

Abstract
We show that a minimalist basic region/leucine zipper (bZIP) hybrid, comprising the yeast GCN4 basic region and C/EBP leucine zipper, can target mammalian and other gene regulatory sequences naturally targeted by other bZIP and basic/helix–loop–helix (bHLH) proteins. We previously reported that this hybrid, wt bZIP, is capable of sequence-specific, high-affinity binding of DNA comparable to that of native GCN4 to the cognate AP-1 and CRE DNA sites. In this work, we used DNase I footprinting and electrophoretic mobility shift assay to show that wt bZIP can also specifically target noncognate gene regulatory sequences: C/EBP (CCAAT/enhancer binding protein, 5′-TTGCGCAA), XRE1 (Xenobiotic response element, 5′-TTGCGTGA), HRE (HIF response element, 5′-GCACGTAG), and the E-box (Enhancer box, 5′-CACGTG). Although wt bZIP still targets AP-1 with strongest affinity, both DNA-binding specificity and affinity are maintained with wt bZIP binding to noncognate gene regulatory sequences: the dissociation constant for wt bZIP in complex with AP-1 is 13 nM, while that for C/EBP is 120 nM, XRE1 240 nM, and E-box and HRE are in the μM range. These results demonstrate that the bZIP possesses the versatility to bind various sequences with varying affinities, illustrating the potential to fine-tune a designed protein’s affinity for its DNA target. Thus, the bZIP scaffold may be a powerful tool in design of small, α-helical proteins with desired DNA recognition properties.
Keywords: bZIP, GCN4, C/EBP, Xenobiotic response element XRE1, Enhancer box E-box, HIF response element HRE
1. Introduction
The basic region/leucine zipper (bZIP) family of transcription factors comprises the simplest motif that Nature uses for targeting specific DNA sites: a pair of short α-helices that recognize the DNA major groove with sequence specificity and high affinity [1,2]. Crystal structures of the bZIP domain of GCN4 bound to two different DNA duplexes, the AP-1 [3] and CRE sites [4,5], and the crystal structure of the Jun-Fos bZIP heterodimer bound to AP-1 [6] show that a continuous α-helix of ~60 amino acids provides the basic region interface for binding to specific DNA sites, as well as the leucine zipper coiled-coil dimerization structure (Fig. 1). The simplicity and tractability of the α-helical bZIP make it an ideal minimalist scaffold for study and protein design.
Fig. 1.
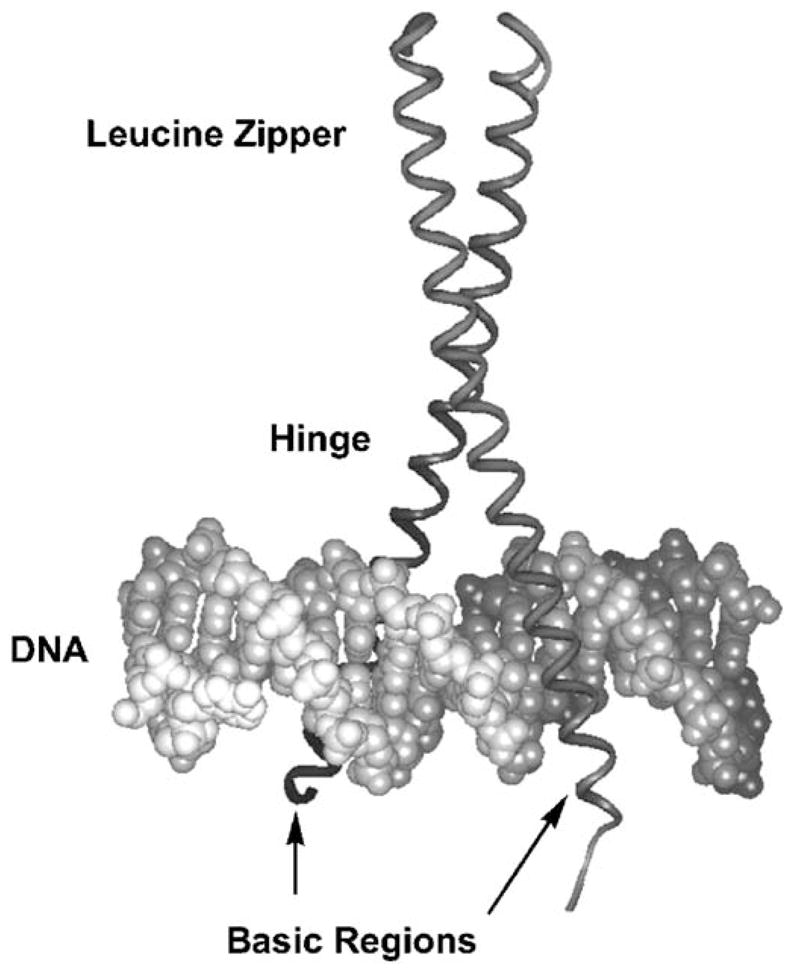
GCN4 bZIP in complex with the AP-1 DNA site [3]. DNA is the double helix at the bottom of the figure, and the bZIP is the vertical ribbon, α-helical dimer. The leucine zipper dimerizes into the coiled-coil structure shown at the top of the figure; the zipper then smoothly forks to either side of the DNA (hinge region), thus allowing the basic region dimer to bind opposite sides of the DNA major groove.
We previously generated proteins with α-helical structure and DNA recognition capabilities from a core scaffold based on the GCN4 bZIP [7,8]. GCN4 is a dimeric transcriptional regulator that governs histidine biosynthesis in yeast under conditions of amino acid starvation [9]. The full-length GCN4 monomer is 281 amino acids, and the bZIP comprises a homodimer of ~60-residue monomers. Our minimalist bZIP derivatives are fusions between two different bZIP proteins: the wt bZIP is the “native” variant comprising the GCN4 basic region and C/EBP leucine zipper. McKnight and coworkers showed that exchanging basic regions and leucine zippers between GCN4 and C/EBP resulted in functional proteins that still targeted cognate DNA sites [10]. Previously, we confirmed that wt bZIP binds specifically and strongly to the cognate pseudosymmetric AP-1 (5′-TGACTCA) and symmetric CRE (5′-TGACGTCA) sites and maintains structure and function comparable to native GCN4 [8,11].
In the current work, we used DNase I footprinting and electrophoretic mobility shift assay (EMSA) to show that not only can wt bZIP bind to AP-1, but also it can bind with sequence specificity and high to modest affinity to C/EBP (CCAAT/enhancer binding protein, 5′-TTGCGCAA), XRE1 (xenobiotic response element, 5′-TTGCGTGA), E-box (Enhancer box, 5′-CACGTG), and HRE (HIF response element, 5′-GCACGTAG) (sites listed in descending order of affinities, Fig. 2 and Table 1). Such promiscuous, yet specific and high-affinity, binding of DNA was not expected, despite the fact that GCN4 can bind to the AP-1 and CRE sites, which differ by one centrally located base pair, with similar affinities. Transcription factors are known for their ability to tolerate variation in DNA target sites. Cognate and noncognate complexes between transcription factors and DNA may therefore be of significance in vivo, for instance, allowing multiple functionality from a single gene.
Fig. 2.

(A) Sequences of the DNA sites used in DNase I footprinting studies. All sequences are duplexes. Core target sequences are in bold. The Partial site actually contains a weak half site, 5′-AGAC-3′, which is italicized, and the full Partial site is in bold. The 3′ and 5′ flanking sequences surrounding the AP-1 and XRE1 target sites are from the his3 and CYP1A1 promoter regions, respectively. Flanking sequences surrounding C/EBP, HRE, and the Partial site are the same as those for EMSA (discussed below). For E-box, the flanking sequences are the same as those used by Agre, Johnson, and McKnight in their work on C/EBP bZIP [10]. (B) Sequences of DNA sites used in EMSA studies. All sequences are 24-mer duplexes. Each duplex contains a core target site surrounded by the same 3′ and 5′ flanking sequences, which were chosen to minimize DNA secondary structure. The core target sequences are in bold. The inserts between flanking sequences are underlined; note that for the Partial site, the actual target is shifted by one base pair. The NS sequence is a nonspecific DNA control. The two thymines at the 3-end of each duplex were P32-labeled.
Table 1.
Dissociation constants for synthetic wt bZIP bound to DNA Sites
| Binding site | Kd (10−9 M) a synthetic wt bZIP |
|---|---|
| AP-1 | 13±0.59 |
| C/EBP | 120±0.16 |
| XRE1 | 240±18 |
| Partial | 280±49 |
| E-box | >800 b |
| HRE | >1000 b |
Average values of dissociation constants were obtained from two independent experiments, R values >0.97.
Saturation protein binding was not achieved in these titrations.
2. Materials and methods
2.1. Materials
Radioactive nucleotides were supplied by Amersham, and radioactivity was monitored on a Beckman LS 6500 scintillation counter. Water was purified through a MilliQ filtration system (Millipore). Reagents were purchased from Aldrich or Acros/Fisher; enzymes were purchased from New England Biolabs and Promega. Electrospray ionization mass spectrometry (ESI-MS) was performed on a Micromass ZQ Model MM1 Mass Spectrometer (Waters) at the University of Toronto at Mississauga. DNA inserts were sequenced at the DNA Sequencing Facility in the Centre for Applied Genomics, Hospital for Sick Children (Toronto, ON) on an ABI 3730XL 96 capillary sequencer using a combination of the Big Dye and dGTP Big Dye Sequencing Kits (Applied Biosystems).
2.2. Preparation of wt bZIP protein
Bacterial expression of wt bZIP was performed as described previously [7]. A brief summary of this procedure follows: the gene for expression of wt bZIP was constructed by mutually primed synthesis, followed by polymerase chain reaction with terminal primers for gene amplification and purification by nondenaturing polyacrylamide gel electrophoresis. Duplex DNA was then cloned into protein expression vector pTrcHis B (Invitrogen) and sequenced; this vector expresses proteins with an amino-terminal six-histidine tag for purification purposes. Recombinant plasmids were transformed into Escherichia coli strain BL21(DE3) (Stratagene) by electroporation (Bio-Rad E. coli Gene Pulser).
Bacterial expression of wt bZIP was performed in Luria-Bertani (LB) medium containing 100 μg/ml ampicillin; induction was initiated with IPTG added to a final concentration of 1 mM. Cells were collected by centrifugation and lysed by sonication. The 6xHis-tagged protein was purified first on TALON cobalt metal-ion affinity resin (Clontech) and further purified by HPLC (Beckman System Gold) on a semipreparative reversed-phase C18 column (Beckman Coulter) with a gradient of acetonitrile–water plus 0.2% trifluoroacetic acid (v/v); the flow rate was 2 ml/min, and the gradient started at 0–20% acetonitrile over 20 min, followed by 20–60% acetonitrile over 80 min. Purity of the final product was confirmed by analytical C18 column (Beckman Coulter) using the same gradient as described above but at a flow rate of 1 ml/min. HPLC-purified protein was concentrated by centrifugation with a Centriplus centrifugal filter (Millipore) and stored at −80 °C. Electrospray ionization mass spectrometric analysis: calculated, 10941.0 g/mol; found, 10939.0 g/mol. The initiating Met at the N-terminus of wt bZIP had been cleaved during posttranslational modification [12].
A chemically synthesized 57-residue peptide (synthetic wt bZIP) was obtained from Biomer Technology (Concord, CA) as a crude product synthesized using standard Fmoc [N-(9-fluorenyl)methoxycarbonyl] chemistry followed by cleavage and deprotection. The peptide was purified as described above. Electrospray ionization mass spectrometric analysis: calculated, 6822.9 g/mol; found, 6821.0 g/mol. Sequences of the expressed and synthetic versions of wt bZIP are provided in the Supplementary information.
2.3. DNase I footprinting analysis
Oligonucleotides with target sequences contained endonuclease restriction sites BamHI and SacI to allow direct incorporation into plasmid. Complementary oligonucleotides were annealed; the DNA duplexes were cloned into vector pUC19, and recombinant plasmids were then transformed into E. coli strain TOP10 (Invitrogen) by electroporation. Incorporation of the insert was confirmed by sequencing.
For 3′-endlabeled footprinting, recombinant pUC19 plasmids were doubly digested with HindIII and SspI. The resulting ~650-bp DNA duplexes were isolated by 1% agarose gel and radiolabeled with [α-32P]-dCTP by DNA polymerase I, Klenow fragment [11]. Radiolabeled DNA duplexes were purified by ethanol precipitation and mini Quick Spin DNA columns (Roche). For 5′-endlabeled footprinting, recombinant pUC19 plasmids were first linearized with HindIII, dephosphorylated with calf intestinal alkaline phosphatase, and phosphatase was removed by phenol/chloroform extraction followed by butanol extraction and ethanol precipitation. The dephosphorylated DNA duplexes were radiolabeled with [γ-32P]-ATP by T4 polynucleotide kinase [11]. Radiolabeled DNA duplexes were purified by ethanol precipitation and mini Quick Spin DNA columns (Roche), followed by SspI digestion. The resulting ~650-bp DNA duplexes were isolated by 1% agarose gel.
The DNase I footprinting reactions (20 μl total volume) were assembled by adding bZIP protein to a mixture containing TKMC buffer (20 mM Tris, pH 7.5, 4 mM KCl, 2 mM MgCl2, 1 mM CaCl2), 100 μg/ml BSA (non-acetylated, DNase/RNase/protease-free, Sigma), 5 mM DTT, 1 μg/ml poly(dI–dC) (Sigma), and 5% glycerol. We emphasize that the expressed wt bZIP protein was added last to the mixture to prevent protein aggregation. We have found the temperature-leap tactic [13] to be critical for correct protein folding and stability, in particular for our His-tagged proteins; therefore, we took great care in applying the temperature-leap tactic, which entailed incubation of protein-containing reactions at 4 °C for >2 h, 37 °C for 1 hr [11,14].
After addition of 32P-endlabeled DNA fragment (~20,000 cpm), footprinting reactions were temperature-leaped one more time (4 °C for 2 h; 37 °C, 1 h) followed by incubation at room temperature for 30 min. Footprinting reactions were initiated by addition of DNase I and allowed to proceed for 3 min at 22 °C. DNase I concentrations varied depending on concentration of wt bZIP: for DNase I cleavage controls with no wt bZIP (lanes 2, 6, 10, 14, 18, and 22 in Figs. 3 and S1), final DNase I concentration was 0.5 mg/ml. Footprinting at lower wt bZIP concentrations (0.5 μM and 1 μM) required 10 mg/ml DNase I (lanes 3, 7, 11, 15, 19, and 23 in Figs. 3 and S1). Footprinting at 3 μM wt bZIP required 50 mg/ml DNase I (lanes 4, 8, 12, 16, 20, and 24 in Figs. 3 and S1). DNase I cleavage activity was hampered at increasing wt bZIP concentrations; we suspect that wt bZIP and DNase I may form soluble aggregates at higher wt bZIP concentrations. Additionally, increasing amounts of wt bZIP may exhibit some nonspecific binding to DNA (positively charged basic regions can nonspecifically interact with DNA) and, therefore, shield DNA from DNase I cleavage. Reactions were terminated by addition of 4 μl DNase stop solution (3 M NH4OAc, 250 mM EDTA), followed by ethanol precipitation, drying, and resuspension in formamide loading buffer. Reaction products were analyzed by electrophoresis on 8% polyacrylamide denaturing gels (Bioshop Gene-gel 8%, 1:19 cross-linked, 7 M urea) run at 2000 V. After electrophoresis, gels were dried, followed by autoradiography on a Molecular Dynamics Storm 840 PhosphorImager System.
Fig. 3.

DNase I footprinting analysis on expressed wt bZIP bound to AP-1, C/EBP, XRE1, Partial site, E-box, and HRE. Data presented for 5′ 32P-endlabeled DNA (~20,000 cpm/lane). Lanes 1, 5, 9, 13, 17, and 21: chemical sequencing G reactions. Lanes 2, 6, 10, 14, 18, and 22: DNase I cleavage control reactions. Lanes 3, 7, 11, and 15: DNase I cleavage reactions with 0.5 μM wt bZIP. Lanes 19 and 23: DNase I cleavage reactions with 1 μM wt bZIP. Lanes 4, 8, 12, 16, 20, and 24: DNase I cleavage reactions with 3 μM wt bZIP. The positions and sequences of the corresponding DNA targets are indicated. See Supplementary information, Figure S1, for the 3′ 32P-endlabeled DNA footprinting analysis. (A) The full DNase I footprinting analysis. (B) The boxed region highlighted above AP-1. A–C indicate possible sites of weak footprinting denoted by vertical bars; X–Z indicate hypersensitivity cleavage (see text and Supplementary information for discussion). (C) The boxed region highlighting footprinting at C/EBP and XRE1. Hypersensitivity cleavage bands within the binding sites are indicated by arrows.
2.4. Electrophoretic gel mobility shift assay (EMSA)
DNA oligonucleotides were purchased from Operon Biotechnologies and used without additional purification. Annealing and 3′-endlabeling of oligonucleotides were performed as described above. 24-mer duplexes were double labeled at the 3′-terminus with [α-32P]-dTTP. The labeled DNA fragments were purified by use of the Mini-Elute DNA Reaction Clean-Up Kit (Qiagen).
Solid wt bZIP was freshly dissolved to 2 μM and sequentially diluted to required concentrations in EMSA buffer (20 mM Tris and 1 mM phosphate, pH 7.5, 5 mM NaCl, 4 mM KCl, 2 mM MgCl2, 1 mM CaCl2, 1 mM EDTA, 100 μg/ml non-acetylated BSA, 2 μg/ml poly(dI–dC), 200 mM guanidine HCl, and 10% glycerol). For expressed wt bZIP, concentrations >2 μM promote formation of insoluble aggregates. EMSA was tried with TKMC buffer described above for footprinting, but wt bZIP solubility could not be maintained in TKMC. We also tried more stringent EMSA conditions on both the cognate (AP-1) and noncognate (XRE1 and E-box) sites. Using TKMC buffer, we varied the concentration of poly(dI–dC) to 2, 10, 20, 30, 40, 60, and 100 μg/ml. There was no significant decrease in binding activity at 2–60 μg/ml poly(dI–dC); at the highest concentration of 100 μg/ml poly(dI–dC), binding was noticeably decreased. We also used a higher salt buffer containing 15 mM Tris, pH 7.5, 75 mM NaCl, 1.35 mM KCl, 5 mM MgCl2, and 2.5 mM CaCl2. However, this buffer caused significant band-broadening and was discontinued.
Protein solutions were boiled at 90 °C for 10 min and slowly cooled to room temperature over 4 h. 3′-Endlabeled DNA duplexes were then added to protein solutions on ice. Each reaction contained wt bZIP and ~3000 cpm labeled DNA duplexes in EMSA buffer. Reactions were incubated at 4 °C overnight, 37 °C for 1 h, and room temperature for 30 min. The temperature-leap tactic was applied to wt bZIP in order to ensure proper protein folding. Other temperature-leap programs were tried, but those above gave the best results. Reactions were loaded onto 10% polyacrylamide, 1:29 cross-linked, nondenaturing gel. Gels were loaded while running at 120 Vand run for 4 h for the qualitative experiments (Fig. 4) and run for 90 min for the Kd determination experiments. Other voltages and gel-running times were tried, but these conditions gave the best results. Gels were dried and analyzed using storage phosphor technology with a Molecular Dynamics Storm 840 PhosphorImager System and ImageQuant software (version 5.2).
Fig. 4.
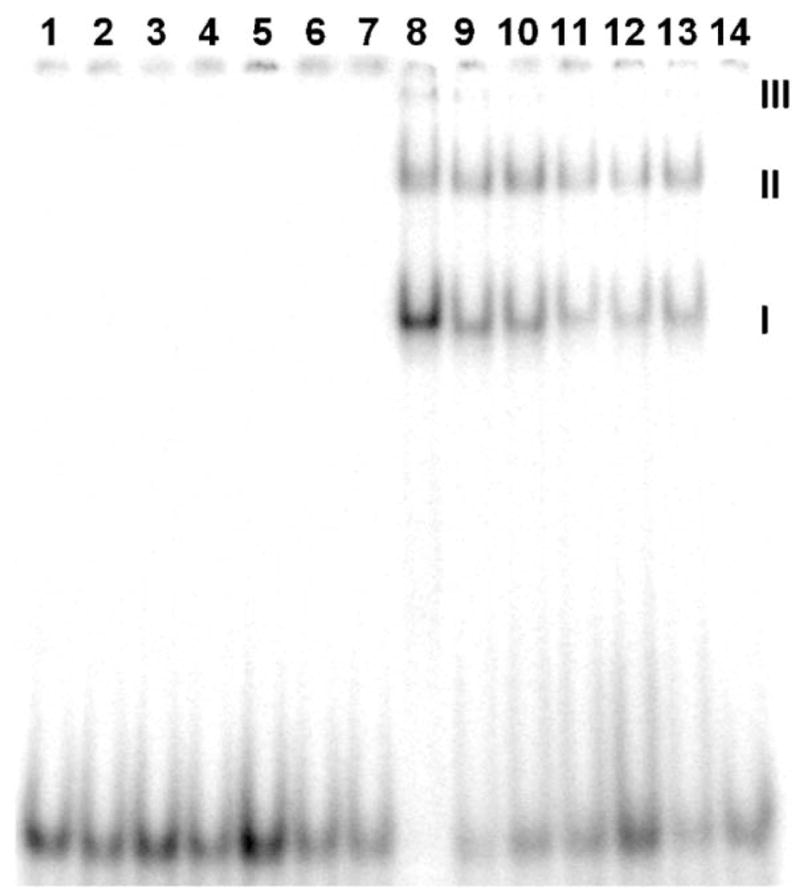
EMSA on wt bZIP bound to AP-1, C/EBP, XRE1, Partial site, E-box, and HRE. Each lane contains ~3000 cpm 32P-endlabeled 24-mer duplex. Lanes 1–7: free DNA. Lanes 8–14: DNA in the presence of 500 nM wt bZIP. Lanes 1 and 8, AP-1; lanes 2 and 9, C/EBP; lanes 3 and 10, XRE1; lanes 4 and 11, E-box; lanes 5 and 12, HRE; lanes 6 and 13, Partial site; lanes 7 and 14, nonspecific control. EMSA was run at 120 V for 4 h. I indicates bandshift from dimeric wt bZIP complexation. II and III indicate wt bZIP aggregates that bind to the duplexes and cause bandshifts (see text for discussion).
2.5. Determination of Kd values
The dissociation constants of the protein–DNA complexes were measured under equilibrium binding conditions. The volumes of the bands corresponding to free and bound DNA were quantified using ImageQuant software (version 5.2). The bound-DNA fraction (θapp) was calculated as the volume of the band corresponding to the bound DNA, divided by the sum of the volumes of the bands corresponding to free and bound DNA. Data were fit to a modified two-state binding equation to determine apparent dissociation constants for each protein–DNA complex as previously described [11]. The treatment of the calculation of dissociation constants is the same as that used by Metallo and Schepartz to determine the dissociation constants of GCN4 basic region derivatives bound to specific DNA sites [15],
where Kd corresponds to the apparent monomeric dissociation constant, [M] is the concentration of monomeric wt bZIP, θmin is the bound DNA fraction when no wt bZIP present, and θmax is the bound DNA fraction when DNA binding is saturated. Curve fits of the fraction of DNA bound vs. protein monomer concentration yielded the apparent monomeric dissociation constants of the protein–DNA complexes. Data were fit to the above equation by KaleidaGraph software (version 3.6.4, Synergy Software). Only data sets fit to the above equation with R values >0.970 are reported. Each dissociation constant was determined from the average of two independent data sets.
3. Results and discussion
3.1. The wt bZIP binds noncognate DNA sequences
We decided to explore the binding of wt bZIP, which is a GCN4 derivative, to sequences other than AP-1 and CRE after we noticed that wt bZIP could target the E-box with specificity and affinity. We then found that wt bZIP could also target the C/EBP, XRE1, and HRE sites. The C/EBP consensus sequence (CCAAT/enhancer binding protein site) is 5′-TTGC·GCAA (black dot separates half sites). C/EBP is a family of bZIP transcription factors, like GCN4. However, C/EBP is found in numerous species including mammals, vertebrates, and invertebrates [16], whereas GCN4 is only native to yeast [17].
The other DNA sites studied are targeted by the basic/helix–loop–helix (bHLH) family of transcription factors. Like bZIP proteins, the bHLH protein family also regulates transcription. Similar to the bZIP, the bHLH uses a dimer of basic α-helices to bind specific DNA sites. Despite their shared structural similarity, the bZIP and bHLH are distinct families of transcription factors with different functions in the cell, and they bind to different DNA sequences. Thus, we did not expect our GCN4 bZIP derivative would bind with sequence specificity and high affinity to gene regulatory sites bound by another class of proteins.
Within the bHLH, there are subfamily variants: the bHLHZ, wherein a leucine zipper contiguous to the bHLH is part of the dimerization domain, as in Max, Mad and Myc [18,19]; and the bHLH/PAS, in which the PAS domain assists in efficient protein dimerization, as in AhR, Arnt, and HIF-1α [20]. The Max, Mad, and proto-oncogenic Myc transcription factor network comprises widely expressed bHLHZ proteins critical for control of normal cell proliferation and differentiation [21,22]. Myc-Max is a transcriptional activator that binds the E-box, which is contained in a number of promoters, including that for p53 tumor suppressor [23]. The E-box core sequence is 5′-CAC·GTG, and the immediate flanking base pairs at the 5′-and 3′-ends of the E-box dictate variants that can modulate protein–DNA binding affinities.
XRE1 and HRE are targeted by AhR, Arnt, and HIF-1α [24,25]. In mammalian cells, AhR (also called the dioxin receptor) [26,27] and Arnt [28,29] heterodimerize in the presence of ligands, including dioxins and polychlorinated biphenyls (PCBs); this activated AhR-Arnt complex [30,31] binds distinct, unrelated half sites in the XRE1 site [32,33], which resides in the 5′-flanking region of the CYP1A1 (cytochrome p450) gene. The XRE1 site (xenobiotic response element 1, 5′-TTGC·GTG) is a member of the xenobiotic response element family that lies in enhancers of target genes and, when recognized by the aryl hydrocarbon receptor (AhR) and Arnt heterodimer, promotes transcription of a battery of xenobiotic-metabolizing enzymes. Note that XRE1 is an asymmetric hybrid containing a C/EBP half site and an E-box half site.
HIF-1α is a transcriptional activator and oxygen sensor. Under normoxia, cells generate ATP and energy via oxidative phosphorylation; under the hypoxic conditions surrounding nascent tumor growth, anaerobic glycolysis becomes the predominant pathway of ATP generation, and this allows development of an oxygen supply network via angiogenesis resulting in cell proliferation [34,35]. This metabolic shift is governed by the HIF-1α/Arnt heterodimer that binds to HRE (HIF response element, 5′-GCAC·GTAG), which is also an asymmetric site containing an E-box half site [36].
Similar to Max, Arnt can homodimerize and bind to specific DNA targets, including the E-box [37]. By heterodimerizing with proteins including AhR and HIF-1α, Arnt regulates their abilities to bind XRE1 and HRE, respectively. Thus, Arnt and Max play similar roles: by heterodimerizing with partner proteins, they can regulate transcription of multiple genes and pathways.
3.2. DNase I footprinting analysis: sequence-specific binding at noncognate sites
Fig. 3 shows DNase I footprinting analysis of wt bZIP targeting AP-1, C/EBP, XRE1, E-box, HRE, and a Partial site. The footprinting analysis was performed with bacterially expressed wt bZIP, which is a 96-mer comprising the GCN4 basic region, residues 226–252; the C/EBP leucine zipper, residues 310–338; extra 31 amino acids from the expression vector at the N-terminus that include a six-His tag, plus a 9-residue GGCGGYYYY linker at the C-terminus (see Supplementary information for sequences) [7]. Binding to AP-1 is clearly the strongest, and we have previously characterized the interaction of wt bZIP to both AP-1 and CRE by footprinting and fluorescence anisotropy titration, which we used to show that the dissociation constant of expressed wt bZIP to AP-1 is 10 nM [8,11]. Qualitatively weaker binding is seen at C/EBP, followed by XRE1 and E-box, and weakest binding at HRE. Despite lower binding affinities, binding is still specific for these sites.
A specific footprint was found at the Partial site. This site contains the 5′-AGAC half site, somewhat similar to the native 5′-TGAC half site in AP-1 and CRE. We suspect that 5′-AGAC is being targeted, for there is no other plausible target in the Partial duplex. Struhl and coworkers did not observe binding at this half site in in vivo yeast assays and in vitro selection experiments [9,38]. However, their in vitro assay conditions are much more stringent than those used in the present study; for instance, their protein concentrations are over three orders of magnitude lower than that used in our EMSA experiments [9]. In their selection experiments, they used much higher concentrations of salt, and 40 of 43 sequences selected were AP-1. The authors state that lower stringency is required to identify the degree and type of degeneracy in the DNA site, and that their in vitro selections were more stringent than required for binding and transcriptional activation in vivo [37]. We were surprised to see a clear footprint at the 5′-AGAC half site, as well as the noticeable bandshift on EMSA (Fig. 4); although it is similar to the native 5′-TGAC half site, it has not been previously characterized as a GCN4 binding site. However, we also observed binding at C/EBP, XRE1, E-box, and HRE, and the sequences of these sites deviate even more from AP-1.
We also note additional weak footprinting and hypersensitivity bands above the binding sites; this region is boxed above AP-1in Fig. 3A. This sequence is the same for all of the other sites (sequence is from pUC19), which also display the same footprinting and hypersensitivity pattern (discussed in Fig. S2 in the Supplementary information). The boxed region in Fig. 3A is detailed in 3B; potential footprints are labeled A, B, and C and marked with vertical bars. A and C appear to be half sites for binding. The sequence at A is 5′-CAC, which is the E-box half site. Since we present data in Fig. 3A showing that wt bZIP binds E-box full site, we suspect wt bZIP is binding weakly at this E-box half site. The sequence at C that appears to be targeted is 5′-TGAC, which is the AP-1 half site. Hollenbeck and Oakley have shown that the GCN4 bZIP can bind as a dimer to the AP-1 half site, wherein one basic region of GCN4 targets the AP-1 half site specifically and the other arm makes nonspecific DNA contacts; the binding affinity is only slightly decreased from that of binding the AP-1 full site [39]. Likewise, we suspect that wt bZIP is binding the E-box and AP-1 half sites, A and C respectively, that fortuitously arise in the DNA fragment in similar fashion. These sites are not random sites; as noted by a reviewer, footprints at these sites are indicative of specific low-affinity protein–DNA complexes that form under our low-stringency experimental conditions (note the low salt concentrations, see Materials and methods).
There appears to be a weak footprint at B, but this sequence is an A tract. We suspect this may be an artifact of DNase I footprinting between the closely spaced A and C sites; note the hypersensitivity cleavage bands at Y and Z flanking the A and C sites. DNase I can bind above A and generate hypersensitivity at Y, and it can bind below C and generate hypersensitivity at Z: what we see at B can be described as a DNase self-footprint. We also suspect that self-footprinting may contribute to the large footprint at A, which may be a combination of binding at the E-box half site, as discussed above, plus DNase I binding above the AP-1 site and generating significant hypersensitivity at X. As one looks further up the footprinting lanes in Fig. 3A, more potential footprints and hypersensitivity can be detected, and these likely originate from the same reasons already discussed.
In Fig. 3C, the footprinting sites at C/EBP and XRE1 are highlighted. Note the hypersensitivity within these two sites indicated by arrows. This is a different footprinting pattern from that displayed at the other sites, notably AP-1, which has been well characterized. We are investigating whether the bZIP employs a different mode of binding at these two sites.
3.3. EMSA: strong binding between wt bZIP and noncognate sites
In the EMSA shown in Fig. 4, binding between wt bZIP and AP-1 is the strongest of all the sites, for no free DNA remains (lowest set of bands). Among the noncognate sites, binding at C/EBP shows the strongest bandshift, followed by XRE1; binding at the Partial site and E-box are weaker, and very weak binding is shown at HRE. These qualitative EMSA results parallel the qualitative footprinting results shown in Fig. 3 and the quantitative EMSA titration results shown in Table 1.
The concentration of wt bZIP is 500 nM for lanes 8–14, Fig. 4, which show a bandshift except for the nonspecific control in lane 14. The bandshift labeled I in Fig. 4 corresponds to dimeric wt bZIP binding to the target site; bandshifts at II and III are higher order multimers or soluble aggregates of wt bZIP. The same EMSA has also been performed with a synthetic version of wt bZIP, which is a 57-mer with no appendages at the N- and C-termini originating from the expression vector (see Supplementary information for sequences); with synthetic wt bZIP, only one bandshift corresponding to I is observed by EMSA (unpublished results). The His tag is known to promote aggregation [39] and is therefore the likely source of bandshifts at II and III. Due to the existence of extra bandshifts at II and III, determination of the dissociation constants of wt bZIP–DNA complexes by EMSA titration experiments would not be accurate. Therefore, synthetic wt bZIP was used in the quantitative EMSA titrations. All dissociation constants shown in Table 1 were obtained by quantitative EMSA titration. Representative EMSA titrations are provided in the Supplementary information.
Because all of the EMSA duplexes have the same flanking sequences (Fig. 2), no bandshift in the nonspecific control demonstrates no binding activity in the flanking regions (lanes 7 and 14, Fig. 4). Incidentally, these same flanking sequences were used in the footprinting experiments for HRE and the Partial site as well. All binding is therefore specific to these centrally located target sites, especially noteworthy for the weakly bound E-box and HRE sites. The C/EBP sequence 5′-TTGCGCAA shows strong binding by wt bZIP with a Kd value of 120 nM (compare with 13 nM Kd for AP-1), whereas the E-box sequence 5′-CACGTG is only weakly bound (Kd >800 nM). The XRE1 sequence 5′-TTGCGTG is a hybrid of the C/EBP and E-box sequences, and its dissociation constant likewise is between the other two values (Kd 240 nM). Thus, we surmise that the 5′-TTGC half site is strongly recognized by wt bZIP; note that in the nonspecific control, there exists a 5′-TTCC sequence, and this very similar sequence is not targeted by wt bZIP.
The HRE sequence 5′-GCACGTAG is the most weakly bound of the sites studied (Kd >1000 nM); HRE contains the E-box half site and the 5′-GTAG site that has not been shown to be targeted by GCN4. We surmise that the weakly bound E-box half site is largely responsible for recognition of HRE by wt bZIP. The Partial site also shows a significant level of binding by the wt bZIP (Kd 280 nM), in agreement with footprinting results. Because the migration of the bandshift indicating binding at the Partial site is the same as that for AP-1 and no binding occurs in the flanking regions, binding must be occurring at a target centrally located on the duplex. In EMSA, we cannot distinguish binding that is offset by a single base pair. Therefore, we would not be able to see a difference in shift when wt bZIP binds to 5′-AGAC in the Partial site duplex, which is offset by one base pair in relative positioning to the other target sites. Both footprinting and quantitative EMSA indicate that binding at this Partial site is specific and significant.
3.4. Protein–DNA recognition: specific, yet promiscuous
For transcription factors, variable levels of promiscuity appear to have evolved as a desirable trait; in contrast, promiscuity of DNA sequence recognition by restriction endonucleases would be harmful to the host. The ability of proteins, especially transcription factors, to bind noncognate DNA sites is known. Two recent analyses of proteins binding cognate and noncognate sites demonstrate that although this type of binding mechanism is prevalent, our ability to understand how Nature uses noncognate binding is difficult to comprehend and predict. Sarai and coworkers examined numerous protein homodimers bound to cognate and noncognate sites and found subtle structural differences lead to marked differences in specificity: it is difficult to determine how these small changes in structure are related to variations in specificity [41]. In one example, their quantitative analysis of the GCN4 bZIP in the asymmetric complex with AP-1 showed that Arg243 in the monomer bound to the cognate half site contributes more to the specific interaction than the other monomer. The Arg243 side chain makes markedly different contacts at the central base pair: in the specifically bound monomer, Arg243 makes bidentate hydrogen bonds to N7 and O6 of the central guanine, whereas Arg243 in the other monomer donates hydrogen bonds to the DNA phosphodiester backbone [3]. Gal4, PPR1, and HPA1 also bind their respective DNA targets as asymmetric homodimers, and this may be a common mechanism for recognition of cognate and noncognate sites [40]. In order to develop a predictive algorithm for searching weak and noncognate binding sites, Locker and coworkers analyzed homeodomain protein HNF1, which exists as a homo- or heterodimer of HNF1α and HNF1β [42]. The HNF1 consensus half site has 7 bp; strongly bound half sites averaged 6.3/7 bp matches, whereas weaker half sites matched 4.5/7 bp. Notably, some half sites matched only 1/7 bp or 7/14 bp in the full site. Thus, weak full and half sites can vary significantly from the consensus sequence [41].
The high-resolution structures of glucocorticoid receptor (GR) bound to cognate and noncognate sites have been known for many years [43]. The GR homodimer is capable of binding to a noncognate sequence in which half-site spacing is enlarged by one base pair. Thus, one GR monomer is bound specifically to the cognate half site, while the other monomer is bound nonspecifically to the noncognate site, similar to the mechanism the homodimeric GCN4 bZIP adopts for binding to a cognate half site. The bZIP protein Maf binding to numerous DNA sites has been examined by in vitro and in vivo techniques [44]. Yoshida and coworkers show the Maf family of proteins binds well to the palindromic full site, as well as a 5′-AT-rich half site, with estimated dissociation constants around 250–500 nM. Like the GCN4 bZIP, the Maf bZIP can bind as monomer or homodimer to the half site, and strong half-site binding is exhibited when the 5′-flanking region is AT-rich. The authors raise the point that the Maf protein is capable of binding to a larger repertoire of sequences than previously recognized, and therefore, there may exist more Maf target genes [43].
Our results with wt bZIP, a GCN4 derivative, corroborate those discussed above: wt bZIP is capable of specific, and even strong binding, to unexpected, noncognate sites. Nature may use this mechanism abundantly for transcription factors, and this observation can make us consider the role of highly conserved amino acids and sequence homology and similarity. For example, in GCN4 and other bZIP proteins, Asn235, Ala238, Ala239, Ser242, and Arg243 are highly conserved. Given the high-resolution information on bZIP–DNA complexes, we might conclude that these residues are indispensable for base-specific contacts: all of the listed amino acids, with the exception of Ser242, make specific contacts to individual bases [3–5]. However, we also know that Arg243 makes completely different contacts to the AP-1 and CRE sites, so these critical contacts responsible for sequence recognition can vary significantly between a given homodimer and different DNA sequences.
This work demonstrates that the bZIP is a far more versatile and flexible protein motif for recognition of specific DNA sequences than previously recognized. Thus, in addition to the well-studied yeast AP-1 and CRE sites, wt bZIP can also target noncognate, noncanonical gene regulatory sequences from invertebrates, vertebrates, and mammals with high to modest affinity. These regulatory sequences include C/EBP, XRE1, E-box, and HRE, which are naturally targeted by other protein families. Additionally, we are pursuing detailed examination of the binding and affinities of wt bZIP at these full and half sites. The data and results presented here may be useful in design of new α-helical proteins capable of targeting specific DNA sites: binding affinities and sequence specificities can be fine-tuned as desired, whether for use as a scientific tool or a possible therapeutic.
Acknowledgments
J.A.S. dedicates this paper to Prof. Peter B. Dervan on the occasion of his 60th birthday: Prof. Dervan was a wonderful doctoral thesis adviser, and J.A.S. had the privilege and pleasure to be part of his research group. The authors are grateful for funding from the US National Institutes of Health (5RO1GM069041), Canadian Foundation for Innovation/Ontario Innovation Trust (CFI/OIT), Premier’s Research Excellence Award (PREA), and the University of Toronto. We also gratefully acknowledge Prof. Tim Westwood for experimental advice and Prof. Barry Saville, who provided us access to the PhosphorImager System.
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbapap.2006.04.009.
References
- 1.Struhl K. Helix–turn–helix, zinc-finger, and leucine-zipper motifs for eucaryotic transcriptional regulatory proteins. Trends Biochem Sci. 1989;14:137–140. doi: 10.1016/0968-0004(89)90145-X. [DOI] [PubMed] [Google Scholar]
- 2.Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 3.Ellenberger TE, Brandl CJ, Struhl K, Harrison SC. The GCN4 basic region leucine zipper binds DNA as a dimer of uninterrupted a helices: crystal structure of the protein–DNA complex. Cell. 1992;71:1223–1237. doi: 10.1016/s0092-8674(05)80070-4. [DOI] [PubMed] [Google Scholar]
- 4.König P, Richmond TJ. The X-ray structure of the GCN4-bZIP bound to ATF/CREB site DNA shows the complex depends on DNA flexibility. J Mol Biol. 1993;233:139–154. doi: 10.1006/jmbi.1993.1490. [DOI] [PubMed] [Google Scholar]
- 5.Keller W, König P, Richmond TJ. Crystal structure of a bZIP/DNA Complex at 2.2 Å: determinants of DNA specific recognition. J Mol Biol. 1995;254:657–667. doi: 10.1006/jmbi.1995.0645. [DOI] [PubMed] [Google Scholar]
- 6.Glover JNM, Harrison SC. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature. 1995;373:257–261. doi: 10.1038/373257a0. [DOI] [PubMed] [Google Scholar]
- 7.Lajmi AR, Wallace TR, Shin JA. Short, hydrophobic, alanine-based proteins based on the bZIP motif: overcoming inclusion body formation and protein aggregation during overexpression, purification, and renaturation. Protein Expr Purif. 2000;18:394–403. doi: 10.1006/prep.2000.1209. [DOI] [PubMed] [Google Scholar]
- 8.Lajmi AR, Lovrencic ME, Wallace TR, Thomlinson RR, Shin JA. Minimalist, alanine-based, helical protein dimers bind to specific DNA sites. J Am Chem Soc. 2000;122:5638–5639. [Google Scholar]
- 9.Hill DE, Hope IA, Macke JP, Struhl K. Saturation mutagenesis of the yeast his3 regulatory site: requirements for transcriptional induction and for binding by GCN4 activator protein. Science. 1986;234:451–457. doi: 10.1126/science.3532321. [DOI] [PubMed] [Google Scholar]
- 10.Agre P, Johnson PF, McKnight SL. Cognate DNA binding specificity retained after leucine zipper exchange between GCN4 and C/EBP. Science. 1989;246:922–926. doi: 10.1126/science.2530632. [DOI] [PubMed] [Google Scholar]
- 11.Bird GH, Lajmi AR, Shin JA. Sequence-specific recognition of DNA by hydrophobic, alanine-scanning mutants of the bZIP motif investigated by fluorescence anisotropy. Biopolymers. 2002;65:10–20. doi: 10.1002/bip.10205. [DOI] [PubMed] [Google Scholar]
- 12.Bird GH, Shin JA. MALDI-TOF mass spectrometry characterization of hydrophobic basic region/leucine zipper proteins. Biochim Biophys Acta. 2002;1597:252–259. doi: 10.1016/s0167-4838(02)00303-5. [DOI] [PubMed] [Google Scholar]
- 13.Xie Y, Wetlaufer DB. Control of aggregation in protein refolding: the temperature-leap tactic. Protein Sci. 1996;5:517–523. doi: 10.1002/pro.5560050314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bird GH, Lajmi AR, Shin JA. Manipulation of temperature improves solubility of hydrophobic proteins and cocrystallization with matrix for analysis by MALDI-TOF mass spectrometry. Anal Chem. 2002;74:219–225. doi: 10.1021/ac010683g. [DOI] [PubMed] [Google Scholar]
- 15.Metallo SJ, Schepartz A. Distribution of labor among bZIP segments in the control of DNA affinity and specificity. Chem Biol. 1994;1:143–151. doi: 10.1016/1074-5521(94)90004-3. [DOI] [PubMed] [Google Scholar]
- 16.Vinson CR, Sigler PB, McKnight SL. Scissors-grip model for DNA recognition by a family of leucine zipper proteins. Science. 1989;246:911–916. doi: 10.1126/science.2683088. [DOI] [PubMed] [Google Scholar]
- 17.Struhl K. Molecular mechanisms of transcriptional regulation in yeast. Annu Rev Biochem. 1989;58:1051. doi: 10.1146/annurev.bi.58.070189.005155. [DOI] [PubMed] [Google Scholar]
- 18.Nair SK, Burley SK. X-ray structure of Myc-Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 19.O’Hagan RC, Schreiber-Agus N, Chen K, David G, Engelman JA, Schwab R, Alland L, Thomson C, Ronning DR, Sacchettini JC, Meltzer P, DePinho RA. Gene-target recognition among members of the Myc superfamily and implications for oncogenesis. Nat Genet. 2000;24:113–119. doi: 10.1038/72761. [DOI] [PubMed] [Google Scholar]
- 20.Gradin K, McGuire J, Wenger RH, Kvietikova I, Whitelaw ML, Toftgård R, Tora T, Gassmann M, Poellinger L. Functional interference between hypoxia and dioxin signal transduction pathways: competition for recruitment of the arnt transcription factor. Mol Cell Biol. 1996;16:5221–5231. doi: 10.1128/mcb.16.10.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amati B, Land H. Myc-Max-Mad: a transcription factor network controlling cell cycle progression, differentiation and death. Curr Opin Gene Dev. 1994;4:102–108. doi: 10.1016/0959-437x(94)90098-1. [DOI] [PubMed] [Google Scholar]
- 22.Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LWM, Cowley SM, Yost C, Pierce S, Edgar BA, Parkhurst SM, Eisenman RN. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003;17:1101–1114. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reisman D, Elkind NB, Roy B, Beamon J, Rotter V. c-Myc transactivates the p53 promoter through a required downstream CACGTG motif. Cell Growth Differ. 1993;4:57–65. [PubMed] [Google Scholar]
- 24.Hogenesch JB, Chan WK, Jackiw VH, Brown RC, Gu YZ, Pray-Grant M, Perdew GH, Bradfield CA. Characterization of a subset of the basic-helix–loop–helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem. 1997;272:8581–8593. doi: 10.1074/jbc.272.13.8581. [DOI] [PubMed] [Google Scholar]
- 25.Pongratz I, Antonsson C, Whitelaw ML, Poellinger L. Role of the PAS domain in regulation of dimerization and DNA binding specificity of the dioxin receptor. Mol Cell Biol. 1998;18:4079–4088. doi: 10.1128/mcb.18.7.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burbach KM, Pol A, Bradfield CA. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc Natl Acad Sci U S A. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ema M, Sogawa K, Watanabe N, Chujoh Y, Matsushita N, Gotoh O, Funae Y, Fujii-Kuriyama Y. cDNA cloning and structure of mouse putative Ah receptor. Biochem Biophys Res Commun. 1992;184:246–253. doi: 10.1016/0006-291x(92)91185-s. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman EC, Reyes H, Chu F, Sander F, Conley LH, Brooks BA, Hankinson O. Cloning of a factor required for activity of the Ah (Dioxin) receptor. Science. 1991;252:954. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- 29.Reyes H, Reisz-Porszasz S, Hankinson O. Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science. 1992;256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 30.Cuthill S, Wilhelmsson A, Poellinger L. Role of the ligand in intracellular receptor function: receptor affinity determines activation in vitro of the latent dioxin receptor to a DNA-binding form. Mol Cell Biol. 1991;11:401–411. doi: 10.1128/mcb.11.1.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitelaw M, Pongratz I, Wilhelmsson A, Gustafsson JA, Poellinger L. Ligand-dependent recruitment of the arnt coregulator determines DNA recognition of the dioxin receptor. Mol Cell Biol. 1993;13:2504–2514. doi: 10.1128/mcb.13.4.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rowlands JC, Gustafsson J-Å. Aryl hydrocarbon receptor-mediated signal transduction. Crit Rev Toxicol. 1997;27:109–134. doi: 10.3109/10408449709021615. [DOI] [PubMed] [Google Scholar]
- 33.Bacsi SG, Reisz-Porszasz S, Hankinson O. Orientation of the heterodimeric Aryl hydrocarbon (Dioxin) receptor complex on its asymmetric DNA recognition sequence. Mol Pharmacol. 1995;47:432–438. [PubMed] [Google Scholar]
- 34.Schmid T, Zhou J, Brüne B. HIF-1 and p53: communication of transcription factors under hypoxia. J Cell Mol Med. 2004;8:423–431. doi: 10.1111/j.1582-4934.2004.tb00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9:10–17. doi: 10.1634/theoncologist.9-90005-10. [DOI] [PubMed] [Google Scholar]
- 36.Kinoshita K, Kikuchi Y, Sasakura Y, Suzuki M, Fujii-Kuriyama Y, Sogawa K. Altered DNA binding specificity of Arnt by selection of partner bHLH-PAS proteins. Nucleic Acids Res. 2004;32:3169–3179. doi: 10.1093/nar/gkh637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swanson HI, Yang JH. Specificity of DNA binding of the c-Myc/Max and ARNT/ARNT dimers at the CACGTG recognition site. Nucleic Acids Res. 1999;27:3205–3212. doi: 10.1093/nar/27.15.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliphant AR, Brandl CJ, Struhl K. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol Cell Biol. 1989;9:2944–2949. doi: 10.1128/mcb.9.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hollenbeck JJ, Oakley MG. GCN4 binds with high affinity to DNA sequences containing a single consensus half-site. Biochemistry. 2000;39:6380–6389. doi: 10.1021/bi992705n. [DOI] [PubMed] [Google Scholar]
- 40.Hammarström M, Hellgren N, Van den Berg S, Berglund H, Härd T. Rapid screening for improved solubility to small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 2002;11:313–321. doi: 10.1110/ps.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selvaraj S, Kono H, Sarai A. Specificity of protein–DNA recognition revealed by structure-based potentials: symmetric/asymmetric and cognate/noncognate binding. J Mol Biol. 2002;322:907–915. doi: 10.1016/s0022-2836(02)00846-x. [DOI] [PubMed] [Google Scholar]
- 42.Locker J, Ghosh D, Luc PV, Zheng J. Definition and prediction of the full range of transcription factor binding sites-the hepatocyte nuclear factor 1 dimeric site. Nucleic Acids Res. 2002;30:3809–3817. doi: 10.1093/nar/gkf484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luisi BF, Xu WX, Otwinowski Z, Freedman LP, Yamamoto KR, Sigler PB. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. 1991;352:497–505. doi: 10.1038/352497a0. [DOI] [PubMed] [Google Scholar]
- 44.Yoshida T, Ohkumo T, Ishibashi S, Yasuda K. The 5′-AT-rich half-site of Maf recognition element: a functional target for bZIP transcription factor Maf. Nucleic Acids Res. 2005;33:3465–3478. doi: 10.1093/nar/gki653. [DOI] [PMC free article] [PubMed] [Google Scholar]