 3b). Protons with this chemical shift only arise from product 3α. After normalization of the non-aromatic protons integration to 7 because of the interference of deuterochloroform in the aromatic region, the integration of the peak at δ 3.6 ppm is about 0.9, indicating about 93 % α attack.
Figure 5.

1H-NMR spectrum of MPN (a) monomer and (b) precipitated homopolymer in CDCl3 (Polymerization conditions: 23 °C; 0.3 wt% ISb as the cationic photoinitiator; 20 mW/cm2, 320–500 nm UV irradiation for 12 min).
Temperature effects on polymerization kinetics
Further studies of reaction kinetics at various photopolymerization temperatures were conducted to determine the apparent activation energy. As shown in Figure 6, temperature has a significant effect on the polymerization rate of MPN, but not as much on the final monomer conversion except for 25 °C. However, polymerization at 25 °C still could reach similar ultimate conversion if given sufficient cure time. These similar ultimate conversion values for curing temperatures ranging from 25 to 90 °C suggest that the resulting polymer from MPN photopolymerization, which is rubbery at room temperature, has a glass transition temperature (Tg) not far below 25 °C, although no measurement of Tg was conducted here. IR spectra of polymers obtained at the various cure temperatures showed no obvious differences that would indicate reaction pathways other than the double ring opening.
Figure 6.

Conversion vs. irradiation time of MPN photopolymerization under various temperatures (Polymerization conditions: 0.3 wt% ISb as the cationic photoinitiator; 20 mW/cm2, 320–500 nm UV irradiation).
With the kinetic profiles available for a series of reaction temperatures, it is possible to calculate the approximate activation energy for this polymerization reaction. To simplify the calculation, an Arrhenius plot (Figure 7) was constructed from cationic polymerization rates measured after a constant photolysis time of 15 s. Presumably, there would be constant cation concentrations in the systems for all temperatures due to the same photolysis time and the relatively slow termination kinetics of cationic polymerizations. In addition, at 15 s, except for 75 °C, when the conversion is about 20 %, the conversion values at all the other plotted temperatures are still below 10 % where the diffusion limitation due to increased viscosity would be minimal. As shown, the plot is linear, according to the Arrhenius relationship, indicating that the apparent activation energy observed for this cationic polymerization is about 13.3 kcal/mol, which is reasonably close to the calculated activation energy of 12.3 kcal/mol. This agreement further confirms the proposed bi-molecular α position attack mechanism.
Figure 7.

Arrhenius Plot of ln k versus 1/T for the cationic polymerization of MPN.
Free radical promoted cationic polymerization of MPN
The previous mechanistic discussion is focused on the opening of the second ring since this has been shown to be the rate determining step in this process14. However, control of the initiation pathway, which involves addition to the SOC vinyl group is critical to the overall reaction kinetics. As shown in Figure 6, the MPN polymerization rate is relatively low at ambient temperature with only cationic photoinitiator ISb, which is partially because ISb only has a weak shoulder absorbance in the UV range over 300 nm as shown in Figure 8. To explore the potential utility of this kind of double ring-opening monomer, instead of raising cure temperature, which would impose limitations on potential applications, the effect of an added free radical initiator (DMPA) on polymerization rate was examined. The utility of free radically-promoted cationic polymerization was initially demonstrated by Bi and Neckers25 based on a dye, an amine, and a diaryliodonium salt initiating system. Later, it was found that free radical-assisted cationic polymerization provides enhanced reactivity in cationic epoxide26 and vinyl ether27 systems.
Figure 8.

UV-Vis absorbance spectra of free radical photoinitiator DMPA and cationic photoinitiator ISb and the UV irradiation spectrum of medium pressure mercury lamp with 320–500 nm filter.
The inclusion of DMPA, which has a significant absorbance over the UV irradiation wavelength range used, helps increase the polymerization rate of MPN (Figure 9) in several ways as shown in Scheme 1. Equations 1 and 2 represent direct photolysis of the cationic and free radical photoinitiators with equations 3 and 4 showing the addition of either the protonic acid or the carbon-based radical to MPN monomer. While primary radicals generated by DMPA can add to the vinyl group, the resulting radical species (equation 4a) does not readily propagate due to the electron-rich nature of the SOC double bond, which is similar to a vinyl ether; however, following oxidation by ISb, the cationic species created (equation 4b) apparently is capable of efficient cationic initiation of MPN polymerization. Finally, radicals produced by the photodecomposition of DMPA (or by other pathways) can also be oxidized by ISb to a carbocation (equation 5a), which can add directly to MPN to form another cationic species (equation 5b) capable of propagation.
Figure 9.

Conversion versus irradiation time of MPN photopolymerization with the addition of free radical photoinitiator DMPA (Polymerization conditions: 23 °C; 20 mW/cm2, 320–500 nm UV irradiation for 12 min).
Scheme 1.

As the concentration of DMPA is increased, the polymerization rate of MPN increases rapidly. Shown more clearly in the plot of normalized polymerization rate versus monomer conversion (Figure 10), DMPA addition results in increases in the rate at the initial stage of polymerization, due to the enhanced cationic center concentration, and it also raises the conversion at which the maximum polymerization rate (Rp, max) occurs. Because of slow cationic termination processes, this is related to the competition between active center generation and deactivation (or trapping)28. Upon irradiation, active centers are generated, while due to the low degree of conversion, there is only negligible diffusion limitation and thus the polymerization rate increases rapidly. As the polymerization goes on, the rate of the active center generation decreases due to the decreased initiator concentration, while the diffusion limitation increases and thus a reduced polymerization rate is obtained. With high DMPA concentrations in the system, the rate of active center generation is fast enough to push the Rp, max to higher conversion values, where a more significant diffusion limitation is expected.
Figure 10.

Normalized (to double bond concentration) rate of polymerization (Rp) for MPN cationic photocuring with the addition of free radical photoinitiator DMPA.
Moisture effect on the polymerization kinetics and mechanism
As mentioned, SOC monomers are susceptible to acid-catalyzed hydrolysis that results in the release of small molecules. This process may help reduce volumetric shrinkage attributed to the ring-opening polymerization; however, it would also be detrimental to the final polymer properties due to the lowered polymer molecular weight and plasticiser effects. A recent study showed that the inclusion of 12 mol% of a tetraoxaspiro undecane in an aliphatic dioxirane resin system significantly reduced the elastic modulus of the polymer29. However, the IR spectrum of the polymer also indicated a very substantial hydroxyl peak around 3500 cm−1. Large OH absorptions have also been observed in the IR spectra of other tetraoxaspiro undecane polymers30. According to the mechanism of cationic polymerization, this suggests significant hydrolysis or chain transfer of propagating species and activated monomer (Figure 11) with or without elimination occurred during the polymerization process when the protonic acid is generated on irradiation. In other words, small molecules and/or very low molecular weight oligomers may be the result of the SOC “polymerization”. Therefore, while the modulus of the polyketocarbonate product expected from efficient double ring-opening polymerization is relatively low, it would not be surprising to see a drastic decrease in modulus with the addition of small amount of SOCs if these types of side reactions are involved. It was reported that water or moisture has great impact on kinetics and mechanism of the cationic photopolymerization in vinyl ethers31 and to a lesser extent, in epoxy systems32. Due to the nature of the spiro orthocarbonate structure, SOCs are rather labile to moisture especially with the presence of strong acids generated after photolysis of cationic photoinitiators. As shown in Figure 12, the two carbonyl peaks around 1756 and 1736 cm−1 are attributed to the linear carbonate and keto groups in the polymer (3α in Figure 3b), respectively. However, in contrast to the polymerization of a laminated specimen, the IR spectrum of the product from the polymerization exposed to the purge gas in the IR chamber, with a relative humidity of only about 12 %, has relatively weak polymer carbonyl peaks and instead, strong peaks near 1800 cm−1 and 3500 cm−1, which are attributed respectively to the cyclic carbonate carbonyl and hydroxyl groups from hydrolysis of MPN.
Figure 11.

(a) Hydrolysis and chain transfer of the propagating species and (b) hydrolysis of the monomer with the presence of moisture after initiation.
Figure 12.

IR spectra of MPN monomer, polymer (laminated specimen between glass slides) and hydrolysis and/or chain transfer product (exposed thin film specimen in 12 % relative humidity during polymerization) (Curing conditions: 23 °C; 0.3 wt% ISb as the cationic photoinitiator; 20 mW/cm2, 320–500 nm UV irradiation for 12 min).
In addition, small amounts of moisture have a great impact on the MPN cationic “polymerization” rate. As shown in Figure 13, the polymerization rate of the laminated thin film is significantly lower than that of the exposed one. Since the viscosities of the two systems should be similar at the initial stage, diffusion limitation should not be a primary cause of the difference in rate and it is likely due to the fast hydrolysis that competes effectively with polymerization. In addition, according to the double ring-opening polymerization mechanism validated here, the rate difference and similar induction time indicate that water is predominately interacting with monomer rather than intercepting the initiating system, which otherwise would result in significant induction time increases with depressed early monomer consumption. In a comparison of the final conversion between these two systems, it was found that polymerization of the laminated specimen reached higher conversion than that of the exposed specimen. Since mobility restrictions are not expected to limit either polymerization significantly, the disparity in final conversion in favor of the laminated specimen may be related to the equilibrium between hydrolysis and ring closure under acidic conditions, which are similar to the conditions used in the synthesis of some SOC and spiro orthoester monomers33. Finally, the thick film polymerization under both laminated and exposed conditions showed much less effect of ambient moisture on the polymerization rate (Figure 14). This result indicates that the diffusion of moisture into the reaction mixture is slower than the rate of monomer consumption due to hydrolysis. The overall slightly higher rate associated with moisture exposure is likely the result of the relatively high monomer consumption rate at the surface because of hydrolysis. However, if comparing the curing rates of laminated thin film and laminated thick film, it is found that the curing in the thick specimen reaches a similar final conversion value but takes a significantly longer time. Since both thin and thick film have the same composition and are polymerized at the same temperature, the significant difference in curing rate is mainly attributed to UV irradiation attenuation in the thick film34.
Figure 13.

Conversion versus irradiation time of laminated and exposed specimens (thin films, mid-IR, 27 % relative humidity) (Curing conditions: 23 °C; 0.3 wt% ISb as the cationic photoinitiator; 20 mW/cm2, 320–500 nm UV irradiation).
Figure 14.

Conversion versus irradiation time of laminated and exposed specimens (1 mm thick films, NIR, 21 % relative humidity) (Curing conditions: 23 °C; 0.3 wt% ISb as the cationic photoinitiator; 20 mW/cm2, 320–500 nm UV irradiation).
Conclusions
Photoinitiated cationic double ring-opening polymerization of 2-methylene-7-phenyl-1,4,6,9-tetraoxaspiro[4.4]nonane (MPN) was successfully monitored using real-time FT-IR. The proposed mechanism based on the simplified propagation step of the ring-opening polymerization of MPN was confirmed by both polymer NMR spectrum and observed apparent activation energy. It was found that α-position attack is the predominant mode for the second ring opening during polymerization of MPN. Cationic photopolymerizations performed in the presence of a suitable free radical initiator or small amounts of moisture showed that free radical promoted cationic polymerization increased the polymerization rate of MPN significantly without altering the polymer structure and that even trace ambient moisture has a profound impact on the course of the polymerization of thin film specimens. In light of the significant structural differences possible in MPN polymers formed under different polymerization conditions, it is reasonable to expect that shrinkage values would also differ considerably; however, substantial volumetric shrinkage and not expansion accompanied the efficient double ring-opening photopolymerization process verified here.
Acknowledgments
This study was supported by NIH/NIDCR R01-DE14227.
Footnotes
Photoinitiated Cationic DROP of MPN
References
- 1.Sakaguchi RL, Wiltbank BD, Murchison CF. Dent Mater. 2005;21:43–46. doi: 10.1016/j.dental.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Hudson AJ, Martin SC, Hubert M, Spelt JK. J Electron Packaging. 2002;124:352–354. [Google Scholar]
- 3.Fuh JYH, Choo YS, Lu L, Nee AYC, Wong YS, Wang WL, Miyazawa T, Ho SH. J Mater Process Tech. 1997;63:887–891. [Google Scholar]
- 4.Francis LF, McCormick AV, Vaessen DM, Payne JA. J Mater Sci. 2002;37:4897–4911. [Google Scholar]
- 5.Ge JH, Trujillo M, Stansbury J. Dent Mater. 2005;21:1163–1169. doi: 10.1016/j.dental.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Chung CM, Kim MS, Kim JG, Jang DO. J Biomed Mater Res. 2002;62:622–627. doi: 10.1002/jbm.10359. [DOI] [PubMed] [Google Scholar]
- 7.Carioscia JA, Lu H, Stanbury JW, Bowman CN. Dent Mater. 2005;21:1137–1143. doi: 10.1016/j.dental.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Yoshikawa T, Burrow MF, Tagami J. Dent Mater. 2001;17:359–366. doi: 10.1016/s0109-5641(00)00095-6. [DOI] [PubMed] [Google Scholar]
- 9.Bailey WJ, Sun RL. Polym Prepr. 1972;13:281. [Google Scholar]
- 10.Hino T, Endo T. Macromolecules. 2003;36:5902–5904. [Google Scholar]
- 11.Takata T, Ariga T, Endo T. Macromolecules. 1992;25:3829–3833. [Google Scholar]
- 12.Takata T, Endo T. Macromolecules. 1988;21:900–904. [Google Scholar]
- 13.Peiffer RW. Photopolymerization. 1997;673:1–14. [Google Scholar]
- 14.Chappelow CC, Pinzino CS, Jeang L, Harris CD, Holder AJ, Eick JD. J Appl Polym Sci. 2000;76:1715–1724. [Google Scholar]
- 15.Moon EJ, Lee JY, Kim CK, Cho BH. J Biomed Mater Res Part B: Appl Biomater. 2005;73B:338–346. doi: 10.1002/jbm.b.30222. [DOI] [PubMed] [Google Scholar]
- 16.Tagoshi H, Endo T. Bull Chem Soc Japan. 1989;62:945–947. [Google Scholar]
- 17.Sakai S, Fujinami T, Sakurai S. J Polym Sci Part C: Polym Lett. 1973;11:631–633. [Google Scholar]
- 18.Bolln C, Frey H, Mulhaupt R. Macromolecules. 1996;29:3111–3116. [Google Scholar]
- 19.Kim MJ, Kim CK. Polym Mater: Sci Eng. 2002;87:106–107. [Google Scholar]
- 20.Ge, J.; Stansbury, J. W. Unpublished results.
- 21.Lovell LG, Berchtold KA, Elliott JE, Lu H, Bowman CN. Polym Adv Tech. 2001;12:335–345. [Google Scholar]
- 22.Stansbury JW, Dickens SH. Dent Mater. 2001;17:71–79. doi: 10.1016/s0109-5641(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 23.Cook WD, Forrest M, Goodwin AA. Dent Mater. 1999;15:447–449. doi: 10.1016/s0109-5641(99)00073-1. [DOI] [PubMed] [Google Scholar]
- 24.Endo T, Sato H, Takata T. Macromolecules. 1988;21:1186–1187. [Google Scholar]
- 25.Bi Y, Neckers DC. Macromolecules. 1994;27:3683–3693. [Google Scholar]
- 26.Crivello JV, Liu SS. Chem Mater. 1998;10:3724–3731. [Google Scholar]
- 27.Lin Y, Stansbury JW. Polymer. 2003;44:4781–4789. [Google Scholar]
- 28.Sipani V, Scranton AB. J Polym Sci Part A: Polym Chem. 2003;41:2064–2072. [Google Scholar]
- 29.Eick JD, Smith RE, Pinzino CS, Kotha SP, Kostoryz EL, Chappelow CC. Dent Mater. 2005;21:384–390. doi: 10.1016/j.dental.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 30.Byerley TJ, Eick JD, Chen GP, Chappelow CC, Millich F. Dent Mater. 1992;8:345–350. doi: 10.1016/0109-5641(92)90016-6. [DOI] [PubMed] [Google Scholar]
- 31.Lin Y, Stansbury JW. J Polym Sci Part A: Polym Chem. 2004;42:1985–1998. [Google Scholar]
- 32.Decker C, Viet TNT, Thi HP. Radtech Report. 2002 December Session [Google Scholar]
- 33.Endo T, Takata T. Sekiyu Gakkaishi. 1989;32:237–247. [Google Scholar]
- 34.Miller GA, Gou L, Narayanan V, Scranton AB. J Polym Sci Part A: Polym Chem. 2002;40:793–808. [Google Scholar]