

Abstract
A simple, direct route to preparation of surface immobilized hydrogel films is described. Specifically, low pressure RF pulsed plasma polymerization of 1-amino-2-propanol and 2-(ethylamino)ethanol monomers produced thin hydrogel films deposited on substrates located in the plasma reactor. The successful syntheses were carried out under plasma conditions which not only yield the hydrogel but are also sufficiently energetic to produce films strongly grafted to the substrates. The polymer films obtained exhibit the thermoresponsive property of hydrogels, as shown by film color change with temperature. Additional evidence for the phase transition properties of these films was obtained using water contact angle and capillary rise measurements. The plasma polymerization approach provides an unusually simple route to synthesis of hydrogels in which the films are pin-hole free and are of easily controlled thickness. An important added advantage, particularly for applications involving biomaterials, is the conformal property of the plasma generated polymer films. The results obtained suggest that this approach should be applicable to a variety of other monomers and, based on differences observed with the present two monomers, suggest synthesis of films which exhibit a range of phase transition temperatures.
Keywords: Hydrogel, thermoresponsive film, plasma polymerization
INTRODUCTION
Hydrogels are three-dimensional hydrophilic polymeric structures able to absorb large quantities of water. They are synthesized by polymerization of hydrophilic monomers. The extent of the reversible swelling and deswelling property of these materials is known to depend on the nature of both intermolecular and intramolecular crosslinking, as well as the degree of hydrogen bonding in the polymer network1. Increasingly, these hydrogels are being utilized in a variety of applications including drug release,2–4 biosensors,5–7, tissue engineering,8–10 and pH sensors.11,12
To date, a variety of compounds have been utilized in synthesis of hydrogels. Examples of monomers employed for this purpose include N-isopropylacrylamide [NIPAM], 13–16 vinyl alcohol, 8,17 ethylene glycol,18,19 and N-vinylpyrrolidone.20 Of these compounds, hydrogels made from NIPAM, including some involving incorporation of co-monomers,21–23 have been extensively studied for their thermoresponsive behavior near physiological temperature. These thermoresponsive hydrogels exhibit phase transitions when subjected to an alteration in the environmental temperature. For example, poly-NIPAM is hydrophilic below ~30°C and becomes hydrophobic above ~35°C. The reported phase transition temperature for NIPAM is 31°–32°C.24–26. Various approaches have been employed to immobilize these thin films on solid surfaces. These techniques include electron beam irradiation,27,28 photoinitiated grafting,8,29,30 use of activated and functionalized substrates,31,32 and plasma polymerization.13,14,33–35
The prior plasma generated films, noted above, involved the use of NIPAM monomer. In the present paper, we report successful extension of the plasma polymerization approach to synthesis of hydrogel films from other monomers. For this purpose, we selected low molecular weight, and thus relatively volatile, monomers containing both amine and hydroxyl functionalities. A variable duty cycle pulsed plasma was employed to control the film compositions obtained during the plasma polymerizations.36 Under pulsed conditions, both the degree of film cross-linking and extent of retention of the monomers functional groups in the resultant plasma films can be controlled, to a relatively high degree. In the present case, our goal was to identify deposition conditions which would provide a compromise between the degree of polymer cross-linking and retention of the hydroxyl and amine groups and thus the extent of hydrogen bonding in the films. The compromise mentioned reflects the fact that under pulsed plasma conditions, higher duty cycles promote increased polymer cross-linking but at the expense of retention of monomer structure.36 A second important consideration in this work was to generate hydrogel films under conditions such that the films were strongly bonded to the substrate supports so as to exhibit sufficient adhesion and abrasion resistance for use in future applications. Thus, both of these goals involved study and careful adjustment of plasma parameters, particularly the average power input.
This paper describes a successful, single step, plasma polymerization synthetic route to prepare uniform, conformal thin films of thermoresponsive hydrogels from the monomers 1-amino-2-propanol (1A2P) and 2-(ethylamino)ethanol (2EAE). In each case, the films exhibit rapid moisture uptake and the thermo responsive behavior characteristic of some hydrogels. The films compositions were characterized by ATR-FTIR and XPS spectroscopy, and physical properties determined using temperature dependent contact angle and capillary rise measurements.13,30
EXPERIMENTAL METHODS
Preparation of the hydrogel films
The 1A2P and 2EAE monomers were obtained from Sigma-Aldrich, St. Luis, MO and had a stated purity of +98%. Prior to film deposition, the monomers were repeatedly freeze-thawed to remove dissolved gases. Monomer vapors were subjected to a radio-frequency (RF) plasma glow discharge, at room temperature, in a bell-shaped reactor chamber (Figure -1). Polished Si wafers were used as substrate for XPS studies and water contact angle measurements,. ATR crystals, made from Si wafers, were employed for the ATR-FTIR spectroscopic studies.37,38 All Si wafers were sonicated with acetone, methanol and hexane to clean the wafer surfaces prior to use. After substrates were placed inside the reactor, a background pressure of 6 mtorr was achieved for each run. Oxygen plasma at 100W average power input was employed to remove any carbonaceous residue left on the substrates. Monomer vapor was introduced into the reactor chamber and an RF glow discharge was maintained at 130 mtorr pressure at 150W peak power input. Three different power input conditions were employed, namely, pulsed discharges at 10/30 and 10/10 (ton/toff, ms) duty cycles, plus runs using a continuous wave (CW) operational mode. All samples were prepared using the 150W power input. It was observed that the polymer film thickness obtained under the 10/30 and 10/10 conditions varied linearly with deposition time. This was not true for the CW films which revealed a decrease in the rate of film formation with increasing deposition times.
Figure 1.
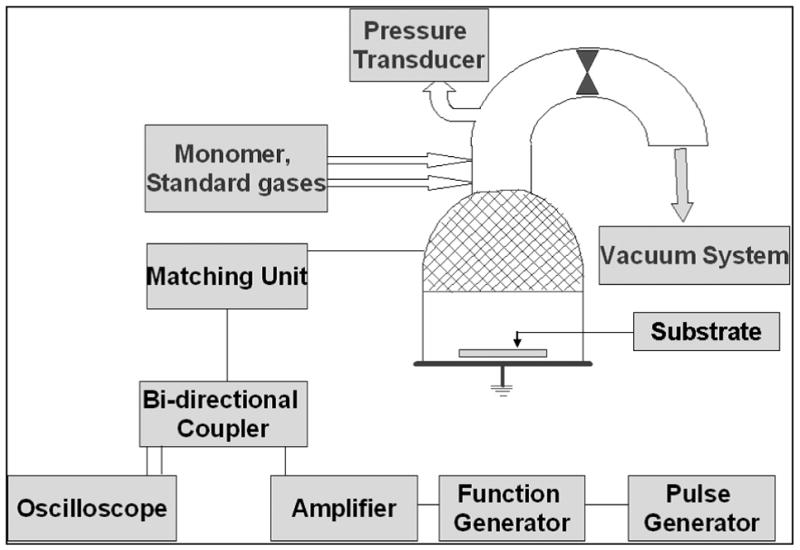
Schematic diagram of the bell-shaped RF plasma polymerization reactor.
Characterization of plasma polymerized films
The 1A2P polymer films were characterized by ATR-FTIR and XPS spectroscopies, water contact angle measurement and capillary rise experiments. The ATR-FTIR spectral analyses were carried out using a Bruker Equinox 55 FT-IR Spectrophotometer. The XPS spectra were obtained using a Perkin-Elmer PSI 5000 series instrument equipped with an X-ray source monochromator. A Rame-Hart sessile drop goniometer, with a small heater and thermocouple attachment, was used to measure the contact angles of the films over a temperature range of 20°C to 60°C. Capillary rise measurements were employed to examine the hydrophilic/hydrophobic transition behavior of the thin films with temperature. Glass capillaries, 1.5 × 50mm in size, were used for this purpose. Ultra-pure water was used for the contact angle and capillary rise measurements.
RESULTS
The initial studies focused on the 1A2P monomer and the following results were obtained.
Energy Efficiency of Film Formation under Pulsed and CW Conditions
It was observed that the polymer film thickness obtained under the 10/30 and 10/10 conditions varied linearly with deposition time. This was not true for the CW films which revealed a decrease in the rate of film formation with increasing deposition times. The decreasing rate with time under CW conditions presumably arises from an increasing contribution from film ablation at the relatively high power employed, an observation which has been made in numerous prior plasma polymerization studies. The variations of film thickness with deposition times are shown in Figure 2. From these data, the energy efficiency of film formation was calculated for each duty cycle and the results are shown in Figure 3, revealing a clear increase in efficiency with decreasing duty cycle. This result, comparable to that observed in other pulsed plasma studies, has been interpreted as evidence for significant film formation during plasma off times.36
Figure 2.
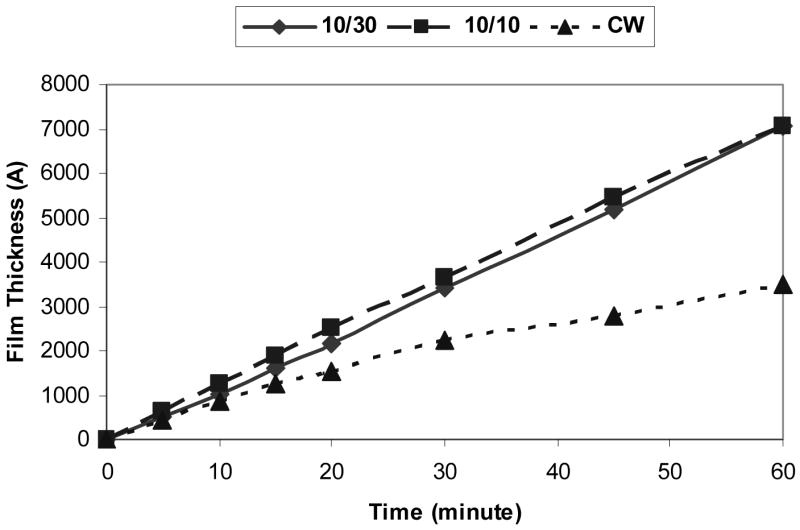
Film thickness with time (deposition rate) of plasma polymerized 1A2P films.
Figure 3.
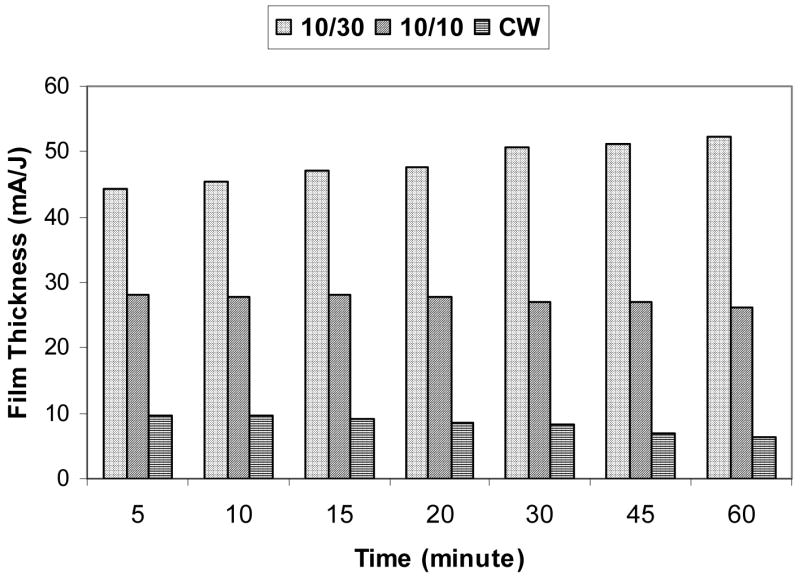
Energy efficiency plot for formation of 1A2P films as a function of plasma duty cycles. Efficiency is expressed in terms of film thickness per J of power input.
ATR-FTIR Characterization
ATR-FTIR spectra of 1A2P films were studied as a function of exposure time to purging by a stream of dry nitrogen. These spectra were recorded after the films had been exposed to the atmosphere sufficiently long to equilibrate with respect to moisture adsorption. The resulting spectra are shown in Figure 4 [a–c] for films synthesized under the two pulsed plasma and CW conditions. FT-IR spectra of each film (thickness ~200nm) were obtained of the initially prepared moisture saturated plasma films, followed by spectra recorded after 2, 5 and 10 minutes of dry nitrogen gas purging. As shown in Figure 4, sequential changes in absorption peak intensities were observed with increasing N2 purging times. Peak assignments for these spectra are as follows: (a) the region from 3600 to ~3000 cm−1 (N-H and O-H stretching modes); (b) 2970 cm−1 (-CH3 asymmetric stretch); (c) 2930 cm−1 (–CH2– asymmetric stretch, (d) 2866 cm−1 (-CH3 symmetric stretch); (e) 2240 cm−1 (–C≡N stretch); (f) 2180 cm−1 (–N≡C stretch); (g) ~1650 cm−1 (amide C=O stretch). Atmospheric absorptions from CO2 and H2O were eliminated by recording a background spectrum on an uncoated ATR crystal which was then subtracted from each of the polymer film absorptions. As shown in Figure 4, substantial decreases in peak intensities were observed in both the 3600–3000 cm −1 and below 1700 cm−1 region with increasing purge times.
Figure 4.
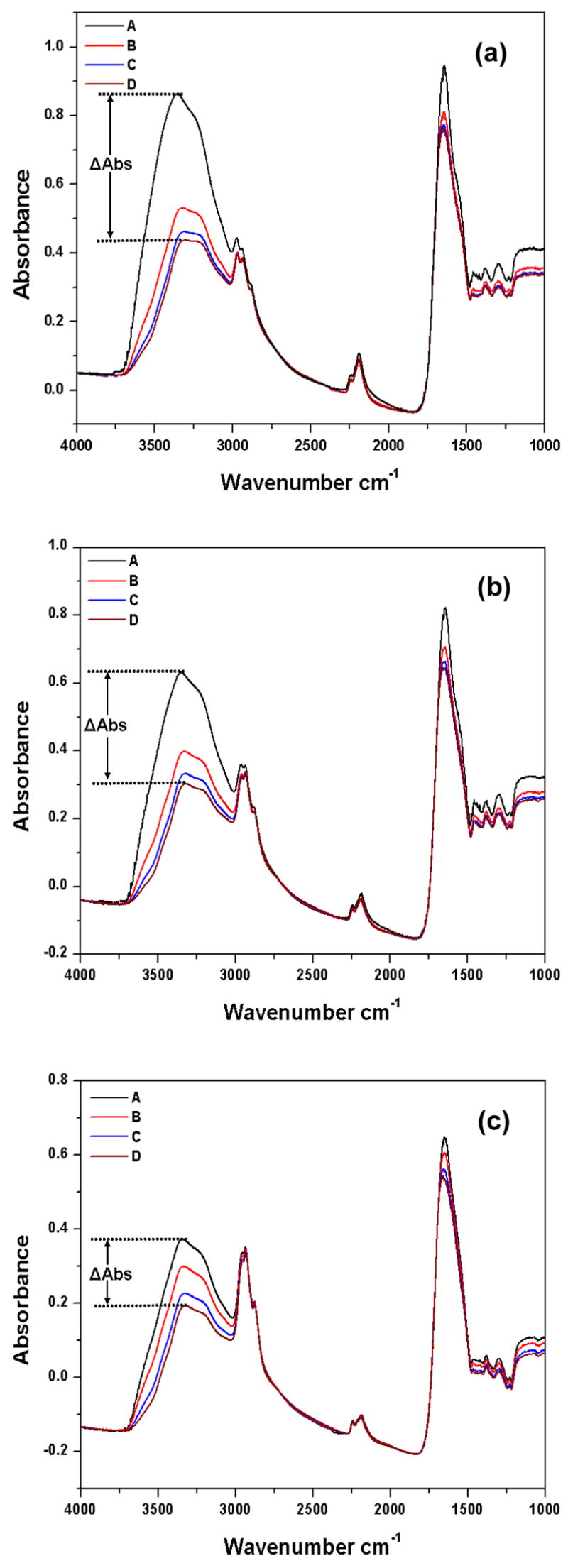
ATR-FTIR spectra of plasma polymerized 1A2P films deposited under (a)10/30 ms; (b) 10/10 ms; (c) CW conditions. The A, B, C., and D spectra shown for each duty cycle represent the sequence as deposited, and after 2, 5, and 10 minute dry N2. purge.]
XPS Characterization
The atomic compositions of the films produced under the different deposition conditions are shown in Table I. The high resolution C(1S) XPS spectra of films obtained from the 1A2P monomer are shown in Figure 5, arranged in order of increasing plasma duty cycle employed during deposition, reading top to bottom. As shown in this Figure, there is a significant decrease in the relative contribution of higher binding energy C(1S) peaks as the plasma duty cycle employed was decreased. Analyses of the deconvoluted C(1S) spectra, shown in Table II, provides a more quantitative measure of the relative increase in the higher binding energy peaks. The peak centered at 284.6 eV represents carbon atoms not bonded to heterogeneous atoms, in this case neither nitrogen nor oxygen. Other peak assignments are to C-N at 285.5 eV, C-O plus C≡N at 286.3 eV and N-C=O at 287.8 eV. The progressive increase in the peak at 284.6 eV, as the plasma duty cycle is increased, is indicative of the increase in polymer cross-linking with increasing average power input. The peak assignments shown in Table II are in accord with known functional group binding energies. 39
TABLE 1.
Percent atomic composition of plasma polymerized 1A2P films produced by varying plasma conditions
| Plasma condition | % C | % N | % O |
|---|---|---|---|
| 10/30 | 70.7 | 17.6 | 11.7 |
| 10/10 | 70.4 | 17.2 | 12.4 |
| CW | 75.6 | 13.7 | 10.7 |
Figure 5.
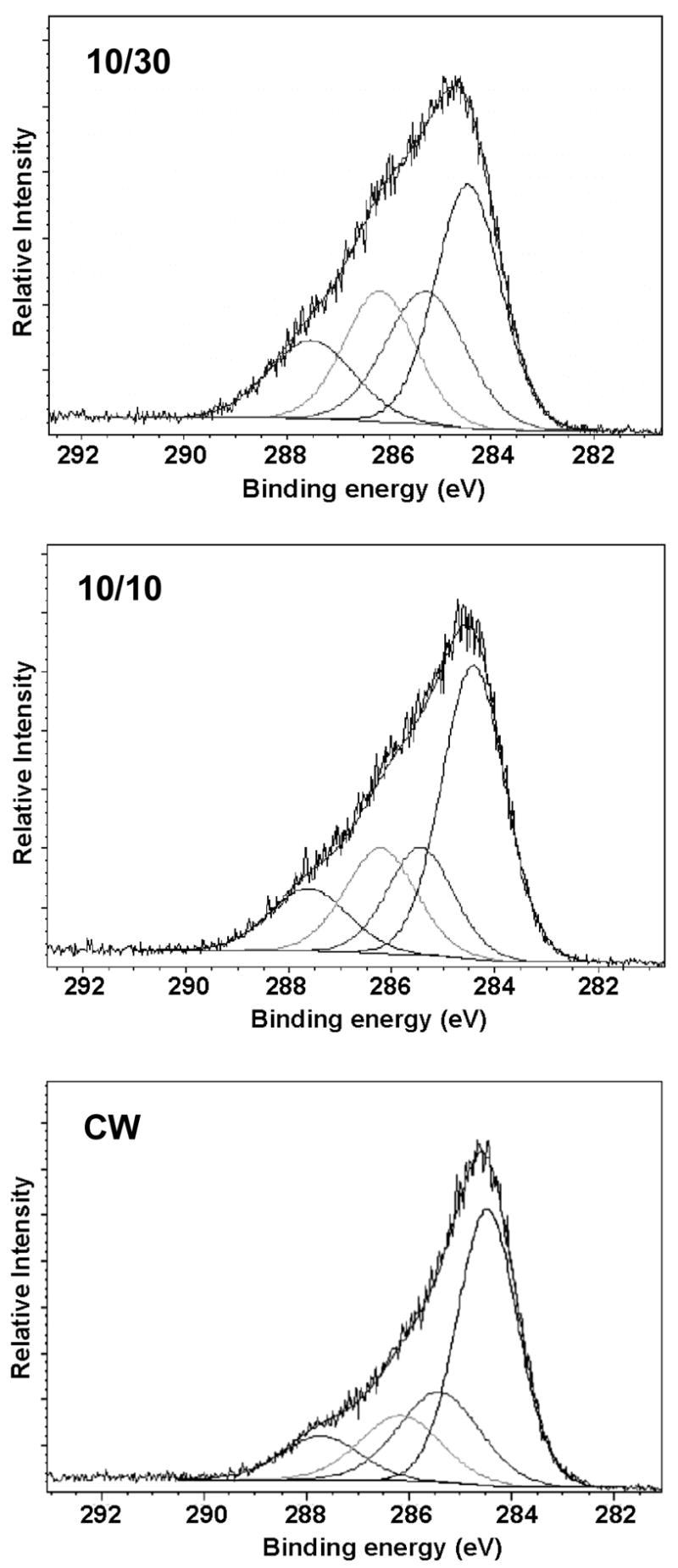
High resolution C(1s) X-ray photoelectron spectra of 1A2P films deposited at 10/30, 10/10 pulsed and CW conditions, as shown..
TABLE II.
Percent carbon surface functionalities from high resolution XPS C(1s) spectra of different 1A2P films
| Plasma condition | C-H 284.6 eV | C-N 285.5 eV | C-O/C≡N 286.3 eV | N-C=O 287.8 eV |
|---|---|---|---|---|
| 10/30 | 37.5 | 24.9 | 21.9 | 15.7 |
| 10/10 | 48.8 | 18.7 | 19.6 | 12.9 |
| CW | 60.2 | 13.9 | 15.6 | 10.2 |
Contact Angle and Capillary Rise Measurements
Static, sessile drop water contact angle measurements were recorded at temperatures ranging from 20 to 60°C for 1A2P films deposited at the three aforementioned plasma duty cycles. The results of these measurements are shown in Figure 6. Notable differences in contact angles, and their temperature dependence, are clearly apparent. The low temperature contact angles increased significantly with higher power input, ranging from 45° for the 10/30 sample, to 55° for the 10/10 sample, to a value of 60° for the film deposited under CW conditions. The increasing film hydrophobicity, with increasing power input during film formation, is in accord with the XPS results which reveal a progressive decrease in hetero atom content, and thus less polar groups, in the film sequence 10/30, 10/10, CW. More significantly, a dramatic increase in contact angle is observed for the 10/30 film, with a sharp change in wettability occurring over the temperature interval from 30 to 35°C. In contrast, no measurable change in film wettabilities were observed with increased temperature for the other two films. A constant time period (30 secs) was employed for each individual measurement to permit equilibration of the film with the sessile drop and, at the same time, to minimize evaporation of water at higher temperature.
Figure 6.
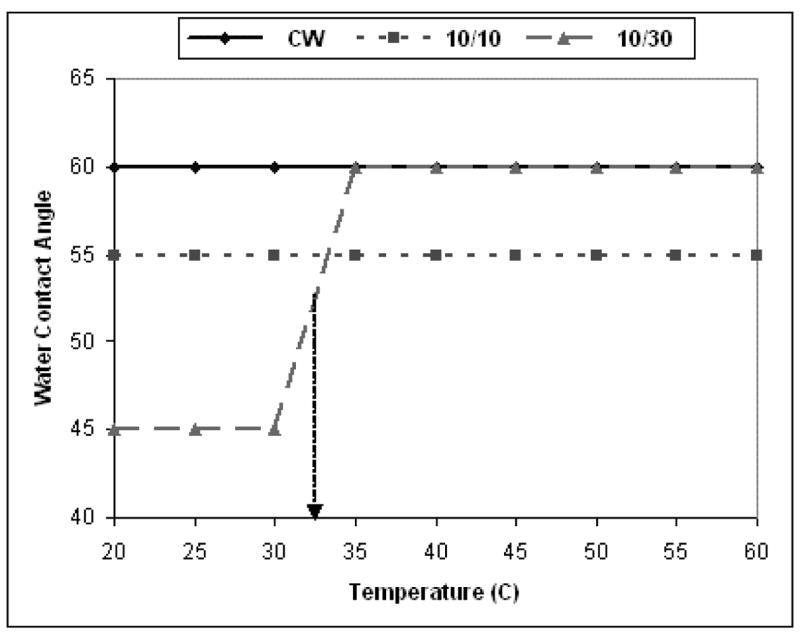
Static contact angle measurements as a function of temperature for plasma polymerized 1A2P films produced under 10/30, 10/10 ms pulsed and CW modes.
The differences in surface wettabilities of the films noted in the contact angle measurements were confirmed by the capillary rise measurements. Figure-7 shows the variation in heights of water column rise in capillaries coated with the three different 1A2P plasma polymers. These measurements were made with the capillary tubes immersed in water in a petridish. As shown in Figure-7, the rise heights clearly show the increased film hydrophobicity as the plasma duty cycle employed during deposition is increased. Additionally, although the 10/10 and CW films exhibit virtually no change in rise height when measurements are made at 40°C, the 10/30 film shows a sharp drop (i.e. increased hydrophobicity) at this higher temperature. The 6.5 mm decrease in capillary rise height level observed is consistent with the sharp increase in water contact angle for this film with increasing temperature, as shown in Figure-6.
Figure 7.

Capillary rise experiment of (1) uncoated capillary, (2) capillary coated with 1A2P (10/30), (3) capillary coated with 1A2P (10/10), (4) capillary coated with 1A2P (CW) at 20°C and 40°C.
Finally, it is interesting to note that a distinctive color change is observed for the 10/30 films but not for the other two films. The color change for the 10/30 samples occur shortly after the samples are removed from the plasma reactor. An example of this color change is shown in Figure-8 for samples deposited on polished silicon substrate. The bright colors shown result from interference effects due to reflection from the polished Si substrate. It is felt that the color changes arise from water absorbed thickness change and not from a change in refractive index. The polymer films, deposited on a transparent substrate, were light yellow in color. The actual color changes observed depend on the film thickness but they always occur spontaneously, over a short time period, with simple exposure of the sample to the atmosphere. This color change is completely reversible with respect to repeated heating and cooling. It is significant to note that no such color change was observed with samples of films produced under the 10/10 and CW conditions.
Figure 8.

Color of plasma polymerized 1A2P (10/30) film at 20°C and 80°C
Swelling studies of 1-amino-2-propanol films
1A2P films of 1 micron thickness were deposited on tared PET cover slips of 22 × 60 mm size and weighed after exposure to the atmosphere to permit water adsorption. Subsequently, each film was placed in a petridish and immersed with 20 ml of deionized distilled water at room temperature for 48 hours. The weight of swollen films was measured after the moisture on their surfaces was carefully wiped with a tissue paper. Swelling ratios (SR) were calculated using SR = Ws/Wd, where Ws and Wd are the weights of the swollen and dry 1A2P films40. The swelling ratio results are summarized in Table III.
TABLE III.
Comparison of swelling ratio of 1A2P films polymerized at different plasma conditions
| Plasma conditions ms/ms | Swelling ratio (Ws/Wd) |
|---|---|
| 10/30 | 28.2 ± 2.2 |
| 10/10 | 12.5 ± 3.2 |
| CW | 7.5 ± 1.3 |
Plasma Polymerization of 2-(ethylamino) ethanol monomer
To further test the generality of hydrogel film synthesis via plasma polymerization, this study was extended to include a limited number of experiments with a second monomer. For this purpose, polymer films were generated using 2-(ethylamino) ethanol (2EAE), monomer. The 2EAE monomer contains structural features (N-H and OH groups) in common with the 1A2P compound. The 2EAE experiments included both pulsed and CW runs, under the same deposition conditions employed with the 1A2P monomer, namely pulsed runs at 10/30 and 10/10 ms, on/off ratios and CW, all runs carried out at 150 W peak RF power. These films were also characterized using ATR-FTIR and static water contact angle measurements.
The IR spectra observed were essentially identical to those obtained from the 1A2P compound, with respect to the absorption bands. Although the same absorption bands are observed with both monomers, the intensities of the high wavenumber 3500 to 3100 cm−1 peaks, relative to the peak centered around 1600 cm−1, were significantly higher in the case of 1A2P than the 2EAE films. As in the case of the 1A2P films, the 2EAE films exhibited the spontaneous absorption of water, as evidenced by the decreased absorption in the high wavenumber region with purging of the samples with dry N2. As shown in table IV, the relative decrease in the high wavenumber absorption intensity, as a function of the plasma deposition conditions with purging, is in the order: 10/30 > 10/10 > CW, the same sequence as observed with the 1A2P compound. The measurement of the temperature dependent water contact angles, shown in Figure-9, reveal a dramatic change in surface wettability for the 10/30 sample but no notable change for the 10/10 and CW samples. Although this same trend was also observed with the 1A2P, the ~55°C transition temperature observed with the 2EAE is significantly higher than that obtained for the 1A2P films.
TABLE IV.
Comparison of change in relative absorbance of the 3600–3100 cm−1 peaks after dry N2 purge, all polymer films deposited at 150W peak power.
| Plasma condition | ΔAbsorbance | ||
|---|---|---|---|
| Duty cycle | Average power (W) | 1A2P | 2EAE |
| 10/30 | 37.5 | 0.44 | 0.17 |
| 10/10 | 75 | 0.33 | 0.14 |
| CW | 150 | 0.18 | 0.08 |
Figure 9.
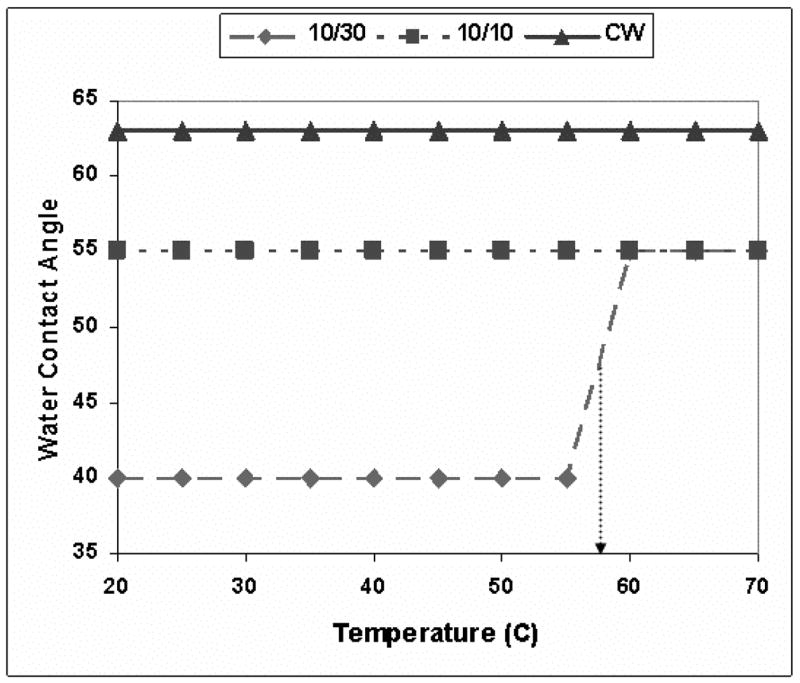
Contact angle measurements with variation of temperature for plasma polymerized 2EAE films produced under 10/30, 10/10 pulsed and CW modes.
DISCUSSION
The films generated from the two monomers employed in this study exhibited similar properties in terms of IR spectra and surface energy changes upon warming. Both sets of polymer films exhibited a pronounced tendency towards spontaneous atmospheric water adsorption, which is subsequently readily released upon exposure to a flow of dry N2. A more quantitative measurement of the variation in extent of water adsorption with duty cycle changes was obtained by measurement of the weight gain by the films, with and without adsorbed water. The weight changes observed for the 1A2P films are shown in table IV, in which the swelling ratio is significantly higher for the 10/30 film than the other two samples. The IR absorption peak frequencies observed are essentially identical with both monomers, although there is some difference in the relative band intensities and the magnitude of water adsorbed upon exposure to air. The extent of spontaneous water adsorption is considerably more pronounced with films from the 1A2P monomer relative to those from 2EAE, as revealed in Table III.
A second important distinction between the 1A2P and 2EAE films, as shown by a comparison of Figures 6 and 9, is the temperature difference required for the hydrophilic to hydrophobic phase transition for the 2EAE film relative to that of the 1A2P. Presumably, the higher transition temperature exhibited by the 2EAE film reflects the somewhat higher cross-linking, and thus the aforementioned less H2O adsorption, in this film compared to the 1A2P sample.
The results obtained in this study provide strong confirmation of the utility of the plasma polymerization technique as a viable approach to synthesis of hydrogel films. Prior work in the area of hydrogel synthesis via plasma polymerizations has focused on NIPAM, including a recent thorough study of the effects of power input and reaction temperatures on the composition and thermal responsive behavior of polymer films produced.35 The present study identifies two new monomers which provide hydrogel films under appropriate plasma conditions. It differs from the NIPAM work in that the volatility of the monomers eliminates the need for heating of the monomer or the reactor chamber. As a demonstrated in the present study, in common with the NIPAM work, the production of films which exhibit thermoresponsive hydrogel behavior is strongly dependent on the plasma conditions employed. With both monomers, the characteristic hydrogel property of a phase transition with temperature change is observed only at the lowest average power inputs employed. Attempts to produce hydrogel films at even lower average power inputs were unsuccessful in that those films exhibited significant solubility in water, even at room temperature. Overall the picture which emerges from this investigation is that with judicious control of the plasma deposition parameters, in this case the plasma duty cycles, it is possible to produce films which possess significant H-bonding to exhibit hydrogel properties, and at the same time, are sufficiently cross-linked to be insoluble in water. For example, the films used in the present study have exhibited long term stabilities with respect to immersion in aqueous solutions as well as to repetitive cycling between wet and dry states.
The film chemistry variations observed with changes in plasma duty cycles are consistent with many prior studies of film chemistry control, covering a wide range of monomers, made available by the variable duty cycle pulsed plasma approach. As revealed by the XPS, IR and water contact angle measurements, a decrease in plasma duty cycle provides an increased retention of the heteroatoms of the monomers in the polymer films, and thus the degree of polarity in these polymers. The increased retention of monomer structure in the polymer films with decreasing plasma duty cycles is believed to arise from the increased importance of film formation under plasma off times, as revealed by the energy efficiency data (Figure-3). The less energetic conditions which prevail during plasma off times limit monomer fragmentation. As illustrated in the present study, this film chemistry control is of pivotal importance in achieving a true thermoresponsive hydrogel state via the plasma polymerization route. At the same time, given the relatively minor variations in atomic compositions (Table I) it is clear that the film properties are determined by the relative amounts of functional groups present, shown in Table II. It is interesting to note that the thermo responsive hydrogel properties are only observed for one of the three samples with each monomer. Clearly, the present results suggest that changes molecular structure can produce very significant film behavior with respect to the thermal induced hydrophilic to hydrophobic phase transitions.
Finally, it seems reasonable to speculate that the film chemistry control provided by the variable duty cycle pulsed plasma technique should provide opportunities to generate hydrogel films from many other monomers. In particular, given the different phase transition temperature observed in the present work, identification of other monomers may well provide a spectrum of hydrogels having transition temperatures covering a wide range of values. We hope to explore this concept in more detail in subsequent studies.
CONCLUSION
A new class of monomers has been used to prepare plasma polymerized hydrogel films. Molecular structure of the films has been characterized by ATR-FTIR and XPS spectroscopies. Variable duty cycle pulsed plasma polymerization has been shown to be an effective route to control moisture uptake and thermoresponsive behavior of the films.
Acknowledgments
The authors are pleased to acknowledge partial support of this work by the National Institutes of Health, under grant RO-1, GM074021-01.
References
- 1.Peppas NA. In: Reflexive Polymers and Hydrogels: Understanding and Designing Fast Responsive Polymeric Systems. Yui Nobuhiko, Mrsny Randall J, Park Kinam., editors. CRC Press LLC; 2004. [Google Scholar]
- 2.Zhang XZ, Wu DQ, Chu CC. Biomaterials. 2004;25:3793. doi: 10.1016/j.biomaterials.2003.10.065. [DOI] [PubMed] [Google Scholar]
- 3.Zhang XZ, Lewis PJ, Chu CC. Biomaterials. 2005;26:3299. doi: 10.1016/j.biomaterials.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 4.Luo Y, Kirker KR, Glenn D, Prestwich GD. Journal of Controlled Release. 2000;69:169. doi: 10.1016/s0168-3659(00)00300-x. [DOI] [PubMed] [Google Scholar]
- 5.Linke B, Kernar W, Kiwit M, Pishko M, Heller A. Biosensors and Bioelectronics. 1994;9:151. doi: 10.1016/0956-5663(94)80107-x. [DOI] [PubMed] [Google Scholar]
- 6.Gajovic N, Beinyamin G, Warsinke A, Scheller FW, Heller A. Analytical Chemistry. 2000;72:2963. doi: 10.1021/ac991021i. [DOI] [PubMed] [Google Scholar]
- 7.Thoniyot P, Cappuccio FE, Gamsey S, Cordes DB, Wessling RA, Singaram B. Diabetes Technology & Therapeutics. 2006;8:279. doi: 10.1089/dia.2006.8.279. [DOI] [PubMed] [Google Scholar]
- 8.Schmedlen RH, Masters KS, West JL. Biomaterials. 2002;23:4325. doi: 10.1016/s0142-9612(02)00177-1. [DOI] [PubMed] [Google Scholar]
- 9.Tana J, Gemeinharta RA, Maa M, Saltzman WM. Biomaterials. 2005;26:3663. doi: 10.1016/j.biomaterials.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 10.Canavan HE, Cheng X, Graham DJ, Ratner BD, Castner DG. Journal of Biomedical Materials Research. 2005;75A:1. doi: 10.1002/jbm.a.30297. [DOI] [PubMed] [Google Scholar]
- 11.Alarc′on CDLH, Twaites B, Cunliffe D, Smith JR, Alexander C. International Journal of Pharmaceutics. 2005;295:77. doi: 10.1016/j.ijpharm.2005.01.037. [DOI] [PubMed] [Google Scholar]
- 12.Gerlach G, Guenther M, Sorber J, Suchaneck G. Sensors and Actuators B. 2005;111–112:555. [Google Scholar]
- 13.Vickie Pan Y, Wesley RA, Luginbuhl R, Denton DD, Ratner BD. Biomacromolecules. 2001;2:32. doi: 10.1021/bm0000642. [DOI] [PubMed] [Google Scholar]
- 14.Cheng X, Canavan HE, Stein MJ, Hull JR, Kweskin SJ, Wagner MS, Somorjai GA, Castner DG, Ratner BD. Langmuir. 2005;21:7833. doi: 10.1021/la050417o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ito Y, Chen G, Guan Y, Imanishi Y. Langmuir. 1997;3:2756. [Google Scholar]
- 16.Hruby M, Subr V, Kucka J, Kozempel J, Lebeda O, Sikora A. Applied Radiation and Isotopes. 2005;63:423. doi: 10.1016/j.apradiso.2005.05.043. [DOI] [PubMed] [Google Scholar]
- 17.Hassan CM, Stewart JE, Peppas NA. European Journal of Pharmaceutics and Biopharmaceutics. 2000;49:161. doi: 10.1016/s0939-6411(99)00056-9. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe J, Ooya T, Nitta KH, Park KD, Kim YH, Yui N. Biomaterials. 2002;23:4041. doi: 10.1016/s0142-9612(02)00122-9. [DOI] [PubMed] [Google Scholar]
- 19.Hahn MS, Taite LJ, Moon JJ, Rowland MC, Ruffino KA, West JL. Biomaterials. 2006;27:2519. doi: 10.1016/j.biomaterials.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 20.Smith LE, Rimmer S, MacNeil S. Biomaterials. 2006;27:2806. doi: 10.1016/j.biomaterials.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 21.Park KH, Yun K. Journal of Bioscience and Bioengineering. 2004;97:374. doi: 10.1016/S1389-1723(04)70221-2. [DOI] [PubMed] [Google Scholar]
- 22.Schmaljohann D, Oswald J, Jørgensen B, Nitschke M, Beyerlein D, Werner C. Biomacromolecules. 2003;4:1733. doi: 10.1021/bm034160p. [DOI] [PubMed] [Google Scholar]
- 23.Gilcreest VP, Carroll WM, Rochev YA, Blute I, Dawson KA, Gorelov AV. Langmuir. 2004;20:10138. doi: 10.1021/la0487996. [DOI] [PubMed] [Google Scholar]
- 24.Park TG, Hoffman AS. Journal of Applied Polymer Science. 1994;52:85. [Google Scholar]
- 25.McPhee W, Tam KC, Pelton R. Journal of Colloid Interface Science. 1993;156:24. [Google Scholar]
- 26.Matsuo ES, Tanaka T. Journal of Chemical Physics. 1988;89:1695. [Google Scholar]
- 27.Hegewald J, Schmidt T, Eichhorn KJ, Kretschmer K, Kuckling D, Arndt KF. Langmuir. 2006;22:5152. doi: 10.1021/la053461c. [DOI] [PubMed] [Google Scholar]
- 28.Alexandre E, Schmitt B, Boudjema K, Merrill EW, Lutz PJ. Macromolecular Bioscience. 2004;4:639. doi: 10.1002/mabi.200400034. [DOI] [PubMed] [Google Scholar]
- 29.Waber LM, He J, Bradley B, Haskins K, Anseth KS. Acta Biomaterialia. 2006;2:1. doi: 10.1016/j.actbio.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 30.Liang L, Feng X, Liu J, Rieke PC, Fryxell GE. Macromolecules. 1998;31:7845. [Google Scholar]
- 31.Kanazawa H, Yamamoto K, Matsushima Y. Analytical Chemistry. 1996;68:100. doi: 10.1021/ac950359j. [DOI] [PubMed] [Google Scholar]
- 32.Go H, Sudo Y, Hosoya K, Ikegami T, Tanaka N. Analytical Chemistry. 1998;70:4086. doi: 10.1021/ac980331i. [DOI] [PubMed] [Google Scholar]
- 33.Schmaljohann D, Beyerlein D, Nitschke M, Werner C. Langmuir. 2004;20:10107. doi: 10.1021/la034653f. [DOI] [PubMed] [Google Scholar]
- 34.Tamirisa PA, Koskinen J, Hess DW. Thin Solid Films. 2006;515:2618. [Google Scholar]
- 35.Bullett NA, Talib RA, Short RD, McArthur SL, Shard AG. Surf Interface Anal. 2006;38:1109. [Google Scholar]
- 36.Timmons RB, Griggs AJ. Plasma Polymer Films. 2004:217. [Google Scholar]
- 37.Chyan OMR, Chen JJ, Xu F, Wu J. Analytical Chemistry. 1997;69:2434. doi: 10.1021/ac970001i. [DOI] [PubMed] [Google Scholar]
- 38.Muller M, Rieser T, Lunkwitz K, Meier-Haack J. Macromolecular Rapid Communications. 1999;20:607. [Google Scholar]
- 39.Beamson G, Briggs D. High Resolution XPS of Organic Polymers - The Scienta ESCA300 Database. John Wiley & Sons; 1992. [Google Scholar]
- 40.Mandracchia D, Pitarresi G, Palumbo FS, Carlisi B, Giammona G. Biomacromolecules. 2004;5:1973. doi: 10.1021/bm0497567. [DOI] [PubMed] [Google Scholar]