

Abstract
Background and Purpose
Current guidelines suggest that cardiac arrest (CA) survivors should be ventilated with 100% O2 after resuscitation. Breathing 100% O2 may worsen neurological outcome after experimental CA. This study tested the hypothesis that graded reoxygenation, with oximetry guidance, can safely reduce FiO2 after resuscitation, avoiding hypoxia while promoting neurological recovery.
Methods
Mature dogs underwent 10 minutes of CA and restoration of spontaneous circulation with100% O2. Animals were randomized to 1-hour additional ventilation on 100% FiO2 or to rapid lowering of arterial O2 saturation to <96% but >94% with pulse oximeter guidance. Animals were awakened at hour 23, and the neurological deficit score (0=normal; 100=brain-dead) was measured. Reanesthetized animals were perfusion-fixed and the brains removed for histopathology.
Results
The neurological deficit score was significantly better in oximetry (O) dogs. O dogs appeared aware of their surroundings, whereas most hyperoxic (H) animals were stuporous (neurological deficit score=43.0±5.9 [O] versus 61.0±4.2 [H]; n=8, P<0.05). Stereological analysis revealed fewer injured CA1 neurons in O animals (cresyl violet: 35.5±4.3% [O] versus 60.5±3.3% [H]; P<0.05). There were also fewer fluoro-Jade B-stained degenerating CA1 neurons in O animals (3320±267 [O] versus 6633±356 [H] per 0.1 mm3; P<0.001).
Conclusions
A clinically applicable protocol designed to reduce postresuscitative hyperoxia after CA results in significant neuroprotection. Clinical trials of controlled normoxia after CA/restoration of spontaneous circulation should strongly be considered.
Keywords: canine model, cerebral ischemia, neurological deficit, oxygen utilization
Oxidative stress plays a critical role in ischemia/reperfusion injury to the brain after cardiac arrest/restoration of spontaneous circulation (CA/ROSC). Evidence that oxidative stress is a primary determinant of outcome after cerebral ischemia comes from correlations of markers of oxidative stress with the extent of neuronal death and neuroprotection by antioxidants.1,2 These findings are supported by genetic animal models demonstrating neuroprotection by knocking out those enzymes that promote oxidative stress or by overexpressing antioxidant enzymes that protect against oxidative injury.3 Virtually every cellular and extracellular molecular component is potentially sensitive to damage caused by oxidative stress. Oxidative modification to DNA, RNA, proteins, lipids, and small metabolites occurs during ischemia/reperfusion.4
There currently exists no evidence-based guideline for postresuscitative O2 therapy after CA. Despite the knowledge that oxidative stress can increase neurological injury after resuscitation, clinicians treating CA survivors rarely consider lowering the FiO2. In fact, current advanced cardiac life support guidelines mandate “To improve oxygenation, healthcare providers should give 100% inspired oxygen during basic life support and advanced cardiovascular life support as soon as it becomes available.”5 Mounting experimental evidence suggests, however, that routine administration of high concentrations of O2 may contribute to significant neurological injury after CA/ROSC.
Supranormal tissue O2 levels during reoxygenation after exposure of rat cortical brain slices to severe hypoxia are correlated with the severity of neuronal damage.6 Gerbils treated with 100% O2 after 15 minutes of bilateral carotid occlusion sustained increased white matter damage compared with those exposed to room air.7 On a neurochemical level, hyperoxic reperfusion worsens the postischemic oxidized shift in tissue redox state8 and exacerbates brain lipid as well as protein oxidation.9,10 One key protein that is preferentially inactivated during hyperoxic resuscitation is pyruvate dehydrogenase, resulting in increased tissue lactic acidosis after resuscitation with 100% O2 compared with normoxic resuscitation.9,10 Finally, animals ventilated with 100% O2 for as little as 1 hour after 9 to 10 minutes of CA demonstrate significantly worse clinical10,11 and histopathological9 outcomes when compared with animals similarly resuscitated with room air.
Although we and others have found that animals are adequately oxygenated with FiO2s in the range of 21% to 30% after CA, use of such low ventilatory O2 is impractical and dangerous for humans undergoing initial resuscitation. Many of these patients require high FiO2 for adequate oxygenation because of existing pulmonary or cardiac dysfunction. Some evidence also suggests that pulmonary compliance is reduced during CA, thus limiting oxygenation during resuscitation even at high FiO2.12 Moreover, preclinical studies do not support a neuroprotective effect of hypoxic resuscitation.13,14
Preclinical evidence suggests that to limit oxidative brain injury, postresuscitative ventilation should be tailored to minimize hyperoxia while simultaneously avoiding hypoxemia. Widespread use of pulse oximetry allows the clinician continuous access to information regarding patient oxygenation. This study was designed to test the hypothesis that postresuscitative pulse oximetry-guided O2 delivery would minimize neurological injury after experimental CA.
Materials and Methods
Surgical Preparation
All animal experiments were approved by the University of Maryland institutional animal care and use committee. Single-source adult female beagles (N=17) were studied with our laboratory’s well-established model of CA/ROSC.10,15,16 A peripheral intravenous line was established, and anesthesia was induced with pentobarbital (12.5 mg/kg) and maintained with α-chloralose (75 mg/kg) during surgery and preparation. Animals were intubated and mechanically ventilated with room air. Core body temperature was continuously monitored and maintained at physiological levels (rectal temperature >37°C and <38.5°C). Left femoral arterial and venous cutdowns were placed for drug delivery, central venous pressure and blood pressure monitoring, and arterial blood gas (ABG) sampling. A left lateral thoracotomy was performed on all animals.
Cardiac Arrest/Return of Spontaneous Circulation
Ventricular fibrillation CA was initiated with a 250-Hz, square-wave DC pulse to the right ventricle and allowed to persist untreated for 10 minutes. At the end of this period, epinephrine (0.02 mg/kg) and sodium bicarbonate (1 mEq/kg) were administered intravenously, simultaneously with the resumption of ventilation with 100% O2 and the performance of open-chest cardiopulmonary resuscitation (CPR) for 3 minutes, followed by internal defibrillation at 5 J.
Experimental Groups
Animals were then randomized to receive either hyperoxic (H) resuscitation (100% O2 for 1 hour after resuscitation) followed by ABG-guided adjustments to physiological O2 levels or pulse oximetry-guided, normoxic ventilation (O). O dogs were similarly resuscitated with 100% O2 during CPR in an attempt to reproduce current clinical resuscitation protocols. However, hemoglobin O2 saturation was continuously monitored in these dogs via pulse oximetry measured on the underside of the tongue. Pulse oximetry measurements were always found to be 99% to 100% immediately after ROSC; inspired O2 in this group was then decreased to 50%. Further reductions of 5% of inspired O2 were performed every 2 minutes until O2 desaturation (96%) was noted. This particular value was chosen because clinical studies in humans have demonstrated that pulse oximetry readings of 96% will ensure that the ABG-measured O2 saturation is >90%, thus avoiding both hypoxia and hyperoxia.17 At this point, ABG was measured, and pulse oximetry was no longer used to guide respiratory parameters. With this protocol, inspired O2 was reduced in the O dogs safely to 21% to 30% within 12 minutes of ROSC.
Postresuscitative Care
All animals received intensive care for 24 hours after ROSC according to a standardized protocol. A morphine drip (0.1 mg · kg-1 · h-1, with additional boluses as needed) provided analgesia during intensive care. Pancuronium (0.1 mg/kg) was used intermittently, only as needed, to facilitate ventilation and only after verification that the animal was adequately sedated. Ventilation was adjusted to maintain aPaCO2 of 30 to 35 and a PaO2 of 80 to 100 torr. At hour 20 analgesia was lightened, and animals were weaned from controlled ventilation. At hour 23 morphine was reversed with naloxone (0.4 mg/kg), and the neurological deficit score (NDS) was measured by 2 examiners blinded to treatment protocol. The NDS used has been validated in other laboratories18 as well as ours10,15 and is a multisystem test of 18 parameters in 5 categories (level of consciousness, respiration, cranial nerve, motor, sensory, behavior), yielding a score between 0 (normal) and 100 (brain death). Animals were reanesthetized with pentobarbital and α-chloralose, intubated, and mechanically ventilated.
Tissue Preparation
At hour 24, dogs were transcardially perfused with cold 1% paraformaldehyde in phosphate buffer for 1 minute followed by cold 4% paraformaldehyde for 10 minutes. After the brain was removed from the skull, it was postfixed for 7 days in 4% paraformaldehyde. Coronal blocks, cut so that they contained the entire hippocampus, were postfixed in 2.5% acrolein (EM grade, Polysciences) solution in 4% paraformaldehyde, pH 6.8, for 2 hours. After 24 hours postfixation in the 4% paraformaldehyde, pH 6.8, the blocks were transferred into 30% sucrose. Once the blocks sunk to the bottom of the containers, they were cut on a sliding microtome while frozen on crushed dry ice. The coronal sections were cut 40 μm thick, and 24 series per animal were produced. These sections were kept in cryoprotectant at -20C° until further processing was initiated.19
Histology
Cresyl Violet Stain
The sections were placed in 50%, 75%, and 95% ethanol for 5 minutes each and in 100% ethanol for 6 hours. The sections were then rehydrated in 95%, 75%, and 50% ethanol and water for 5 minutes each and treated with cresyl violet solution (FD NeuroTechnologies) for 2 minutes. Subsequently, the sections were placed in 0.1% acetic acid in 75% ethanol for 2 minutes, rinsed in water, dehydrated, and mounted in DPX.
Fluoro-Jade B Stain
The sections were immersed in 100% ethanol for 3 minutes, followed by 1 minute in 70% ethanol and a 1-minute rinse in water. Slides were then transferred to 0.06% potassium permanganate for 15 minutes. After 2 more rinses, sections were placed in 0.001% fluoro-Jade B (Chemicon International) in 0.1% acetic acid for 30 minutes at room temperature in the dark. After being stained, the sections were rinsed 3 times with distilled water. The slides were dried, immersed in xylene, and mounted with DPX. Sections were examined under a fluorescence microscope with fluorescein isothiocyanate filter sets.
Stereology
Materials included in the quantitative analyses were taken from the dorsal hippocampus. One hemisphere per animal and 1 coronal block containing the entire hippocampus were selected for quantitative analyses. From a random starting point, a 1-in-24 series of 40-μm-thick sections was stained with cresyl violet or fluoro-Jade B. Cresyl violet-stained sections were used for estimating total neuronal numbers of normal and dying (apoptotic or necrotic) neurons in the dorsal part of hippocampal CA1 regions. Quantitative analyses were performed by a blinded investigator (I.B.) with a computer-assisted image analysis system consisting of a Nikon Eclipse E800 microscope equipped with a MicroBrightField 3-axis, computer-controlled motorized stage; an Optronics 1-CCD digital video camera; a PC workstation; and StereoInvestigator program (MicroBrightField), a custom-designed morphology and stereology software package. Tracings were made from the rostral through the caudal extent of the dorsal hippocampus. A total of 6 tracings were made per hippocampus, per hemisphere, for each animal. After outlining the boundaries of the CA1 fields at low magnification (×4) on the computer graphics display in each section separately, the software placed within each subfield boundary a set of optical counting frames (55×55 μm) in a systematic-random fashion. Cresyl violet-stained normal and dying neurons (ie, necrotic and apoptotic cells) were then counted in optical dissectors 7 μm in depth, according to stereological principles.20,21 All analyses were performed with a Nikon Plan Apo ×100/1.40 oil objective. Cresyl violet-stained normal neurons were defined as large cells with identifiable nuclei and discrete nucleoli. Neurons showing a clear accumulation of dense, globular material in the cytoplasm with evidence of nuclear fragmentation were considered apoptotic, whereas necrotic neurons were defined as cells showing shrunken perikarya and darkly stained nuclei of reduced size. In each region, at least 500 neurons were sampled to ensure robustness of the data.22 Fluoro-Jade B-stained sections were used for estimating the total number of dying neurons in a 0.1-mm3 volume of the dorsal CA1 region with the StereoInvestigator program.
Statistical Analysis
All data are expressed as mean±SE. Physiological parameters were compared with the 2-sample t test. NDS scores were compared with a 2-tailed Mann-Whitney U test. Differences in quantitative histopathology were analyzed with the Mann-Whitney rank-sum test. In all cases, P<0.05 was considered significant.
Results
All animals were successfully resuscitated from CA with a single shock. However, 1 animal was excluded before randomization because of profound postresuscitative hypotension. Careful physiological monitoring was performed for the entire postresuscitative period; comparative analysis focused on the period before and shortly after CA. Detailed results are provided in the Table. There were no baseline differences in any hemodynamic or respiratory parameters between groups before CA. Hemodynamic parameters were also compared at 30 and 60 minutes after ROSC. Although no differences were noted between groups at 60 minutes, mean arterial pressure was significantly higher in the O group at 30 minutes. At 60 minutes (end of hyperoxia), PO2 was markedly increased in the H group, as expected, whereas O-group dogs demonstrated PO2 values comfortably within designated physiological parameters. PO2 remained mildly elevated at 120 minutes in H dogs despite the ABG-guided lowering of inspired O2.
Physiological Data for H and O-Guided Normoxic Animals Before and After CA.
| Time Relative to CA, min | O/Normoxia | Hyperoxia | Significance (P Value) | |
|---|---|---|---|---|
| Temperature | -10 | 37.7±0.2 | 38±0.1 | 0.27 |
| MAP | -10 | 111±5 | 108±7 | 0.75 |
| 30 | 88±4 | 75±3 | 0.027 | |
| 60 | 93±5 | 87±3 | 0.30 | |
| CVP | -10 | 1±1 | -1±1 | 0.65 |
| 30 | 2±1 | 3±1 | 0.34 | |
| 60 | 0±1 | 1±1 | 0.73 | |
| pH | -10 | 7.36±0.02 | 7.38±0.02 | 0.56 |
| 60 | 7.35±0.04 | 7.32±0.01 | 0.35 | |
| PCO2 | -10 | 36±2 | 34±2 | 0.53 |
| 60 | 34±2 | 35±2 | 0.58 | |
| PO2 | -10 | 102±5 | 106±3 | 0.58 |
| 15 | 94±4 | 564±36 | 0.00 | |
| 60 | 82±2 | 488±61 | 0.00 | |
| 120 | 83±4 | 148±18 | 0.004 | |
| 180 | 101±9 | 94±3 | 0.47 |
MAP indicates mean arterial pressure; CVP, central venous pressure.
Animals were weaned from controlled ventilation beginning at hour 20 after ROSC. A standardized NDS was then measured by 2 blinded investigators. Graphical representation of individual NDS values is provided in Figure 1. Animals resuscitated with the O protocol demonstrated significantly better neurological outcome than those resuscitated with 100% O2 (43±6 [O group] versus 61±4 [H group]; P<0.02). Six of 8 dogs in group O demonstrated better neurological outcome than any group H animal. In clinical terms, several animals in the O group attempted to stand and drink. In contrast, none of the animals in the H group attempted to right themselves. Several demonstrated purposeless repetitive running motions or opisthotonos.
Figure 1.
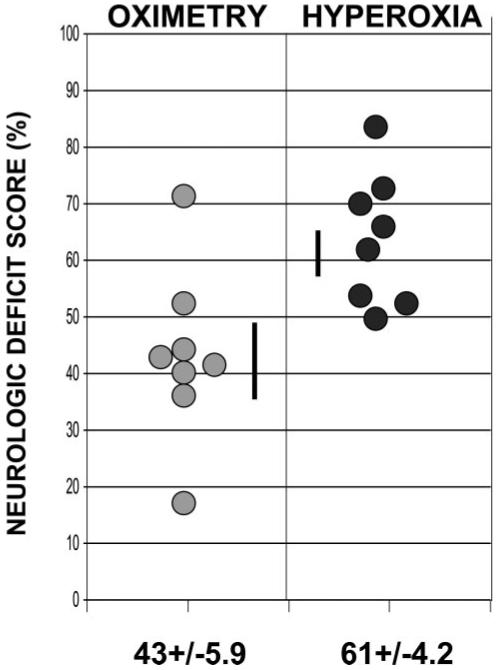
Individual variation of NDSs 24 hours after CA/ROSC in animals ventilated with H or O protocols.
We used unbiased stereological methods to estimate the difference in neuronal damage between treatment groups. Twenty-four hours after CA/ROSC, we assessed neuronal damage in the dorsal hippocampal CA1 region. At high resolution, dying neurons appear shrunken, with condensed nuclei lacking a nucleolus (Figure 2). Cresyl violet-stained neurons in representative nonischemic control animals appeared normal, as expected. StereoInvestigator analysis of the cresyl violet-stained sections revealed 63.7%±3.1 dying neurons in the dorsal CA1 region after H resuscitation (Figure 3). Although there were abnormal neurons (37.8%±3.7) in the dorsal CA1 region after O-based resuscitation, the fraction of cells exhibiting abnormal morphology was much smaller in the O group (P<0.001). Fluoro-Jade B staining confirmed these results (Figure 2). The number of fluoro-Jade B-positive (dying) neurons in a 0.1-mm3 volume of the dorsal CA1 region of the H dogs was significantly higher than for similar O-reoxygenated animals (6633±356 versus 3320±267; P≤0.001; Figure 4).
Figure 2.
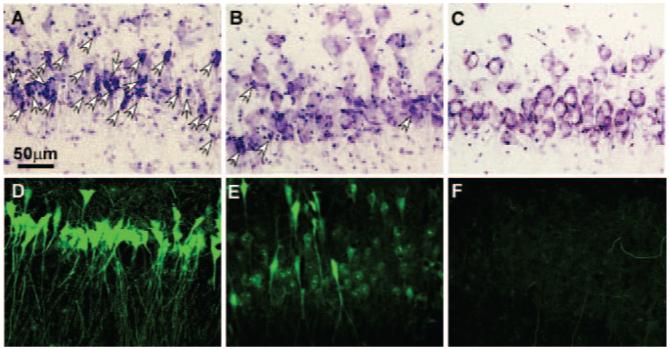
Cresyl violet (upper panel; white arrows denote necrotic neurons) or fluoro-Jade B (lower panel) staining demonstrates dying or degenerating neurons in the CA1 region of the dorsal hippocampus of sham (C, F), H (A, D), and O (B, E) ventilated dogs 24 hours after reperfusion.
Figure 3.
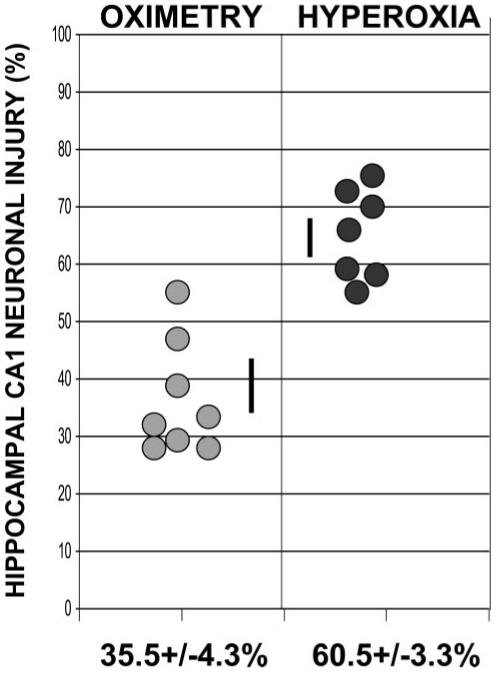
Individual variation of neuronal injury in the dorsal CA1 region of the cresyl violet-stained hippocampus 24 hours after CA/ROSC.
Figure 4.
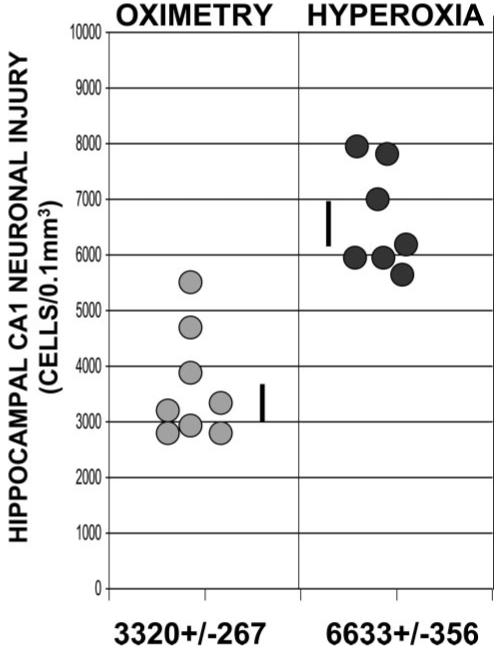
Individual variation of neuronal injury in the dorsal CA1 region of the fluoro-Jade B-stained hippocampus 24 hours after CA/ROSC.
Discussion
The results of this study validate and expand the results previously reported in our clinically relevant model of CA/ROSC. We have demonstrated significant clinical10 as well as histological9 neuroprotection afforded to animals resuscitated with room air after 10 minutes of CA compared with similar animals resuscitated with 100% O2 for as little as 1 hour. In addition, normoxic animals showed significantly less protein oxidation in the selectively vulnerable hippocampal CA1 region. In particular, animals resuscitated with room air showed significantly greater enzyme activity and immunoreactivity for pyruvate dehydrogenase,9,23 a key enzyme of metabolism that may contribute to cerebral metabolic derangements for 24 hours or longer after CA/ROSC.24,25
The goal of any preclinical translational trial, however, must be to provide the background information necessary to develop safe, human clinical trials with a reasonable expectation of positive outcomes. Although our earlier studies have been extremely promising and although our large-animal model closely mimics the clinical scenario of human CA/ROSC, the protocol used in those studies would not easily translate to human trials. The animals used were healthy at baseline; as such, we were able to successfully resuscitate all animals without supplemental O2 and then provide minimal O2 support in the postresuscitative phase. In contrast, most humans undergoing CA have underlying cardiopulmonary disease, which would make initial resuscitation difficult, if not impossible, in the absence of supplemental O2.
We thus attempted to design a clinical paradigm whereby supplemental O2 would be provided to all CA victims during CPR in an attempt to maximize initial resuscitation, followed by rapid pulse oximetry-guided lowering of inspired O2 to physiological levels, thus simultaneously avoiding the potentially damaging extremes of both hyperoxia and hypoxia. Although this protocol has proven experimentally feasible, we were concerned at the outset that even a short exposure to supplemental O2 would worsen neurological outcomes because of the well-described respiratory burst of O2 free radicals that occurs almost immediately on reperfusion after complete global ischemia.26
We chose to use pulse oximetry to guide O2 titration rather than ABGs because of its ease of use as well widespread availability in both prehospital and hospital settings. With pulse oximetry guidance, we were able to safely and rapidly lower inspired O2 after resuscitation, reaching physiological levels of arterial O2 within 12 minutes. Animals receiving O2 titration in this manner demonstrated improved clinical as well as histological outcomes. The utility of pulse oximetry in the critical patient is sometimes questioned, with early studies showing a disparity between pulse oximeter-measured O2 saturations and blood gas values.27,28 New pulse oximeter technology,29 however, as well as alternative probe sampling sites,30 have been shown to greatly increase the utility of pulse oximetry in the critically ill patient, making translation of these study results to human trials clinically feasible.
Several limitations of this study should be noted. We used only healthy adult female animals to study CA, a condition that occurs in humans of both sexes with advanced cardiopulmonary disease. The implications of studying cerebral ischemia in female animals only must be considered, especially in light of mounting evidence for the role of sex-defining steroids in the modulation of cerebral ischemic injury.31 Although a similar model of CA/ROSC found no sex-related differences in clinical neurological outcome at 24 hours,32 trials are planned to extend these experiments to male dogs in an attempt to demonstrate neuroprotection independent of sex. We also examined neurological deficit and histological damage at only 1 time point, 24 hours. Although significant neuroprotection was demonstrated at 24 hours, the question must be raised whether oximetry-guided normoxia provides permanent neuroprotection or whether this treatment protocol merely delays the onset of further neurological injury. In an attempt to answer this question, studies are ongoing in our laboratory in an established rat model of complete global ischemia to examine the long-term implications of normoxic versus hyperoxic resuscitation.
Conclusions
Pulse oximetry-guided O2 administration after CA/ROSC safely and rapidly lowers arterial O2 concentration to physiological levels, providing significant short-term neurological protection. Clinical trials of pulse oximetry-guided reoxygenation after CA are indicated.
Acknowledgment
The authors thank Kyni Jones for expert technical assistance.
Source of Funding
This work was supported by National Institutes of Health grants NS34152 and NS49425.
Footnotes
Disclosures
None.
References
- 1.Nakai A, Kuroda S, Kristian T, Siesjo BK. The immunosuppressant drug FK506 ameliorates secondary mitochondrial dysfunction following transient focal cerebral ischemia in the rat. Neurobiol Dis. 1997;4:288–300. doi: 10.1006/nbdi.1997.0146. [DOI] [PubMed] [Google Scholar]
- 2.Rosenthal RE, Chanderbhan R, Marshall G, Fiskum G. Prevention of post-ischemic brain lipid conjugated diene production and neurological injury by hydroxyethyl starch-conjugated deferoxamine. Free Radic Biol Med. 1993;14:96–97. doi: 10.1016/0891-5849(92)90055-l. [DOI] [PubMed] [Google Scholar]
- 3.Klein MB, Chan PH, Chang J. Protective effects of superoxide dismutase against ischemia-reperfusion injury: development and application of a transgenic animal model. Plast Reconstr Surg. 2003;111:251–255. doi: 10.1097/01.PRS.0000034938.58120.12. [DOI] [PubMed] [Google Scholar]
- 4.Lewen A, Matz P, Chan PH. Free radical pathways in CNS injury. J Neurotrauma. 2000;17:871–890. doi: 10.1089/neu.2000.17.871. [DOI] [PubMed] [Google Scholar]
- 5.Advanced Cardiac Life Support guidelines: adjuncts for airway control and ventilation. Circulation. 2005;112(suppl IV):IV-51–IV-57. [Google Scholar]
- 6.Halsey JH, Jr, Conger KA, Garcia JH, Sarvary E. The contribution of reoxygenation to ischemic brain damage. J Cereb Blood Flow Metab. 1991;11:994–1000. doi: 10.1038/jcbfm.1991.166. [DOI] [PubMed] [Google Scholar]
- 7.Mickel HS, Vaishnav YN, Kempski O, von Lubitz D, Weiss JF, Feuerstein G. Breathing 100% oxygen after global brain ischemia in Mongolian gerbils results in increased lipid peroxidation and increased mortality. Stroke. 1987;18:426–430. doi: 10.1161/01.str.18.2.426. [DOI] [PubMed] [Google Scholar]
- 8.Feng ZC, Sick TJ, Rosenthal M. Oxygen sensitivity of mitochondrial redox status and evoked potential recovery early during reperfusion in post-ischemic rat brain. Resuscitation. 1998;37:33–41. doi: 10.1016/s0300-9572(98)00031-8. [DOI] [PubMed] [Google Scholar]
- 9.Vereczki V, Martin E, Rosenthal RE, Hof PR, Hoffman GE, Fiskum G. Normoxic resuscitation after cardiac arrest protects against hippocampal oxidative stress, metabolic dysfunction, and neuronal death. J Cereb Blood Flow Metab. 2006;26:821–835. doi: 10.1038/sj.jcbfm.9600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Rosenthal RE, Haywod Y, Miljkovic-Lolic M, Vanderhoek JY, Fiskum G. Normoxic ventilation after cardiac arrest reduces oxidation of brain lipids and improves neurological function. Stroke. 1998;29:1679–1686. doi: 10.1161/01.str.29.8.1679. [DOI] [PubMed] [Google Scholar]
- 11.Zwemer CF, Whitesall SE, D’Alecy LG. Cardiopulmonary-cerebral resuscitation with 100% oxygen exacerbates neurological dysfunction following nine minutes of normothermic cardiac arrest in dogs. Resuscitation. 1994;25:159–170. doi: 10.1016/0300-9572(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 12.Ornato JP, Bryson BL, Donovan PJ, Farquarson RR, Jaeger C. Measurement of ventilation during cardiopulmonary resuscitation. Crit Care Med. 1983;11:79–82. doi: 10.1097/00003246-198302000-00004. [DOI] [PubMed] [Google Scholar]
- 13.Agardh CD, Zhang H, Smith ML, Siesjo BK. Free radical production and ischemic brain damage: influence of postischemic oxygen tension. Int J Dev Neurosci. 1991;9:127–138. doi: 10.1016/0736-5748(91)90003-5. [DOI] [PubMed] [Google Scholar]
- 14.Zwemer CF, Whitesall SE, D’Alecy LG. Hypoxic cardiopulmonary-cerebral resuscitation fails to improve neurological outcome following cardiac arrest in dogs. Resuscitation. 1995;29:225–236. doi: 10.1016/0300-9572(94)00848-a. [DOI] [PubMed] [Google Scholar]
- 15.Rosenthal RE, Williams R, Bogaert YE, Getson PR, Fiskum G. Prevention of postischemic canine neurological injury through potentiation of brain energy metabolism by acetyl-l-carnitine. Stroke. 1992;23:1312–1317. doi: 10.1161/01.str.23.9.1312. [DOI] [PubMed] [Google Scholar]
- 16.Rosenthal RE, Silbergleit R, Hof PR, Haywood Y, Fiskum G. Hyperbaric oxygen reduces neuronal death and improves neurological outcome after canine cardiac arrest. Stroke. 2003;34:1311–1316. doi: 10.1161/01.STR.0000066868.95807.91. [DOI] [PubMed] [Google Scholar]
- 17.Seguin P, Le Rouzo A, Tanguy M, Guillou YM, Feuillu A, Malledant Y. Evidence for the need of bedside accuracy of pulse oximetry in an intensive care unit. Crit Care Med. 2000;28:703–706. doi: 10.1097/00003246-200003000-00017. [DOI] [PubMed] [Google Scholar]
- 18.Bircher N, Safar P. Cerebral preservation during cardiopulmonary resuscitation. Crit Care Med. 1985;13:185–190. doi: 10.1097/00003246-198503000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Watson RE, Jr, Wiegand SJ, Clough RX, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–159. doi: 10.1016/0196-9781(86)90076-8. [DOI] [PubMed] [Google Scholar]
- 20.West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rev. 1991;23:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 21.Nimchinsky EA, Young WG, Yeung G, Shah RA, Gordon JW, Boom FE, Morrison JH, Hof PR. Differential vulnerability of oculomotor, facial and hypoglossal nuclei in G86R superoxide dismutates transgenic mice. J Comp Neurol. 2000;416:112–125. doi: 10.1002/(sici)1096-9861(20000103)416:1<112::aid-cne9>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 22.Schmitz C, Hof PR. Recommendations for straightforward and rigorous methods of counting neurons based on a computer simulation approach. J Chem Neuroanat. 2000;20:93–114. doi: 10.1016/s0891-0618(00)00066-1. [DOI] [PubMed] [Google Scholar]
- 23.Richards EM, Rosenthal RE, Kristian T, Fiskum G. Postischemic hyperoxia reduces hippocampal pyruvate dehydrogenase activity. Free Radic Biol Med. 2006;40:1960–1970. doi: 10.1016/j.freeradbiomed.2006.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogaert YE, Rosenthal RE, Fiskum G. Postischemic inhibition of cerebral cortex pyruvate dehydrogenase. Free Radic Biol Med. 1994;16:811–820. doi: 10.1016/0891-5849(94)90197-x. [DOI] [PubMed] [Google Scholar]
- 25.Bogaert YE, Sheu KF, Hof PR, Brown AM, Blass JP, Rosenthal RE, Fiskum G. Neuronal subclass-selective loss of pyruvate dehydrogenase immunoreactivity following canine cardiac arrest and resuscitation. Exp Neurol. 2000;161:115–126. doi: 10.1006/exnr.1999.7250. [DOI] [PubMed] [Google Scholar]
- 26.Dirnagl U, Lindauer U, Them A, Schreiber S, Pfister HW, Koedel U, Reszka R, Freyer D, Villminger A. Global cerebral ischemia in the rat: online monitoring of oxygen free radical production using chemiluminescence in vivo. J Cereb Blood Flow Metab. 1995;15:929–940. doi: 10.1038/jcbfm.1995.118. [DOI] [PubMed] [Google Scholar]
- 27.Smatlak P, Knebel AR. Clinical evaluation of noninvasive monitoring of oxygen saturation in critically ill patients. Am J Crit Care. 1998;7:370–373. [PubMed] [Google Scholar]
- 28.Secker C, Spiers P. Accuracy of pulse oximetry in patients with low systemic vascular resistance. Anaesthesia. 1997;52:127–130. doi: 10.1111/j.1365-2044.1997.32-az0062.x. [DOI] [PubMed] [Google Scholar]
- 29.Durbin CG, JR, Rostow SK. Advantages of new technology pulse oximetry in adults in extremis. Anesth Analg. 2002;94:S81–S83. [PubMed] [Google Scholar]
- 30.Branson RD, Mannheimer PD. Forehead oximetry in critically ill patients: the case for a new monitoring site. Respir Care Clin North Am. 2004;10:359–367. vi–vii. doi: 10.1016/j.rcc.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 31.Hurn PD, Vannucci SJ, Hagberg H. Adult or perinatal brain injury: does sex matter? Stroke. 2005;36:193–195. doi: 10.1161/01.STR.0000153064.41332.f6. [DOI] [PubMed] [Google Scholar]
- 32.Zwemer CF, O’Connor EM, Whitesall SE, D’Alecy LG. Gender differences in 24-hour outcome after resuscitation after 9 minutes of cardiac arrest in dogs. Crit Care Med. 1997;25:330–338. doi: 10.1097/00003246-199702000-00023. [DOI] [PubMed] [Google Scholar]