

Abstract
Interdisciplinary investigation at the interface of chemistry, engineering, and medicine has enabled the development of self-assembled nanomaterials with novel biochemical and electro-optical properties. We have recently shown that emissive polymersomes, polymer vesicles incorporating porphyrin-based fluorophores, feature large integrated-emission oscillator strengths and narrow emission bands; these nanoscale assemblies can be further engineered to fluoresce at discrete wavelengths throughout the visible and near-infrared (NIR) spectral domains. As such, emissive polymersomes effectively define an organic-based family of soft-matter quantum-dot analogs that possess not only impressive optical properties, but also tunable physical and biomaterial characteristics relative to inorganic fluorescent nanoparticles.
Here, we expand upon our initial studies on poly(ethyleneoxide)-block-poly(butadiene)-based vesicles to examine fluorophore membrane-loading in other polymersome systems. Through modulation of fluorophore ancilliary group substituents and choice of polymer chain chemistries, we are able to predictably control intramembranous polymer-fluorophore interactions; these phenomena, in turn, influence the nature of fluorophore solvation, local dielectric environment, and emission quantum yield within emissive polymersome assemblies. By utilizing different classes of vesicle-generating diblock copolymers, including bioresorbable poly(ethyleneoxide)-block-poly(ε-caprolactone) (PEO-b-PCL) and poly(ethyleneoxide)-block-poly(γ-methyl-ε-caprolactone) (PEO-b-PMCL), we ascertain general principles important for engineering nanoscale optical vesicles. Further, this work heralds the first generation of fully-biodegradable fluorescent nanoparticles suitable for deep-tissue in vivo imaging.
Introduction
While conventional visible fluorophores are routinely utilized for in vitro biological research, there has been increasing interest in the development of near-infrared (NIR) emissive agents suitable for deep-tissue in vivo imaging.1–6 To date, many investigators have focused on the development of quantum dots (inorganic semiconductor nanocrystals) for both in vitro and in vivo applications.7–11 Although quantum dots exhibit vastly superior optical characteristics when compared to traditional organic fluorophores,12,13 they may be of limited clinical utility since their metallic composites are inherently toxic.14,15 As such, the design of alternative fluorescent nanoparticles is an area of active investigation and aims to generate novel organic-based agents that possess large magnitude irradiance and high photostability.
Meso-to-meso ethyne-bridged oligo(porphinato)zinc(II)-based supermolecules (PZnn compounds) define a family of near-infrared fluorophores (NIRFs) that exhibit high molar absorptivities throughout the visible and NIR regions,16–19 large NIR fluorescence quantum yields,20,21 high photobleaching thresholds,21,22 and no chemical or photo-based in vitro toxicity.23 Through cooperative self-assembly with amphiphilic diblock copolymers, these electronically-conjugated porphyrinic NIRFs can be dispersed, non-covalently, at high concentrations (> 10 mol/wt%) within the thick synthetic membranes of polymersomes24–26 (50 nm to 50 μm diameter polymer vesicles).27–29 These NIR-emissive polymersomes define a family of organic-based, soft matter quantum dot analogues that are ideally suited for in vivo optical imaging.27 Previous studies have focused upon the delineation of a rich photophysical diversity,28 and quantification of fluorophore membrane-loading,29 in these emissive assemblies; membrane incorporation of a wide-range of related multi-porphyrinic fluorophores has enabled precise emission energy modulation over a broad domain of the visible and near-infrared spectrum (600–900 nm).27,28 Here, we expand upon our initial studies involving poly(ethyleneoxide)-block-poly(butadiene)-based polymersomes that disperse PZnn fluorophores, to develop a generalized methodology for engineering the bulk optical properties of other emissive vesicle assemblies. Through rational design of fluorophore ancilliary group substituents and choice of polymer chain chemistries, we show that intramembranous polymer-fluorophore interactions can be extensively modulated to influence the nature of fluorophore solvation, mean electronic environment, and thus, the PZnn emission quantum yield.
We focus herein upon the meso-to-meso ethynyl-bridged tris-[porphyrinato]zinc(II)-based (PZn3) fluorophore and examine the effects of substituent-driven PZn3-dispersion in various polymersome environments, differing in both membrane core-thickness (ℓ) and chemical composition (Figure 1), via steady-state absorption and emission spectroscopies. Five related PZn3-based fluorophores that differ with respect to ancillary group substituents (PZn3 A–E, Figure 1B) were utilized. By comparing each vesicular nanoscale compositions’ integrated steady-state emission intensity as a function of membrane-loading concentration, the role of polymer-chain/PZn3-fluorophore local solvation effects in determining the bulk optical properties of emissive polymersomal assemblies were elucidated.
Figure 1.
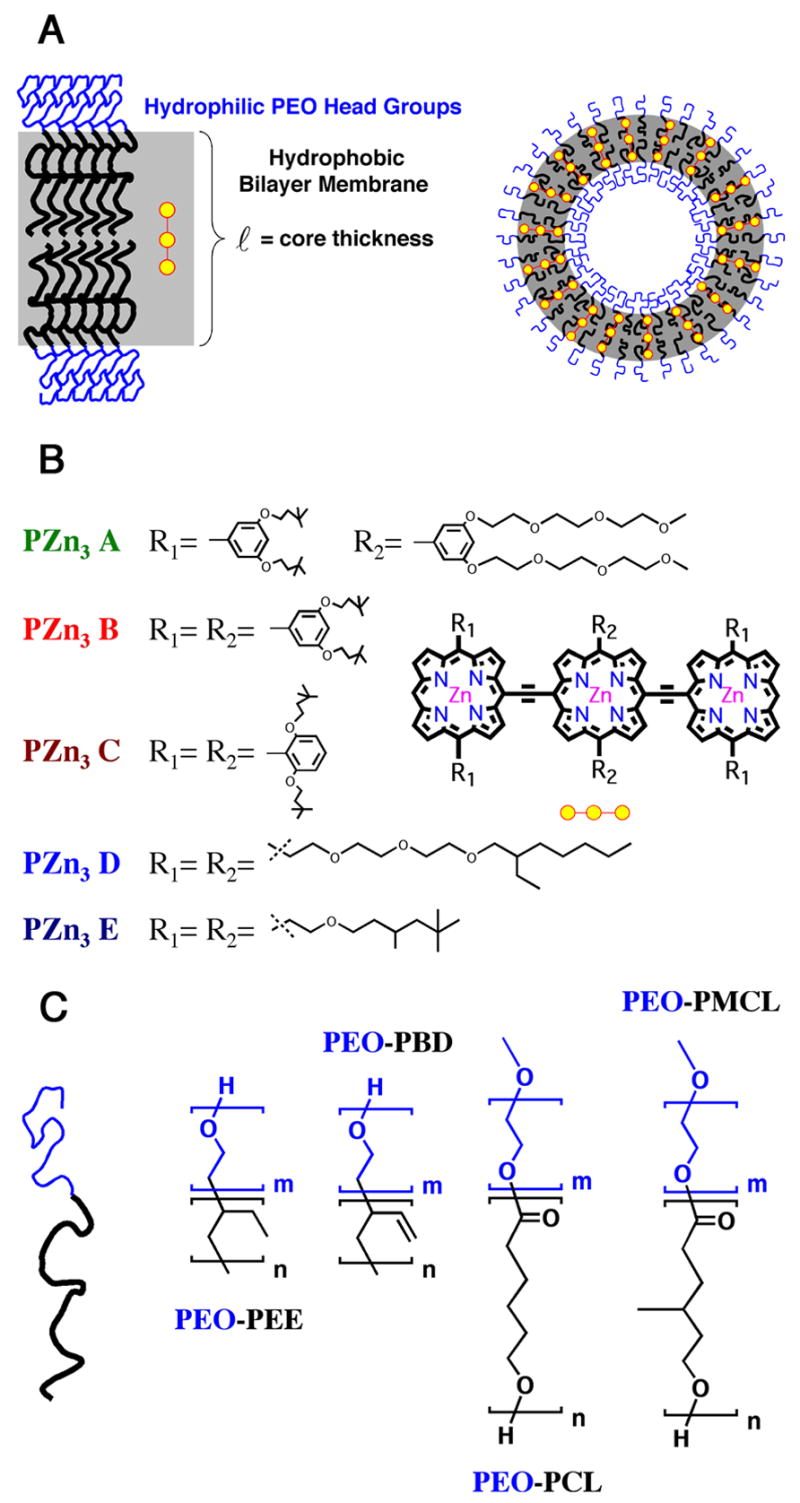
Schematic of various meso-to-meso ethynyl-bridged tris[porphyrinato]zinc(II)-based (PZn3) fluorophores incorporated within different polymersome environments. A) Illustration of PZn3 dispersion within the vesicle’s hydrophobic bilayer membrane. B) Chemical structures of five PZn3-based fluorophores (PZn3 A–E) differing in ancilliary pendant-group substituents. C) Chemical structures of biocompatible (PEO-b-PEE and PEO-b-PBD) and biodegradable (PEO-b-PCL and PEO-b-PMCL) vesicle-generating diblock copolymers.
Several different classes of vesicle-generating diblock copolymers were utilized (Figure 1C), including biocompatible poly(ethyleneoxide)-block-poly(butadiene) (PEO-b-PBD) and poly(ethyleneoxide)-block-poly(ethylethylene) (PEO-b-PEE),24,30 as well as biodegradable poly(ethyleneoxide)-block-poly(ε-caprolactone) (PEO-b-PCL) and poly(ethyleneoxide)-block-poly(γ-methyl-ε-caprolactone) (PEO-b-PMCL) compositions.31,32 Membrane-loading of the PZn3–based fluorophores in PEO30-b-PBD46 (ℓ= 9.6 nm) vs. PEO80-b-PBD125 (ℓ= 14.8 nm)-based vesicles was compared in order to assess the effects of increasing membrane core-thickness on relative fluorophore distribution and intermolecular spacing, while maintaining a uniform PZn3 solvation environment. Similar experiments comparing PZn3-loading in PEO30-b-PBD46- and PEO40-b-PEE37–based vesicles were conducted in order to ascertain the roles that local solvation and interchain packing effects play in influencing the extent of fluorophore-fluorophore π–π interactions and the degree to which the emitter is dispersed in the bilayer. Finally, membrane-loading studies of PZn3–based fluorophores in two biodegradable vesicle systems (PEO45-b-PCL105, and PEO43-b-PMCL66) were preformed in order to evaluate how membrane crystallinity influences PZn3-solvation and mean electronic environment.
Materials and Methods
Materials
PEO30-b-PBD46 diblock copolymers under study were purchased from Polymer Source Inc, (Dorval, Quebec Canada). PEO80-b-PBD125, PEO40-b-PEE37, PEO45-b-PCL105, and PEO43-b-PMCL66 diblock polymers30–32 as well as PZn3 NIRF species A–C19,23 were synthesized following procedures described previously.
5,15-Bis[[5′,-10′,20′-bis([11-ethyl-3,6,9-trioxapentadecane-1-yl]porphinato)zinc(II)]ethynyl]-10,20-bis([11-ethyl-3,6,9-rioxapentadecane-1-yl]porphinato)zinc(II) (PZn3 D)
(5-Ethynyl-10,20-bis(11-ethyl-3,6,9-trioxapentadecane-1-yl)porphinato)zinc(II) (46.2 mg, 5.21 × 10−5 mol) and (5,15-dibromo-10,20-bis(11-ethyl-3, 6, 9-trioxapentadecane-1-yl)porphinato)zinc(II) (24.2 mg, 2.37 × 10−5 mol) were charged into a 100 mL Schlenk tube along with Pd2dba3 (3 mg, 3.55 × 10−6 mol) and AsPh3 (10 mg, 2.84 × 10−5 mol). A previously degassed solution of THF:TEA (9:1) was cannula-transferred into the reaction flask; the reaction mixture was then heated to 60 °C and stirred overnight under Ar. The reaction was quenched with water, diluted with CHCl3, and washed three times with aq. NH4Cl. The organic layer was collected, dried with Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography on silica using CHCl3:pyridine (99:1) as the eluent, followed by gravimetric size exclusion chromatography (Biobeads, SX-1, THF). The fastest moving band was collected. The product was purified a final time by flash chromatography on silica. Yield = 48 mg (77 mol%, based on (5,15-dibromo-10,20-bis(11-ethyl-3, 6, 9-trioxapentadecane-1-yl)porphinato)zinc(II)). 1H NMR (500 MHz, CDCl3: 1 drop pyridine-d5): δ 10.56 (d, 4H, J = 4.45 Hz), δ 10.50(d, 4H, J = 4.45 Hz), δ 9.98 (s, 2H), δ 9.87 (d, 4H, J = 4.50 Hz), δ 9.80 (d, 4H, J = 4.50 Hz), δ 9.66 (d, 4H, J = 4.40 Hz), δ 9.33 (d, 4H, J = 4.35 Hz), δ 5.12 (t, 4H, J = 7.50 Hz), δ 4.37 (t, 4H, J = 7.26 Hz), δ 4.14 (s, 2H), δ 3.55 (t, 4H, J = 4.01 Hz), δ 3.46 (t, 4H, J = 4.84 Hz), δ 3.08 (t, 4H, J = 4.63 Hz), δ 2.89 (t, 4H, J = 4.64 Hz), δ 1.50(m, 42H), δ 1.28 (m, 69H), δ 0.89 (m, H). Vis (THF) λmax (log ε): 414 (5.17), 494 (5.42), 574 (4.17), 774 (5.03) nm. MALDI-TOF MS m/z: 2623.3485 (M+) (calculated for C148H200N12O18Zn3 2625.2978).
5,15-Bis[[5′,-10′,20′-bis([3-(3′,5′,5′-trimethylhexyloxy)-ethyl]porphinato)zinc(II)]ethynyl]-10,20-bis([3-(3′,5′,5′-trimethylhexyloxy)-ethyl]porphinato)zinc(II) (PZn3 E)
(5,15-Dibromo-10,20-bis(3-(3′,5′,5′-trimethylhexyloxy)-ethyl)porphinato)zinc(II) (71 mg, 8.13 × 10−5 mol) and (5-ethynyl-10,20-bis(3-(3′, 5′, 5′-trimethylhexyloxy)-ethyl)porphinato)zinc(II) (132 mg, 1.79 × 10−4 mol) were charged into a 100 mL Schlenk tube along with Pd2dba3 (25 mg, 2.69 × 10−5 mol) and AsPh3 (70 mg, 2.15 × 10−4 mol). A previously degassed solution of THF:TEA (9:1) was cannula-transferred into the reaction flask; the reaction mixture was then heated to 60 °C and stirred overnight under Ar. The reaction was quenched with water, diluted with CHCl3, and washed three times with aq. NH4Cl. The organic layer was collected, dried with Na2SO4, filtered, and evaporated. The residue was purified by flash chromatography on silica using CHCl3:pyridine (99:1) as the eluent, followed by gravimetric size exclusion chromatography (Biobeads, SX-1, THF). The fastest moving band was collected. The product was purified a final time by flash chromatography on silica. Yield = 137 mg (77 mol%, based on (5,15-Dibromo-10,20-bis(3-(3′,5′,5′-trimethylhexyloxy)-ethyl)porphinato)zinc(II)). 1H NMR (500 MHz, CDCl3): δ 10.56 (d, 4H, J = 4.15 Hz), δ 10.49 (d, 4H, J = 4.05 Hz), δ 9.96 (s, 2H), δ 9.84 (d, 4H, J = .3 85 Hz), δ 9.77 (d, 4H, J = 4.45 Hz), δ 9.63 (d, 4H, J = 4.30 Hz), δ 9.32 (d, 4H, J = 4.10 Hz), δ 5.38 (br-m, 12H), δ 4.63 (t, 12H, J = 8.05 Hz), δ 4.57 (t, 12H, J = 8.06 Hz), δ 3.74 (t, 12H, J = 6.55 Hz), δ 3.69 (t, 12H, J = 5.47 Hz), δ 1.62 (m, 12H), δ 1.45 (m, 12H), δ 1.30 (m, 12H), δ 1.19 (m, 12H), δ 1.09 (m, 6H), δ 0.87 (m, 54H). Vis (THF) λmax (log ε): 414.00 (5.17), 494 (5.42), 574 (4.22), 774 (5.03) nm. MALDI-TOF MS m/z: 2182.9642 (M +) (calculated for C130H164N12O6Zn3 2181.0771).
Vesicle preparation
Formation of small (< 300 nm diameter) emissive polymersomes followed procedures described previously.28 Briefly, 1 mM diblock copolymer solutions and 1 mM solutions of NIRF species PZn3 A–E in 10% THF:CH2Cl2 were combined in various molar ratios and uniformly coated on the surfaces of roughened Teflon plates, followed by evaporation of the solvent under vacuum for > 12 h. The Teflon plate consisted of a 1/16″ thick sheet cut in 1×1″ squares and machine-roughened with industrial grade sandpaper (60 grit, aluminum oxide) to create macroscopically uniform digitations. Each dry Teflon plate, containing the polymer:fluorophore sample, was placed in a 20 mL glass vial; 2 mL of aqueous solution (e.g., DI water) were then added. The vial was subsequently sealed, immersed in a bath sonicator, and heated at 60 °C for 1 hr, leading to spontaneous budding of medium sized (~300 nm diameter) emissive polymersomes (membrane loaded at the prescribed polymer:fluorophore molar ratio) off of the Teflon and into the aqueous surroundings. Small unilamellar polymersomes that possess appropriately narrow size distributions were prepared via procedures analogous to those used to formulate small lipid vesicles (sonication, freeze-thaw extraction, and extrusion).27
Steady-State Electronic Absorption Spectroscopy of Emissive Polymersomes that Disperse PZn3 A–E Fluorophores
Aqueous suspensions (in DI water) of small, unilamellar NIR-emissive polymersomes (loaded at a 20:1 polymer:NIRF molar ratio; n = 5 for each unique polymer:fluorophore formulation) were placed in 10 mm quartz optical cells and electronic absorption spectra for each of the membrane-incorporated fluorophore species were recorded using an OLIS UV/Vis/NIR spectrophotometry system that is based on the optics of a Cary 14 spectrophotometer (On-line Instrument Systems Inc, Bogart, GA). The solutions were then transferred to a glass vial, frozen in liquid N2, and lyophilized (FreeZone 4.5 L Benchtop Freeze Dry System, Labconco Corporation, Kansas City, MO; Model 77500) for 24 hours to destroy the vesicles and dry the polymer and fluorophore species. The dry samples were then taken up in the same volume of THF and their absorption spectra recorded. NIRF concentrations in the original polymersome solutions were calculated via Beer’s Law using the THF absorption spectra and the previously determined molar extinction coefficients (ε, M−1 cm−1) for each emitter in this solvent. These calculated concentrations, as well as the original NIRF absorption spectra in aqueous polymersome solutions, were then utilized in order to determine the ε values for each polymersome-loaded fluorophore.
Steady-State Fluorescence Spectroscopy of Emissive Polymersomes that Disperse PZn3 A–E Fluorophores
Aqueous suspensions (in DI water) of small, unilamellar NIR-emissive polymersomes (loaded at a 20:1 polymer:NIRF molar ratio; n = 5 for each unique polymer:fluorophore formulation) were placed in 10 mm quartz optical cells and steady-state emission spectra were obtained using a Spex Fluorolog-3 spectrophotometer (Jobin Yvon Inc, Edison, NJ) that utilized a dual S- and T-channel configuration and PMT/InGaAs/Extended-InGaAs detectors (λex = 510 nm). Sample concentrations were modulated so that the optical density at the excitation wavelength was < 0.05 O.D. All spectra were collected with the single excitation and emission apertures set at 5 nm. Fluorescence spectra were corrected to account for the wavelength-dependent efficiency of the detection system that was determined using the spectral output of a calibrated light source obtained from the National Bureau of Standards.
Comparison of Relative Concentration-Dependent-Fluorescence Intensities of PZn3 A and B
Suspensions (in DI water) of small (< 300 nm diameter) PEO30-b-PBD46-, PEO80-b-PBD125-, PEO40-b-PEE37-, PEO45-b-PCL105-, and PEO43-b-PMCL66-based emissive polymersomes were generated by combining the respective diblock copolymers with NIRFs PZn3 A and B in various molar ratios of polymer:NIRF (500:1, 250:1, 100:1, 40:1, 20:1, 10:1, 5:1; note, n = 3 for each unique polymer:NIRF combination). Steady-state absorption spectra for each of the solutions were recorded, and 250 μL of each sample were transferred to black 96-well plates (n = 3 plates; Becton Dickinson Co,).
Near-infrared fluorescence from each well in the plate was then detected and quantified with an eXplore Optix Imaging System (GE Healthcare). The instrument used the following experimental parameters: 785 nm excitation laser, 785 ± 5 nm band-pass excitation filter, 850 ±50 nm band-pass emission filter, 0.1 s integration time, 1 mm step size, and 25 °C stage temperature. The fluorescence intensity from each solution well in the plate was normalized by the laser power using analysis software provided by the instrument manufacturer (Analysis WorkStation 1.1.3.0, Advanced Research Technologies, Quebec, Canada). Finally, the normalized integrated-emitted-photon counts from each sample well were divided by the NIRF concentration at the excitation wavelength (determined from the steady-state absorption spectra obtained for each emissive polymersome sample), compared, and divided by the largest sample value. The average values for normalized emission from PZn3 A and B are plotted against the calculated fluorophore concentration in the polymersome membrane and reported in Figures 4–6; note, the individual values determined for each sample in a plate varied by less than 10% from the reported average-normalized-emission intensity for each unique emissive polymersome formulation.
Figure 4.
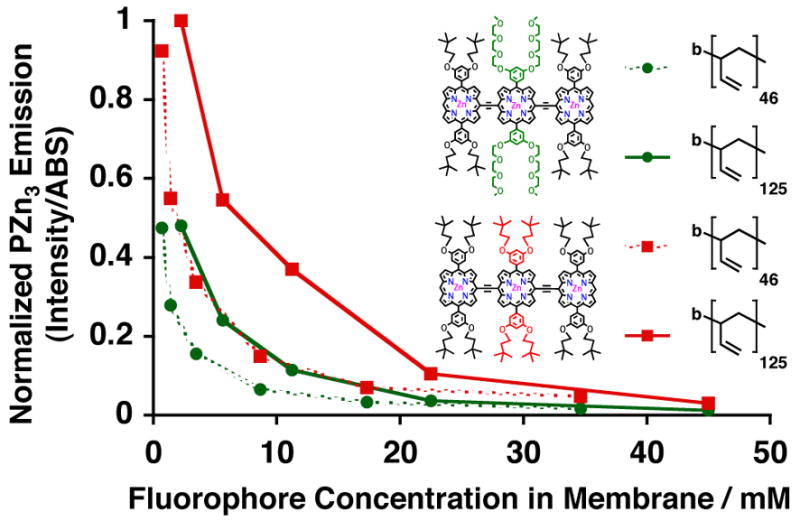
Relative steady-state normalized-fluorescence-emission intensities of PZn3-based fluorophores A (green) and B (red), as a function of membrane-loading concentration, in aqueous suspensions of PEO30-b-PBD40 (dotted) and PEO80-b-PBD125 (solid) vesicles. Experimental conditions: T = 25 °C, DI water, λex = 785 nm.
Figure 6.
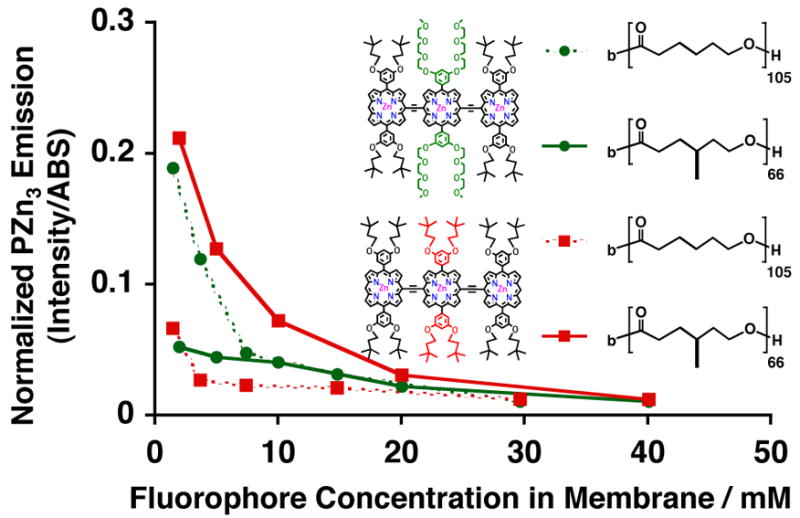
Relative steady-state normalized-fluorescence-emission intensities of PZn3-based fluorophores A (green) and B (red), as a function of membrane-loading concentration, in aqueous suspensions of PEO45-b-PCL105 (dotted) and PEO43-b-PMCL66 (solid) vesicles. Experimental conditions: T = 25 °C, DI water, λex = 785 nm.
Results and Discussion
Design of PZn3 A–E
We have previously demonstrated that meso-to-meso ethynyl-bridged tris[porphinato]zinc(II) (PZn3) fluorophores possess not only strong near-infrared absorption bands (ε > 100,000 at λ = 767 nm)16,19 but also large NIR fluorescence quantum yields (λemmax = 806 nm, φF = 22 +/− 3%)21 in tetrahydrofuran (THF) solvent. Moreover, these hydrophobic PZn3-based fluorophores are readily dispersed in aqueous solution when incorporated within the thick membranes of synthetic polymer vesicles. Emissive polymersomes, dispersing PZn-based fluorophores at precise membrane concentrations, are self-assembled through a simple and quantitative method involving aqueous hydration of dry, uniform thin-films of amphiphilic polymer and fluorophore deposited in prescribed molar ratios on roughened Teflon surfaces (see Experimental Section).27,29 Notably, our earlier work determined that PZn3 ancillary-aryl-group substituents influence not only interplanar torsional-angle distribution of fluorophore component-PZn macrocyles, and thus the magnitude of PZn-to-PZn conjugative interactions,28 but also the relative fluorophore distribution within the hydrophobic bilayer of PEO-b-PBD-based vesicles. As a result, we focused our current efforts on designing several new PZn3-based fluorophores with differing pendant ancillary groups in order to identify key intramembranous molecular interactions that would enable optimization of polymersome bulk optical properties.
While PZn3 A–E each possess an identical conjugated chromophoric core, their ancilliary pendant groups vary (Figure 1), impacting the nature of the molecular interactions of these fluorophores with the polymer chains composing the vesicles’ membranes. For example, we have previously shown that modulating the amphiphilicity of these PZn-based fluorophores affords control over their optical properties within PEO-b-PBD-based polymersomes.28 Although both PZn3 A and B possess 3′, 5′-disubstituted pendant aryl groups, replacement of PZn3 A’s 9-methoxy-1,4,7-trioxanonyl substituents with 3,3-dimethyl-1-butyloxy units augments the fluorophore’s hydrophobicity. As a result, PZn3 B’s relative membrane localization is shifted away from the environment defined by the interface of the hydrophilic PEO and hydrophobic PBD block fractions (location of PZn3 A), and facilitates its predominant dispersion into the hydrophobic core of the PBD membrane.28 Further, varying aryl-group substitution from a 3′, 5′-(PZn3 B) to a 2′, 6′-substitution pattern (PZn3 C) converts overall chromophore geometry from a biconcave wedge to a cylinder and influences the local arrangement of surrounding polymer chains within the vesicle membrane.28
Given these earlier observations with PEO-b-PBD-based emissive polymersomes, we synthesized two new PZn3-fluorphores (PZn3 D and E) that lack 10- and 20-pendant aryl groups; these species possess respectively 11-ethyl-3,6,9-trioxapentadecane-1-yl (PZn3 D) and 3-(3′,5′,5′-trimethylhexyloxy)-ethyl (PZn3 E) ancilliary substitutents, directly linked to the PZn3 component macrocycle 10- and 20-positions. These design variations were intended to potentially augment fluorophore-fluorophore intermolecular spacing by minimizing both the disruption of polymer chain packing and the required PZn3-dissolution volume within the vesicle membrane. Likewise, the differences between PZn3 D and E’s ancilliary substitutent groups were expected to affect their relative membrane-dispersion in a fashion similar to that evinced in earlier experiments involving the comparison of PZn3 A and B.28
Electronic Absorption Spectra of PZn3 A–E in Various Polymersome Environments
Although PZn3-based fluorophores A–E differ with respect to the nature and location of their ancilliary group substitutents, they display nearly identical absorptive and emissive spectral characteristics in THF solvent (Figure 2). The same fluorophores, however, show marked spectral differences depending upon the polymersome environment in which they are non-covalently incorporated (Figure 3). When comparing the absorptive and emissive signatures of PZn3 A–E in aqueous suspensions of PEO-b-PBD (Figure 3A) and PEO-b-PEE-based (Figure 3B) vesicles to their THF solution spectra (Figure 2), it is readily apparent that ancilliary group substituents alter the nature of PZn3-dissolution in a polymersome-specific manner. For example, although PZn3-based fluorophores D (light blue) and E (dark blue), which lack pendant aryl groups, display spectral features consistent with extensive fluorophore aggregation within PBD-based membranes (Figure 3A), they are more effectively dispersed in the fully saturated PEE-based bilayers (Figure 3B); in particular, PZn3 D shows a significant decrease in spectral heterogeneity,19–21,28 signaling a narrower PZn-PZn torsional angle distribution about a diminished mean torsional angle, within these PEO-b-PEE-based vesicles (Figure 3B) relative to that manifest within other polymersome environments (Figure 3 A, C, and D).
Figure 2.
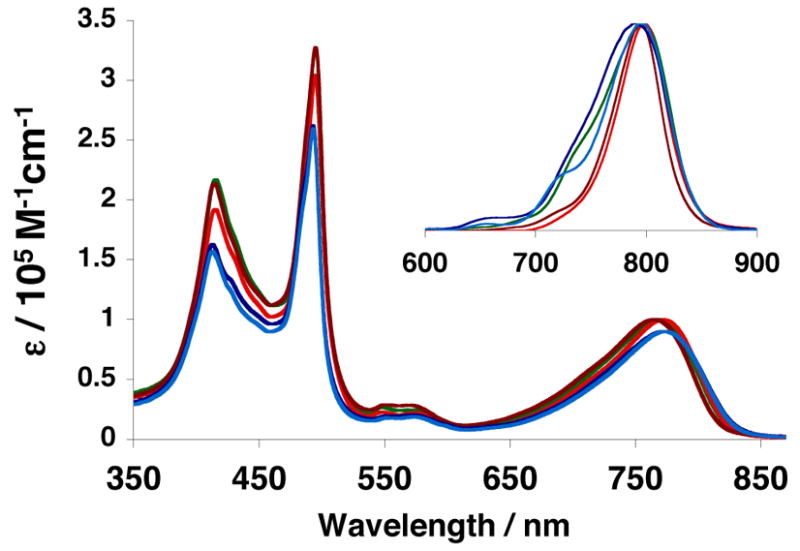
Steady-state electronic absorption and emission (inset) spectra of PZn3-based fluorophores A–E in tetrahydrofuran solvent. Compound color scheme corresponds to that displayed in Figure 1B. λex = 510 nm.
Figure 3.

Steady-state electronic absorption and emission (inset) spectra of PZn3-based fluorophores A–E when incorporated within different polymersome environments. Compound color scheme corresponds to that displayed in Figure 1B. A) Aqueous suspensions of PEO-b-PBD-based vesicles. B) Aqueous suspensions of PEO-b-PEE-based vesicles. C) Aqueous suspensions of PEO-b-PCL-based vesicles. D) Aqueous suspensions of PEO-b-PMCL-based vesicles. Experimental conditions: T = 25 °C, DI water, λex = 510 nm.
As seen in Figure 3B, relative dissolution of PZn3 A (green), B (red) and C (crimson) within PEE-based membranes is similar to that observed previously for the same fluorophores dispersed in PBD (Figure 3A). Namely, the relative proportion of ancillary 3,3-dimethyl-1-butyloxy and 9-methoxy-1,4,7-trioxanonyl aryl-substituents affects the emission energy, as well as the spectral breadth of the low-energy absorptive and emissive transitions, of these supermolecular fluorophores. Hydrophobic 3,3-dimethyl-1-butyloxy groups preferentially disperse PZn3-based fluorophores into the core of the bilayer membrane (PZn3 B), while increasing numbers of 9-methoxy-1,4,7-trioxanonyl groups enhance the extent to which these chromophores are dispersed at the membrane environment defined by the interface of the hydrophilic (PEO) and hydrophobic (PBD or PEE) block fractions (PZn3 A).28,29
In dilute THF solution, the position of the ancillary aryl 3,3-dimethyl-1-butyloxy substituent does not impact fluorophore optical properties: chromophores having 2′,6′-disubstituted aryl rings (PZn3 C, Figure 2 crimson) exhibit the same absorptive and emissive signatures as those that feature 3′,5′-disubstituted aryl groups (PZn3 B, Figure 2 red). By comparison, however, within PEO-b-PEE and PEO-b-PBD-based polymersome environments, chromophores bearing 2′,6′-di(3,3-dimethyl-1-butyloxy)phenyl rings display fluorescence-band hypsochromic shifts (PZn3 C, Figure 3A and B crimson), while the same chromophores possessing a 3′, 5′ substitution pattern exhibit bathochromically shifted emission bands (PZn3 B, Figure 3A and B red) relative to that observed in THF solvent (Figure 2). Note that PZn3 B and C display low energy-absorptive and emissive bands with decreased oscillator strengths and augmented spectral breadths within PEO-b-PEE-, relative to that observed in PEO-b-PBD-, based vesicles likely due to PEE polymer-chain-packing effects that drive an augmented PZn-PZn torsional angle distribution about a larger mean torsional angle; such a perturbation to the conformational heterogeneity disrupts PZn3 symmetry as manifest in PBD-based bilayers (Figure 3A),28 and makes more probable fluorophore aggregation within PEE membrane environments.
In comparing PZn3 membrane-incorporation in the two biodegradable vesicle systems, PEO-b-PCL (Figure 3C) and PEO-b-PMCL (Figure 3D), the PZn3 B absorption and emission spectra differ substantially with respect to those determined in biocompatible PEO-b-PBD- (Figure 3A) and PEO-b-PEE- (Figure 3B) based polymersomes. In the caprolactone-based membranes, PZn3 B, as well as PZn3 C–E, exhibit reduced dissolution, broadened spectral manifolds, and substantially bathochromically-shifted emission bands indicative of aggregate formation.33 Within all polymersome environments (Figure 3 A–D), PZn3 A displays uniquely conserved spectral properties that are largely identical to those that it exhibits in THF solvent (Figure 2). This is attributed to PZn3 A’s relative localization at the membrane hydrophobic-block/hydrophilic-block interface, where its dissolution and dispersion are less sensitive to PZn3-substitutent-group/hydrophobic-polymer-chain interactions that normally dictate fluorophore solvation and the nature of the local dielectric environment within the bilayer core.
PZn3 Fluorescence as a Function of Membrane-Loading Concentration
We next compared the relative concentration-dependent fluorescence intensities of two PZn3-based fluorophores (A and B) within a variety of different polymersome environments. PZn3 A was selected because it universally disperses within each vesicle system examined, independent of membrane chemical composition. Due to its similar 3′, 5′-aryl substitution pattern, PZn3 B was also examined in order to help elucidate the importance of PZn3 A’s 9-methoxy-1,4,7-trioxanonyl substituents, which localize PZn3 A fluorophores within a smaller membrane volume element relative to those sampled by PZn3 B–E emitters, in determining the steady-state integrated emission intensity as a function of membrane-loading concentration. Fluorophore membrane-loading was varied between 0.2 – 20 mol% (corresponding to vesicles formed from 500:1 and 5:1 polymer-to-fluorophore molar ratios, respectively). For each unique emissive polymersome formulation (n = 3 experiments for each polymer:fluorophore combination), steady-state integrated-emission intensities were measured (λex = 785 nm, λem = 800–900 nm), normalized by total emitter numbers in suspension (determined via a Beer’s law calculation), and plotted on a relative scale vs. the calculated concentration of emitters within the polymersome membrane.
The fluorophore concentration (mM) within the membrane’s polymer matrix was calculated from the following derived expression:
Here, χ is the molar ratio of fluorophore/polymer, d is the vesicle’s diameter (~300 nm +/− 100 nm as determined by dynamic light scattering), Sp is the interfacial area per polymer chain (~1 nm2 for each of the examined polymers)34, NA is Avogadro’s number, and ℓ is the membrane core-thickness (nm). Key metrical parameters of each of the vesicle-forming diblock copolymers examined in this study are compiled in Table I.
Table I.
Polymersome-Forming Diblock Copolymers
| Polymer Composition | Mw | ℓ(nm) |
|---|---|---|
| PEO30-b-PBD46 | 3800 | 9.6 |
| PEO80-b-PBD125 | 10400 | 14.8 |
| PEO40-b-PEE37 | 3900 | 8.0 |
| PEO45-b-PCL105 | 14000 | 22.5 |
| PEO43-b-PMCL66 | 10300 | 16.6 |
Effects of Increasing Membrane Core-Thickness of Fluorophore Dissolution
We first examined membrane-loading of PZn3 A and B within two different poly(ethyleneoxide)-block-poly(butadiene)-based polymersomes in order to assess the effects of increasing membrane core-thickness (ℓ) on relative fluorophore distribution and inter-fluorophore spacing. Although PZn3 A and B display nearly identical steady-state absorbance and fluorescence spectra within both PEO30-b-PBD46 (ℓ = 9.6 nm) and PEO80-b-PBD125 (ℓ= 14.8 nm)-based vesicles, they exhibit different concentration-dependent fluorescence intensities (Figure 4). In both PEO30-b-PBD46 (dotted) and PEO80-b-PBD125 -based polymersomes (solid), PZn3 B (red) manifests more intense fluorescence at a given membrane concentration relative to PZn3 A (green) (Figure 4). Further, at a specific intramembranous fluorophore concentration, the effects of increasing membrane core-thickness upon augmentation of integrated fluorescence intensity (comparing dotted to solid lines for each fluorophore in Figure 4) are more profound for PZn3 B. These results are consistent with previous observations28 that hydrophobic 3,3-dimethyl-1-butyloxy groups are expected to preferentially disperse these PZn-based fluorophores into the core of the bilayer membrane (PZn3 B), while increasing numbers of 9-methoxy-1,4,7-trioxanonyl groups should enhance the extent to which these fluorochromes are dispersed at the membrane interface of the hydrophilic PEO and hydrophobic PBD block fractions (PZn3 A). Thus, increasing membrane core-thickness tends to augment the available volume for PZn3 B dissolution to a greater extent relative to that for the PZn3 A fluorophore, thereby enlarging mean fluorophore-fluorphore intermolecular distances and membrane-localized emissive output for this fluorochrome.
Fluorophore Loading Within Saturated and Fully Unsaturated Branched-Polymer Membranes
We next compared vesicle-loading of PZn3 A and B within related branched poly(butadiene) and poly(ethylethylene)-based membranes. Diblock copolymers of PEO30-b-PBD46 and PEO40-b-PEE37 were utilized in order to generate polymersomes of similar membrane core-thickness (ℓ= 9.6 vs. 8 nm, respectively).30 By comparing relative normalized emission intensities for PZn3 A (green) and B (red) within these two related vesicle systems (Figure 5), the importance of inter-chain packing effects and fluorophore-fluorophore π-π interactions in determining vesicular PZn3 emissive output can be assessed. Reexamining the Figure 3 steady-state absorbance and emission spectra shows that PZn3 A exhibits nearly identical spectral features within both PEO-b-PBD (Fig 3A, green) and PEO-b-PEE-based vesicles (Fig 3B, green), while PZn3 B (red) shows a slight decrease in its Q-state absorption manifold intensity and a relative bathochromically-shifted fluorescence band λmax within PEE (λmax = 801 nm) vs. PBD (λmax = 818 nm)-based membranes. Moreover, PZn3 B’s relative normalized emission intensities were lower when incorporated within PEO-b-PEE-(solid) relative to that observed for PEO-b-PBD- (dotted) based polymersomes (Figure 5).
Figure 5.
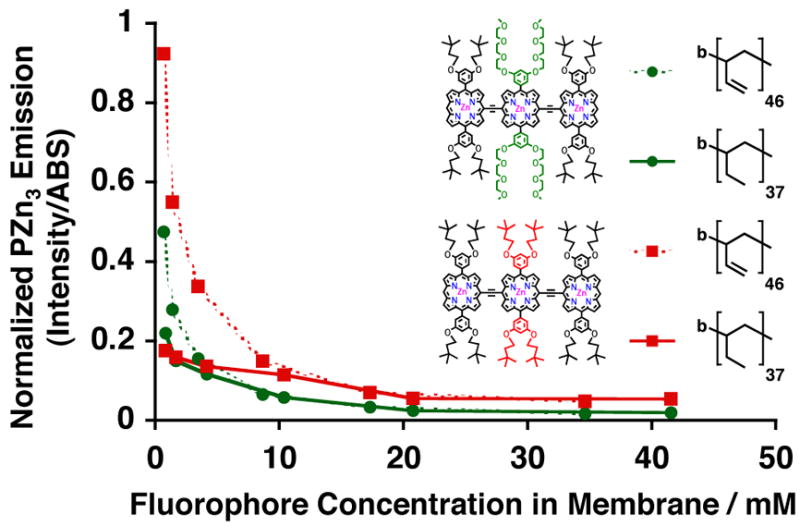
Relative steady-state normalized-fluorescence-emission intensities of PZn3-based fluorophores A (green) and B (red), as a function of membrane-loading concentration, in aqueous suspensions of PEO36-b-PBD40 (dotted) and PEO40-b-PEE37 (solid) vesicles. Experimental conditions: T = 25 °C, DI water, λex = 785 nm.
While it is difficult to isolate the etiology of these phenomena, it could potentially implicate differences in local intramembranous dielectric environments between PEE- and PBD-based vesicles. PZn3 A’s relative localization near the membrane hydrophobic-block/hydrophilic-block interface is consistent with the fact that its steady-state spectral features are unchanged when dispersed in either PEO-b-PBD- or PEO-b-PEE-based vesicles (Figure 3A and B, green). At high membrane-loading concentrations (> 5 mM), PZn3 A (green) exhibits nearly identical concentration-dependent emission intensities within both PEO-b-PBD (dotted) vs. PEO-b-PEE-based polymersomes (solid) (Figure 5). As a result, its local dielectric environment can be assumed to be relatively unchanged and the fluorophore is likely similarly solvated when incorporated within either vesicle membrane. At lower membrane-loading concentrations (< 5 mM), however, PZn3 A shows interestingly a significant difference in the integrated fluorescence intensity between the two membrane environments (Figure 5, green dotted vs. solid).
These results suggest that changes in local membrane dissolution of the PZn3 A fluorophore occur with increased loading. At low membrane-loading concentrations (<5mM), PZn3 A may be more homogeneously distributed throughout the bilayer core, in a fashion similar to that previously established for PZn3 B. At higher loading concentrations (> 5 mM), while PZn3 A’s emission intensities are identical within both polymersomal environments (Figure 5, green dotted and green solid lines), the integrated fluorescence is less than that observed for PZn3 B (Figure 5, red dotted and red solid lines). This is consistent with other data that indicate that PZn3 B is dissolved uniformly throughout the hydrophobic core at all membrane loading concentrations; in contrast, increasing membrane-dispersed PZn3 A concentrations results in localization of greater numbers of fluorochromes to a more limited dissolution volume near the vesicles’ aqueous/membrane interface. At low membrane loading levels, as the steady-state integrated-emission intensities are larger for both PZn3 A and B in PEO-b-PBD- relative to PEO-b-PEE-based membranes, the PBD solvation environment more effectively minimizes aggregation of emitters relative to that which exists in the more electronically-insulating PEE-based bilayers.
Fluorophore Loading Within Biodegradable Caprolactone-based Vesicle Membranes
We have recently described the generation of biodegradable vesicles formed through spontaneous self-assembly of pure diblock copolymers of poly(ethyleneoxide)-block-poly(ε-caprolactone) (PEO-b-PCL).31 When compared to degradable polymersomes formed from blending “bio-inert” and hydrolysable components (where τ1/2 release35,36 ≈ τ1/2 circulation37 ~ tens of h), PEO45-b-PCL105-based vesicles possess much slower release kinetics (τ1/2 release ~ days), offering potential advantages for future in vivo applications. Moreover, their large membrane core thicknesses (22.5 +/− 2.3 nm) afford the opportunity for facile incorporation of PZn-based fluorophores, enabling the generation of NIR-emissive polymersomes that are not only biocompatible but also fully biodegradable. Vesicles generated from pure diblock copolymers of PEO-b-PCL possess solid (crystalline-phase) membranes31 while those assembled from poly(ethyleneoxide)-block-poly(γ-methyl-ε-caprolactone) (PEO-b-PMCL) have fluid-phase32 bilayers. As such, we compared relative concentration-dependent emission intensities of PZn3 A and B within two biodegradable polymersome systems based on PEO45-b-PCL105 and PEO43-b-PMCL66, in order to evaluate how membrane crystallinity influences PZn3 solvation and mean electronic environment (Figure 6).
As polycaprolactone-based vesicles possess membranes that have dielectric environments that differ substantially from those derived from branched poly(ethylethylene) and poly(butadiene), intramembranous PZn3 distributions for a given fluorophore need not be identical within these two classes of vesicles. PZn3 A’s (green) steady-state absorption and emission spectra look identical within both PEO-b-PCL (Figure 3C) and PEO-b-PMCL-based vesicles (Figure 3D), resembling those observed in THF and other polymersome environments. PZn3 B’s spectra in both biodegradable environments (Figure 3C and D, red), however, are consistent with significant intermolecular fluorophore interactions, and notably possess fluorescence bands that are substantially broadened and bathochromically shifted as compared to those observed for the same fluorophore in THF or within other emissive vesicle systems (Figure 3A and B). Further, at low loading levels (< 5 mM) PZn3 A exhibited significantly greater concentration-dependent emission intensities within crystalline PCL-based membranes (Figure 6, green dotted), relative to those derived from fluid PMCL (Figure 6, green solid). At higher membrane-loading levels (> 5 mM), PZn3 A ‘s relative concentration-dependent fluorescence was identical within both membrane environments. PZn3 B’s emission, however, was uniformly greater within fluid PMCL-based vesicles (Figure 6, red solid) relative to that within PCL polymersomes (Figure 6, red dotted). Finally, although PEO45-b-PCL105–based polymersomes possess a larger membrane core-thickness (ℓ= 22.5 nm) than those comprised of PEO43-b-PMCL66 (ℓ= 16.6 nm), PZn3 displayed relatively greater fluorescence within the smaller-volume PMCL-based environment (Figure 6, red solid).
These differences in PZn3 A and B membrane-distribution and dissolution, within the two different biodegradable vesicle systems, likely derive again in large part from substituent-driven intermembraneous interactions involving fluorophore-aryl-group substituents and polymer chains that dictate fluorophore solvation. Replacement of PZn3 B’s central meso-aryl ring 3′- and 5′-3,3-dimethyl-1-butyloxy substituents with 9- methoxy-1,4,7-trioxanonyl groups (PZn3 A) augments chromophore amphiphilicity and drives its dissolution into relatively more polar environments. At low fluorophore loading levels within PCL-based membranes (< 5 mM), PZn3 A is likely more homogeneously distributed throughout the bilayer core as attributed to its larger relative concentration-dependent emission intensities when compared to its dissolution within PMCL-based bilayers (Figure 6, dotted green vs. red). At higher fluorophore loading levels, incorporation of PZn3 A within PCL membranes resembles that within PMCL-based systems, consistent with incremental incorporation of larger numbers of fluorophores predominantly to the vesicles’ aqueous/membrane interface. As compared to the more hydrophilic PZn3 A, P Z n3 B is uniformly distributed throughout the membrane core of the relatively apolar PMCL-based membrane (Figure 6, solid red vs. green lines); as a result, its concentration-dependent fluorescence is larger than that observed for PZn3 A within PEO-b-PCL-based systems at all loading levels (Figure 6 red solid vs. green dotted lines). Finally, PZn3 B’s fluorescence is predominantly quenched within PCL-based membranes at all but the most dilute concentrations (Figure 6, dotted red line), consistent with significant aggregation of this hydrophobic fluorophore within this more polar caprolactone-based membrane environment (Figure 3C, red).
Comparison of PZn3-loading Within Various Polymersome Systems
In considering the entire set of steady-state spectra for PZn3 A–E (Figures 2 and 3), as well as the relative concentration-dependent emission intensities for PZn3 A and B within the various polymersome systems (Figures 4–6), it is evident that the PZn3-based fluorophore A displays robust optical properties regardless of its solvation environment. Figure 7 compares PZn3 A’s relative concentration-dependent fluorescence in aqueous suspensions of biocompatible PEO30-b-PBD46-, and PEO40-b-PEE37-, as well as biodegradable PEO45-b-PCL105- and PEO43-b-PMCL66-based polymersomes. Consistent with earlier discussion, at high intramembranous PZn3 A concentrations (> 10 mM), the normalized emission is nearly identical within each vesicle environment. Along with its conserved steady-state spectral features, these data indicate that the fluorophore is similarly solvated, likely located predominantly in a region near the vesicles’ hydrophobic-block/hydrophilic-block interface. At lower loading levels (< 10 mM), fluorescence emission is highly dependent upon the dielectric environment of the membrane matrix. Low-level, nearly concentration-independent emission (over the range of examined fluorophore loadings) is evinced in apolar PMCL-based bilayers incorporating PZn3 A (Figure 7, square). Markedly greater normalized integrated-emission intensity is observed for PZn3 A dispersed in polymersomes derived from electron-rich PBD (Figure 7, diamond). Finally, its relative concentration-dependent fluorescence intensities in aqueous suspensions of PCL- (Figure 7, circle) and PEE-based vesicles (Figure 7, triangle) are similar, indicative of its incorporation within isotropic electronic environments.
Figure 7.
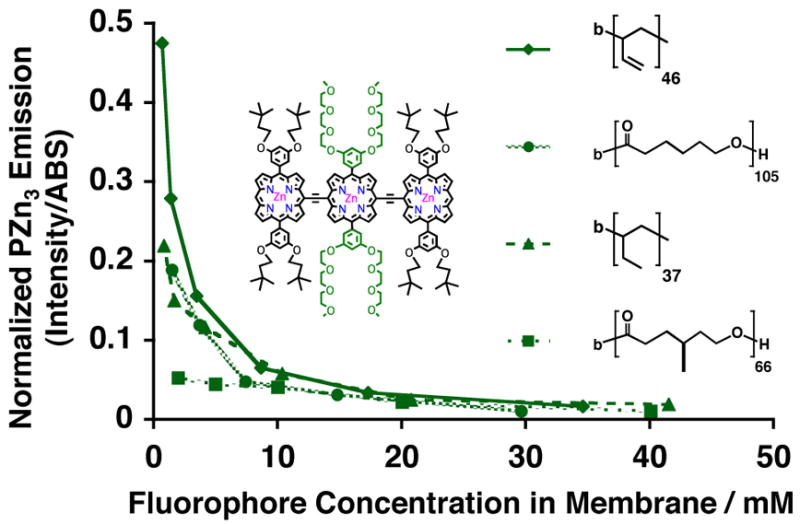
Relative steady-state emission intensities of PZn3-based fluorophore A, as a function of membrane-loading concentration, in aqueous suspensions of PEO30-b-PBD46 (diamond), PEO45-b-PCL105 (circle), PEO40-b-PEE37 (triangle), and PEO43-b-PMCL66 (square) vesicles. Experimental conditions: T = 25 °C, DI water, λex = 785 nm.
Optimization of Steady-State Fluorescence from NIR-Emissive Polymersomes
For future biological applications, it is necessary to optimize the total-integrated vesicular-emission of polymersomes incorporating PZnn-based fluorophores. Choosing the appropriate polymer:fluorphore molar loading level, which dictates the effective intramembranous fluorophore concentrations and hence mean intermolecular spacing, involves a trade-off between larger relative-emission per fluorophore and greater fluorophore-copy-numbers per vesicle. Figure 8 displays the total-integrated-vesicular emission of PZn3 A- and PZn3 B-based biocompatible and biodegradable emissive polymersomes. Emission of PEO80-b-PBD125–based vesicles incorporating PZn3 B (Figure 8, solid red line with squares) is greatest and is maximized at an approximately 2.5 mol% loading level. In general, total-integrated-emission intensities of polymersomes incorporating the more hydrophobic PZn3–based fluorophore B (Figure 8, red) are greater than identical vesicles incorporating PZn3 A (Figure 8, green). Further, amongst vesicles incorporating the same PZn3 fluorophore (either A or B), emissive output tends to correlate with membrane-core thickness: emission from PEO80-b-PBD125–based polymersomes (Figure 8, solid lines with square) is greater than those vesicles comprised of PEO30-b-PBD46 (Figure 8, dotted lines with square) or PEO40-b-PEE37 diblock copolymers (Figure 8, dotted lines with diamond), which possess thinner bilayers. Finally, the total-vesicular-emissive outputs from PEO45-b-PCL105–based polymersomes incorporating PZn3 A (Figure 8, dotted green line with circle) are nearly identical to those of PEO43-b-PMCL66-based vesicles dispersing PZn3 B (Figure 8, dotted red line with circle), due to appropriately matched fluorophore substituents driving polymer membrane solvation; consistent with these results, PZn3 A displays a low-level of concentration-independent emission within apolar PMCL-based membranes (Figure 6, solid green line), while hydrophobic PZn3 B displays similar aggregate-like behavior (Figure 6, dotted red line) within more polar PCL-derived polymersomes.
Figure 8.
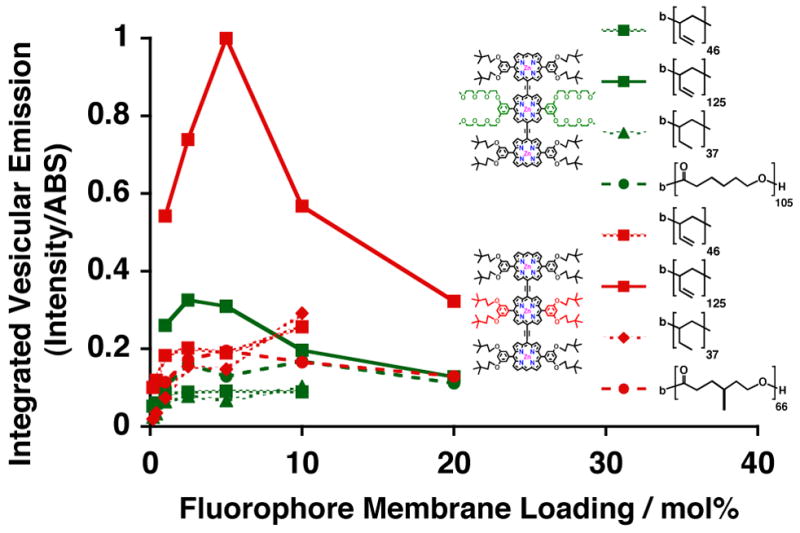
Comparison of the relative integrated-steady-state emission of various emissive polymersome systems, as a function of mol% membrane-loading with PZn3 A (green) and B (red). Experimental conditions: T = 25 °C, DI water, λex = 785 nm.
CONCLUSIONS
We have demonstrated that the nature of intramembranous polymer-fluorophore physicochemical interactions profoundly influences the bulk optical properties NIR-emissive polymersomes. Specifically, appropriate selection of fluorophore ancilliary-aryl-group substituents, and choice of polymer chain chemistries, modulate fluorophore solvation, mean electronic environment, and concentration-dependent fluorescence. PZn3-based fluorophores with 3′, 5′-disubstituted pendant aryl groups (PZn3 A and B) are more effectively membrane-dispersed than identical fluorophores that possess a 2′, 6′-substitution pattern (PZn3 C) or those that lack 10′, 20′-phenyl rings (PZn3 D and E). Further, appropriate selection of ancilliary aryl group substitutents drives fluorphore solvation into membranous regions with matching dielectric properties.
Within biocompatible PEO-b-PBD- and PEO-b-PEE-based polymersomes, hydrophobic PZn3 B exhibited the highest per molecule fluorescence at all membrane-loading levels. Emissive output was further augmented within thicker vesicle membranes comprised of the more electronic-rich PBD-based matrix, relative to that manifest in polymersomes featuring PEE-derived hydrophobic block fractions. PZn3 emission within the two biodegradable vesicle systems, PEO-b-PCL and PEO-b-PMCL, was also similarly incumbent on specific ancilliary aryl group substitution. Hydrophobic PZn3 B exhibited larger normalized emission intensities within the relatively apolar PMCL-based membrane environment, while PZn3 A’s increased amphiphilicity enabled its effective dissolution within more polar PCL-based bilayers.
The optimization of NIR-emissive polymersomes requires: 1) elucidation of fluorophore photophysics in order to design emitters with appropriately large NIR extinction coefficients and fluorescence quantum yields, 2) the regulation of the average fluorophore-fluorophore interspatial separations in order to facilitate fluorophore emission, and 3) the loading of appropriate numbers of fluorophores within a single vesicle composite in order to maximize signal sensitivity. Moreover, incorporation of PZnn-based near-infrared fluorophores within poly(caprolactone)-based membranes heralds the introduction of NIR-emissive polymersomes that are not only biocompatible, but now also fully biodegradable. Development of these new self-assembled, colloidal nanoparticles offers insights into the design of other high-irradiance, nanoscale organic probes for in vivo optical imaging applications.
Acknowledgments
This work was supported by a grant from the National Cancer Institute. M.J.T. and D.A.H. thank the MRSEC Program of the National Science Foundation (DMR05-20020) for infrastructural support. D.A.H. also thanks the National Institutes of Health (EB003457-01), and P.P.G. acknowledges fellowship support from the NIH Medical Scientist Training Program and the Whitaker Foundation.
References
- 1.Weissleder R, Tung CH, Mahmood U, Bogdanov A., Jr Nat Biotechnol. 1999;17:375–378. doi: 10.1038/7933. [DOI] [PubMed] [Google Scholar]
- 2.Achilefu S, Dorshow RB, Bugaj JE, Rajagopalan R. Inv Rad. 2000;35:479–485. doi: 10.1097/00004424-200008000-00004. [DOI] [PubMed] [Google Scholar]
- 3.Becker A, Hessenius C, Licha K, Ebert B, Sukowski U, Semmler W, Wiedenmann B, Grotzinger C. Nat Biotechnol. 2001;19:327–331. doi: 10.1038/86707. [DOI] [PubMed] [Google Scholar]
- 4.Sevick-Muraca EM, Houston JP, Gurfinkel M. Curr Opin Chem Biol. 2002;6:642–650. doi: 10.1016/s1367-5931(02)00356-3. [DOI] [PubMed] [Google Scholar]
- 5.Frangioni JV. Curr Opin Chem Biol. 2003;7:626–634. doi: 10.1016/j.cbpa.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Weissleder R, Ntziachristos V. Nat Med. 2003;9:123–128. doi: 10.1038/nm0103-123. [DOI] [PubMed] [Google Scholar]
- 7.Larson DR, Zipfel WR, Williams RM, Clark SW, Bruchez MP, Wise FW, Webb WW. Science. 2003;300:1434–1436. doi: 10.1126/science.1083780. [DOI] [PubMed] [Google Scholar]
- 8.Wu XY, Liu HJ, Liu JQ, Haley KN, Treadway JA, Larson JP, Ge NF, Peale F, Bruchez MP. Nat Biotechnol. 2003;21:41–46. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- 9.Voura EB, Jaiswal JK, Mattoussi H, Simon SM. Nat Med. 2004;10:993–998. doi: 10.1038/nm1096. [DOI] [PubMed] [Google Scholar]
- 10.Kim S, Lim YT, Soltesz EG, De Grand AM, Lee J, Nakayama A, Parker JA, Mihaljevic T, Laurence RG, Dor DM, Cohn LH, Bawendi MG, Frangioni JV. Nat Biotechnol. 2004;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao XH, Cui YY, Levenson RM, Chung LWK, Nie SM. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 12.Dabbousi BO, RodriguezViejo J, Mikulec FV, Heine JR, Mattoussi H, Ober R, Jensen KF, Bawendi MG. J Phys Chem B. 1997;101:9463–9475. [Google Scholar]
- 13.Chan WCW, Nie SM. Science. 1998;281:2016–2018. doi: 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- 14.Jaiswal JK, Simon SM. Trends Cell Biol. 2004;14:497–504. doi: 10.1016/j.tcb.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 15.Frangioni JV. Nat Biotechnol. 2006;24:326–328. doi: 10.1038/nbt0306-326. [DOI] [PubMed] [Google Scholar]
- 16.Lin VSY, DiMagno SG, Therien MJ. Science. 1994;264:1105–1111. doi: 10.1126/science.8178169. [DOI] [PubMed] [Google Scholar]
- 17.Lin VSY, Therien MJ. Chem-Eur J. 1995;1:645–651. [Google Scholar]
- 18.Angiolillo PJ, Lin VSY, Vanderkooi JM, Therien MJ. J Am Chem Soc. 1995;117:12514–12527. [Google Scholar]
- 19.Susumu K, Therien MJ. J Am Chem Soc. 2002;124:8550–8552. doi: 10.1021/ja0203925. [DOI] [PubMed] [Google Scholar]
- 20.Rubtsov IV, Susumu K, Rubtsov GI, Therien MJ. J Am Chem Soc. 2003;125:2687–2696. doi: 10.1021/ja021157p. [DOI] [PubMed] [Google Scholar]
- 21.Duncan TV, Susumu K, Sinks LE, Therien MJ. J Am Chem Soc. 2006;128:9000–9001. doi: 10.1021/ja061897o. [DOI] [PubMed] [Google Scholar]
- 22.Susumu K, Frail PR, Angiolillo PJ, Therien MJ. J Am Chem Soc. 2006;128:8380–8381. doi: 10.1021/ja0614823. [DOI] [PubMed] [Google Scholar]
- 23.Wu SP, Lee I, Ghoroghchian PP, Frail PR, Zheng G, Glickson JD, Therien MJ. Bioconjugate Chem. 2005;16:542–550. doi: 10.1021/bc0497416. [DOI] [PubMed] [Google Scholar]
- 24.Discher BM, Won YY, Ege DS, Lee JCM, Bates FS, Discher DE, Hammer DA. Science. 1999;284:1143–1146. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- 25.Discher DE, Eisenberg A. Science. 2002;297:967–973. doi: 10.1126/science.1074972. [DOI] [PubMed] [Google Scholar]
- 26.Antonietti M, Forster S. Adv Mater. 2003;15:1323–1333. [Google Scholar]
- 27.Ghoroghchian PP, Frail PR, Susumu K, Blessington D, Brannan AK, Bates FS, Chance B, Hammer DA, Therien MJ. Proc Natl Acad Sci U S A. 2005;102:2922–2927. doi: 10.1073/pnas.0409394102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghoroghchian PP, Frail PR, Susumu K, Park TH, Wu SP, Uyeda HT, Hammer DA, Therien MJ. J Am Chem Soc. 2005;127:15388–15390. doi: 10.1021/ja055571b. [DOI] [PubMed] [Google Scholar]
- 29.Ghoroghchian PP, Lin JJ, Brannan AK, Frail PR, Bates FS, Therien MJ, Hammer DA. Soft Matter. doi: 10.1039/b604212k. In Press. [DOI] [PubMed] [Google Scholar]
- 30.Bermudez H, Brannan AK, Hammer DA, Bates FS, Discher DE. Macromolecules. 2002;35:8203–8208. [Google Scholar]
- 31.Ghoroghchian PP, Li G, Levine DH, Davis KP, Bates FS, Hammer DA, Therien MJ. Macromolecules. 2006;39:1673–1675. doi: 10.1021/ma0519009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zupancich JA, Bates FS, Hillmyer MA. Macromolecules. 2006;39:4286–4288. [Google Scholar]
- 33.Turro NJ. Modern Molecular Photochemistry. New Ed. University Science Books; Sausalito, CA: 1991. [Google Scholar]
- 34.Won YY, Brannan AK, Davis HT, Bates FS. J Phys Chem B. 2002;106:3354–3364. [Google Scholar]
- 35.Ahmed F, Hategan A, Discher DE, Discher BM. Langmuir. 2003;19:6505–6511. [Google Scholar]
- 36.Ahmed F, Discher DE. J Controlled Release. 2004;96:37–53. doi: 10.1016/j.jconrel.2003.12.021. [DOI] [PubMed] [Google Scholar]
- 37.Photos PJ, Bacakova L, Discher B, Bates FS, Discher DE. J Controlled Release. 2003;90:323–334. doi: 10.1016/s0168-3659(03)00201-3. [DOI] [PubMed] [Google Scholar]