

Abstract
Mitochondria contribute to cytosolic Ca2+ homeostasis through several uptake and release pathways. Here we report that 1,2-sn-diacylglycerols (DAGs) induce Ca2+ release from Ca2+-loaded mammalian mitochondria. Release is not mediated by the uniporter or the Na+/Ca2+ exchanger, nor is it attributed to putative catabolites. DAGs-induced Ca2+ efflux is biphasic. Initial release is rapid and transient, insensitive to permeability transition inhibitors, and not accompanied by mitochondrial swelling. Following initial rapid release of Ca2+ and relatively slowreuptake, a secondary progressive release of Ca2+ occurs, associated with swelling, and mitigated by permeability transition inhibitors. The initial peak of DAGs-induced Ca2+ efflux is abolished by La3+ (1mM) and potentiated by protein kinase C inhibitors. Phorbol esters, 1,3-diacylglycerols and 1-monoacylglycerols do not induce mitochondrial Ca2+ efflux. Ca2+-loaded mitoplasts devoid of outer mitochondrial membrane also exhibit DAGsinduced Ca2+ release, indicating that this mechanism resides at the inner mitochondrial membrane. Patch clamping brainmitoplasts reveal DAGs-induced slightly cation-selective channel activity that is insensitive to bongkrekic acid and abolished by La3+. The presence of a second messenger-sensitive Ca2+ release mechanism in mitochondria could have an important impact on intracellular Ca2+ homeostasis.
Keywords: Mitochondria, calcium, diacylglycerol, mitoplast, cation channel, permeability transition pore, protein kinase C, transient receptor potential, OAG
INTRODUCTION
A plethora of cellular signaling cascades converge to the production of diacylglycerols (DAGs), leading to the activation of various target proteins regulating cellular processes of extraordinary diversity (Brose et al., 2004). Depending on the identity of the stimulated receptor, different PLC isoforms are activated resulting in the immediate formation of DAG from inositol phospholipids, most rapidly from P1-4,5-bisphosphate (PIP2). This DAG molecule disappears quickly; however, a second wave of DAG emerges with a relatively slow onset persisting for minutes or hours. This latent increase in DAG is partially attributed to the subsequent action of phospholipase D (PLD) on phosphatidylcholine (PC) to yield phosphatidate plus choline followed by phosphatidate phosphatase generating DAG and orthophosphate (Nishizuka, 1992,1995). An additional pathway contributing to this latency is substantiated by the slow degradation of DAG by DAG lipase; this DAG is produced from PC hydrolysis by phosphatidylcholine-specific phospholipase C and is a poor substrate for the fast-acting enzyme DAG kinase (Ford and Gross, 1990; Lee et al., 1991). DAG kinase rapidly catabolizes DAG produced from phosphatidylinositol hydrolysis converting it to phosphatidate at the expense of ATP (Florin-Christensen et al., 1992).
Downstream effects of DAGs are commonly attributed to activation of PKCs. There are, however, additional important targets of DAGs including protein kinase D (PKD), DAG kinases α, β, and γ, RasGRPs, chimaerins, Munc13s, and channels of the transient receptor potential (TRP) family, namely TRPC3, 6, and 7 (Brose et al., 2004; Brose and Rosenmund, 2002; Hofmann et al., 1999; Kazanietz, 2002; Okada et al., 1999; Yang and Kazanietz, 2003).
Upon PIP2 hydrolysis, DAGs are formed in a membrane-delimited manner both on the plasma membrane and on intracellular membranes (Irvine, 2002; Panagia et al., 1991; Rebecchi and Pentyala, 2000), serving as hydrophobic anchors to recruit their targets to the membrane (Brose et al., 2004). The other product, IP3, diffuses in the cytosol to activate IP3 receptors on the ER releasing Ca2+ to the cytoplasm followed by triggering of Ca2+ influx from the extracellular space (Mikoshiba and Hattori, 2000). Within the context of intracellular Ca2+ homeostasis, mechanisms and effectors downstream of the IP3 signaling pathway received the vast majority of attention, culminating in a spark of interest on the socalled “store-operated Ca2+ entry” (SOCE) (Nilius, 2004; Penner and Fleig, 2004; Venkatachalam et al., 2002). SOCE is a process whereby the depletion of intracellular Ca2+ stores (likely ER or SR) activates plasma membrane Ca2+ permeable channels (Putney, 1986). Members of the TRP family are candidates for SOCE; however, unequivocal evidence demonstrating that TRP channels account for this phenomenon is yet to be reported (Clapham, 2003; Nilius, 2004). Conversely, it has been shown that at least three members of the TRP family are activated by DAGs but not through PKC (Clapham et al., 2003). These channels (TRPC3, 6, and 7) do not possess the DAG-binding C1 domain (Gudermann et al., 2004), while TRPC3 plus two additional TRP channels, TRPC4 and TRPC5, are inhibited upon activation of PKC (Venkatachalam et al., 2003).
Enzymes and macromolecules participating in the formation of DAGs have been reported to reside or translocate to mitochondria. PIP2 is found in mitochondria (Watt et al., 2002) and a brain-specific isomer may exist (Bothmer et al., 1992); PLCδ is found in mitochondria from liver (Knox et al., 2004), yeast (Vaena de et al., 2004), and kidney (Nishihira and Ishibashi, 1986). PLD and phosphatidate phosphatase exist in intestinal and myocardial mitochondria (Freeman and Mangiapane, 1989; Liscovitch et al., 1999). The same is true for DAG targets: mitochondrial translocation of PKCδ is a critical proapoptotic event in cardiac responses following ischemia and reperfusion (Murriel et al., 2004), as well as in phorbol ester-induced apoptosis in human myeloid leukemia cells (Majumder et al., 2000) and keratinocytes (Li et al., 1999). An alternative isoform PKCε, confers beneficial effects upon translocation to mitochondria by inhibiting the permeability transition (Baines et al., 2003), while others, such as PKCα, phosphorylate mitochondrial Bcl-2 suppressing apoptosis (Ruvolo et al., 1998) or promote cardioprotection at least partially due to modulation of mitochondrial K+ATP channels (Korge et al., 2002; Sato et al., 1998). With the exception of mitochondrial PKD (Storz et al., 2000), the presence of other non-PKC targets of DAG has not been reported.
Based on the involvement of DAGs on multiple mitochondrial functions, we tested the hypothesis that this second messenger affects mitochondrial Ca2+ handling. We show that DAGs release Ca2+ from Ca2+-loaded mitochondria in conjunction with activation of novel channel(s) present on the inner mitochondrial membrane.
MATERIALS AND METHODS
Isolation of Mitochondria
All animal procedures were carried out according to the National Institutes of Health and the University of Maryland, Baltimore animal care and use committee guidelines. Adult male Sprague-Dawley rats and C57BL/6 mice were used. Nonsynaptic adult rat brain mitochondria were isolated on a Percoll gradient exactly as described previously (Chinopoulos et al., 2003). Mouse heart, kidney, and liver mitochondria were prepared as described previously (Starkov and Fiskum, 2001). Mitochondria from sweet potato (Ipomoea batatas) were isolated as detailed previously (Chen and Lehninger, 1973).
Mitochondrial Ca2+ Uptake in Mammalian Mitochondria
Rat Brain
Mitochondrial or mitoplast-dependent (0.275 mg/mL) removal of medium Ca2+ was followed using the impermeant pentapotassium salt of the ratiometric dye Fura 6F (Molecular Probes, Portland, OR, USA) as previously described in a KCl-based medium containing malate plus glutamate as respiratory substrates (Chinopoulos et al., 2003). All experiments were performed at 37°C.
Rat Liver
The experiments were conducted as for rat brain mitochondria, with the exception that these mitochondria (0.5 mg/ml) were suspended in 250 mM sucrose, 20 mM Hepes, 2 mM KH2PO4, 1mM MgCl2, 5 mM succinate, 1 μM rotenone, 1 mg/ml bovine serum albumin (BSA, fatty acid-free), pH 7.2.
Mouse Heart and Kidney
These experiments were performed similar to the ones for rat brain, but at room temperature (∼23°C), the fluorescent dye used was the hexapotassium salt of Calcium Green-5N (200 nM, Kd = 4.3 μM) (Rajdev and Reynolds, 1993), and the mitochondria (0.25 mg/mL) were suspended in 225 mM mannitol, 75 mM sucrose, 2mM KH2PO4 5 mM HEPES, 0.2 mg/mL BSA (fatty acid-free) 5 mM glutamate, 5 mM malate, pH 7.4.
Mitochondrial Ca2+ Uptake in Sweet Potato Mitochondria
1 mg/mL of mitochondria was used; the experimental conditions were similar to those for mammalian mitochondria but performed at room temperature (∼23°C) in 300 mM mannitol, 10 mM Hepes, 2 mM KH 2PO4,10mM succinate, 0.5 mM ADP, 2 mM ATP, plus 1 mg/mL BSA (fatty acid-free), pH 7.4.
Preparation of Mitoplasts From Rat Brain Mitochondria for Ca2+ Uptake Studies
Mitoplasts were prepared as described previously (Schnaitman et al., 1967), with minor modifications: Upon isolation of mitochondria, protein determination was performed (Biuret); afterwards, 450-500 μL of 20mg/mL mitochondria were suspended in 2.5 mL of buffer “A” containing 10 mM TRIS and 2 mM MgCl2, pH = 7.8 for 20 min in ice while shaking. Subsequently, 3 mL=of 1.8 M Sucrose plus 2 mM MgCl2 was added and left in ice while shaking for an additional 20 min. Next, 12 mL of buffer A plus 45 μg of digitonin (>99% pure) was added to the suspension and left in ice while shaking for 15 min. At the end of this step, the suspension was diluted 5-fold with buffer “B” containing: 225 mM mannitol, 75 mM sucrose, 5 mM Hepes, 1 mM EGTA, 0.5 mg/mL BSA (fatty acid-free), pH = 7.4, and were centrifuged at 12, 000 × g for 10 min. This centrifugation step was repeated once, and the resulting pellet was suspended in buffer B without EGTA. To evaluate the efficiency of the removal of the outer mitochondrial membranes, mitoplasts were tested for the presence of adenylate kinase, as described previously (Schmidt et al., 1984); >95% of adenylate kinase activity was lost with this method. These mitoplasts retained a respiratory control ratio of 5 ± 1 over a period of 45 min (in the presence of externally added cyt c).
Preparation of Mitoplasts From Rat Brain Mitochondria for Patch Clamp Studies
Upon isolation of mitochondria, the organelles were suspended in a hypertonic buffer consisting of 460 mM Mannitol, 140 mM Sucrose, 10 mM HEPES, pH 7.4 for 10 min. Subsequently, they were subjected to 1500 psi with a French press (American Instruments Company, Silver Springs, MD, USA). The suspension was diluted with 2 volumes of 230 mM Mannitol, 70 mM Sucrose, 5 mM HEPES, pH 7.4. Mitoplasts were pelleted at 12,000× g for 10 min and resuspended in 100 μL of the same buffer.
Oxygen Consumption
Mitochondrial respiration was recorded at 37°C with a Clark-type oxygen electrode (Hansatech, UK) as described previously (Chinopoulos et al., 2003).
Mitochondrial Swelling
Swelling of isolated liver mitochondria was assessed by measuring light scatter at 540 nm (37°C) in a Beckman Coulter DU 7500 spectrophotometer (Fullerton, CA, USA) as detailed previously (Bernardi et al., 1992).
Patch Clamp of Rat Brain Mitoplasts
Membrane patches were excised from rat brain mitoplasts after formation of a giga-seal (2-5 G Ω) using glass electrodes made of borosilicate and resistances of 10-30 MΩ. The solution in the electrodes and bath was 150 mM KCl, 5 mM HEPES, pH 7.4. This buffer contained 10 μM free Ca2+. Experiments were carried out at room temperature (∼23°C). Voltage clamp was established with the inside-out excised configuration and currents were amplified using an Axopatch 200 amplifier (Union city, CA, USA). Current traces were sampled at 5 kHz with 2 kHz filtration and analyzed with WinEDR version v2.4.3 program (Strathclyde Electrophysiological Software courtesy of J. Dempster, Univ. of Strathelyde, UK). Ion selectivity was measured by a 1:5 gradient by perfusion of the bath with 30 mM KCl, 184 mM mannitol, 56 mM sucrose, 5 mM HEPES, pH 7.4.
Reagents
Standard laboratory chemicals were from Sigma. 2-APB (Sigma), CGP 37157 (Calbiochem), cyclosporin A (Sigma), bongkrekic acid (Calbiochem), alamethicin (Sigma), BSA (Sigma), Digitonin (Spectrum, New Brunswick, NJ, USA), PMA (Sigma), RHC 80267 (Calbiochem), DAG kinase inhibitor II (Sigma), all PKC inhibitors (Calbiochem, PKC inhibitor set), all 1,2-DGs and MGs were from Sigma or Biomol, all 1,3-DGs were from ICN Biomedicals (Aurora, OH, USA).
RESULTS
Release of Sequestered Ca2+ in Mammalian but Not Sweet Potato Mitochondria by OAG
Ca2+-loaded mitochondria from rat brain (Fig. 1(A)), mouse kidney (Fig. 1(B)), mouse heart (Fig. 1(C)), and rat liver (Fig. 1(E)) release Ca2+ upon exposure to the putative, cell-permeable diacylglycerol analogue, OAG. After the initial rapid release, Ca2+ was re-accumulated, followed by a secondary slow net release. In contrast to OAG, phorbol 12-myristate 13-acetate (PMA) failed to induce Ca2+ efflux (Fig. 1(A), trace b). Sweet potato mitochondria loaded with Ca2+ did not exhibit OAG-induced Ca2+ release (Fig. 1(D), trace b). Liver mitochondria pretreated with Cys A (Fig. 1(E), trace b) display an almost identical OAG-induced Ca2+ release and a moderate extent in the lag time until the spontaneous secondary efflux. Using identical conditions, swelling was not observed upon addition of OAG to Ca2+-loaded liver mitochondria (Fig. 1(G)); however, they exhibit spontaneous large amplitude swelling coincident with the secondary Ca2+ efflux in the absence (trace a) or presence (trace b) of Cys A. Cys A moderately extends the threshold of PTP induction. In the absence of OAG, Cys A elevated the threshold for PTP induction by CaCl2 in rat liver mitochondria from 160 nmol/mg protein to 1280 nmol/mg protein. PTP inhibitors didn’t affect the first or the second pulse of OAG-induced Ca2+ efflux (Fig. 1(F)); however, Cys A partially inhibited the secondary Ca2+ rise, while BKA and 2-APB conferred substantial protection (Fig. 1(F) and (H)).
Fig. 1.
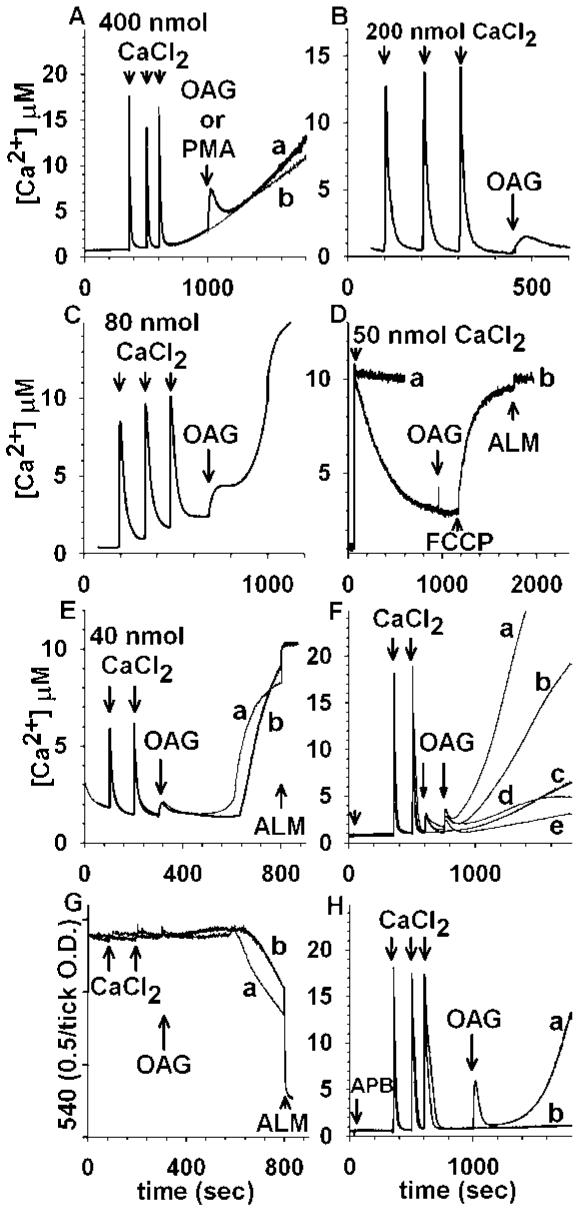
OAG induces mitochondrial Ca2+ efflux and its relation to the permeability transition. Release of sequestered Ca2+ from rat brain (A, trace a), mouse kidney (B), mouse heart (C), and rat liver (E), but not sweet potato (D) mitochondria by the diacylglycerol analogue, OAG (100 μM). The phorbol ester PMA (1 μM) fails to induce Ca2+ release (A, trace b). In (D) trace a, Ru360 (165 nM) was present before addition of CaCl2; in trace b,1 μM FCCP was added at 1200 s. (E): Rat liver mitochondria are loaded with two pulses of CaCl2 (100 and 200 s, 40 nmol each) in the absence (trace a) or presence of Cys A (1 μM, trace b), followed by addition of 100 μM OAG at 300 s. At approximately 600 s, mitochondria spontaneously release Ca2+. Alamethicin (40 μg/ml) was added at 800 s. (F): PTP inhibitors do not affect the initial OAG-induced Ca2+ efflux, but modulate the secondary rise: Rat brain mitochondria were loaded with two pulses of CaCl2 (350 and 500 s, 400 nmol each) followed by addition of OAG (50 μM) in the presence and absence of Cys A (2 μM) or BKA (20 μM) or 2-APB (100 μM) (each added at 50 s, small arrow). In trace a, mitochondria were not pretreated with either PTP inhibitor; trace b, Cys A is present; trace c, mitochondria were not challenged by OAG (replaced with vehicle); trace d, mitochondria were pretreated with 2-APB; trace e, mitochondria were pretreated with BKA. (G): OAG does not evoke an immediate large-amplitude swelling in Ca2+ -loaded rat liver mitochondria: Mitochondria were loaded with Ca2+ exactly as in (A) in the absence (trace a) or presence of Cys A (1 μM, trace b), followed by addition of 100 μM OAG at 300 s, and light scatter was followed spectrophotometrically. At approximately 600 s, mitochondria spontaneously swell. Alamethicin (40 μg/mL) was added at 800 s. (H): 2-APB does not inhibit the initial OAG-induced Ca2+ release. Rat brain mitochondria were loaded with three pulses of CaCl2 (350, 500, and 600 s, 400 nmol each) followed by addition of OAG at 1000 s in the presence of 2-APB (100 μM, added at 50 s). Trace a,OAG (100 μM) was added at 1000 s; in trace b, no OAG was added (replaced by vehicle). Traces for this and all subsequent figures are representative of at least four independent experiments unless otherwise indicated.
OAG-Induced Ca2+ Release Is Not Mediated by the Uniporter or the Mitochondrial Na+/Ca2+ Exchanger
Inhibition of the uniporter by Ru360 fails to inhibit OAG-induced mitochondrial Ca2+ release and actually potentiates it (Fig. 2(A), trace d). Inhibition of the Na+/Ca2+ exchanger by CGP-37157 had no effect on modulating the OAG-induced Ca2+ release mechanism (Fig. 2(A), trace e). Addition of FCCP (Fig. 2(A), trace c), or antimycin A3 plus oligomycin (trace b) in Ca2+-loaded mitochondria pretreated with Ru360 caused only a minor increase in the rate of Ca2+ efflux. La3+ abolishes OAG-induced Ca2+ release completely, but only moderately affects the reversal of the uniporter (Fig. 2(B)). Fura 6F reacts to La3+ similar to Ca2+, however the Kd of the dye for the trivalent is different (not quantified). Its presence also alters the Kd for Ca2+; therefore only qualitative measures of Ca2+ flux are possible after the addition of LaCl3. La3+ is sequestered very slowly by mitochondria, competing with Ca2+ (Reed and Bygrave, 1974) and blocking the uniporter (Gunter and Pfeiffer, 1990). In trace a, addition of antimycin A3 plus oligomycin induced slow Ca2+ efflux followed by faster release upon subsequent addition of FCCP. In trace b where no mitochondrial inhibitors were added, OAG-induced Ca2+ release was inhibited by LaCl3. To address the directionality of the OAG-induced Ca2+ flux, mitochondria were not preloaded with CaCl2, and Ru360 was added to eliminate Ca2+ uptake through the uniporter (Fig. 2(C), trace a). After subsequent addition of CaCl2, there was no mitochondrial Ca2+ uptake and subsequent addition of OAG (trace a) had no effect on Fura 6F fluorescence. In contrast, Ca2+-loaded mitochondria subsequently exposed to Ru360 still exhibited OAG-induced Ca2+ efflux (trace b).
Fig. 2.
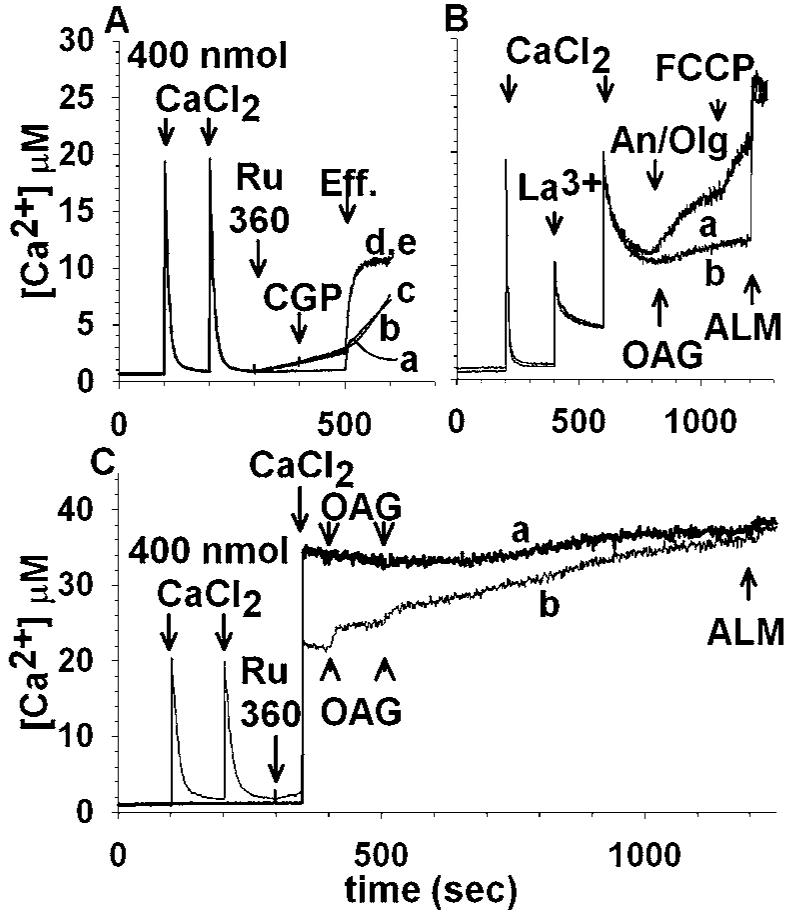
OAG-induced Ca2+ efflux from rat brain mitochondria is not mediated by known Ca2+ uptake and release pathways. (A): Mitochondria are loaded by exogenously added Ca2+ (each pulse is 400 nmol CaCl2) and the following compounds are administered: trace a, OAG (100 μM) at 500 s; trace b, 165 nM Ru360 at 300 s, 1 μM antimycin A3 plus 2 μM oligomycin at 500 s; trace c, 165 nM Ru360 at 300 s, 0.5 μM FCCP at 500 s; trace d, 165 nM Ru360 at 300 s, 100 μM OAG at 500 s; trace e, 165 nM Ru360 at 300 s, 20 μM CGP-37157 at 400 s, 100 μM OAG at 500 s; traces d and e are superimposed. “Eff.”: effectors. (B): 1 mM LaCl3 abolishes OAG-induced Ca2+ efflux, without affecting the reverse function of the uniporter: mitochondria are treated with 400 nmol CaCl2 at 350 and 600 s and 1 mM LaCl3 in between (500 s). Subsequently 1 μM antimycin A3 plus 2 μM oligomycin are administered at 800 s (trace a) or 100 μM OAG at 800 s (trace b). In trace a 0.5 μM FCCP was also added at 1100 s. In both traces, alamethicin (40 μg/ml) is added at 1200 s. (C): In trace a, rat brain mitochondria are not preloaded with Ca2+, and Ru360 (165 nM) is added at 300 s; subsequently, 800 nmol of CaCl2 is added at 350 s, followed by two pulses of OAG (100 μM each) at 400 and 500 s. In trace b, mitochondria are preloaded with two pulses of exogenously added CaCl2 (100 and 200 s, 400 nmol each); 165 nM Ru360 is added at 300 s, followed by an additional pulse of 400 nmol CaCl2 at 350 s. Subsequently OAG (two pulses of 100 μM each) are added at 400 and 500 s. In both traces, alamethicin (40 μg/mL) is added at 1200 s.
Effect of OAG on Mitochondrial Sr2+Handling
Rat brain mitochondria were loaded with SrCl2 (Fig. 3(A)) and flux of Sr2+ was monitored with Fura 6F. This dye responds to Sr2+ similarly to Ca2+; however, the Kd for Sr2+ is different (not quantified). In the absence of added OAG (trace a) mitochondria retain their Sr2+ load for the duration of the experiment. Addition of OAG (trace b) leads to an abrupt elevation of Fura 6F fluorescence, indicating release of sequestered Sr2+. The increase in Fura 6F fluorescence is not due to efflux of endogenous mitochondrial Ca2+ as addition of OAG (or FCCP, not shown) to freshly isolated mitochondria that have not been loaded with Ca2+ does not lead to an increase in Fura 6F fluorescence (Fig. 4(D)). Similar to the effect of La3+ on Ca2+ efflux, La3+ completely blocked OAG-induced Sr2+ release (Fig. 3(B)).
Fig. 3.
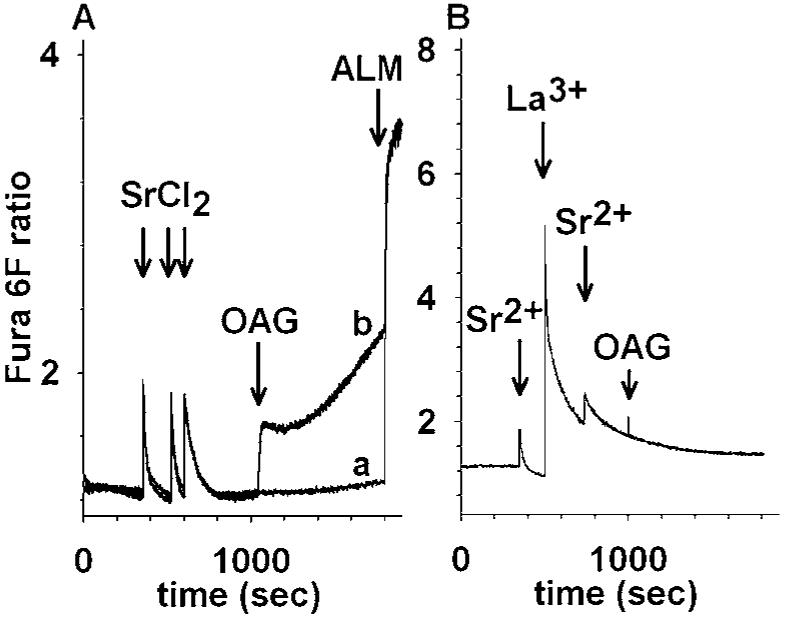
OAG-induced Sr2+ efflux from rat brain mitochondria and inhibition by La3+. (A): Mitochondria are loaded with three pulses of SrCl2 (400 nmol each, trace a). In trace b, subsequent to Sr2+ loading, OAG is added (100 μM) at 1000 s causing an immediate increase in Fura 6F ratio fluorescence. In both traces, alamethicin (40 μg/ml) is added at 1800 s. (B): Mitochondria are treated with 400 nmol SrCl2 at 350 and 750 s and 1 mM LaCl3 in between (500 s). OAG is added (100 μM) at 1000 s causing no further increase in Fura 6F ratio fluorescence.
Fig. 4.
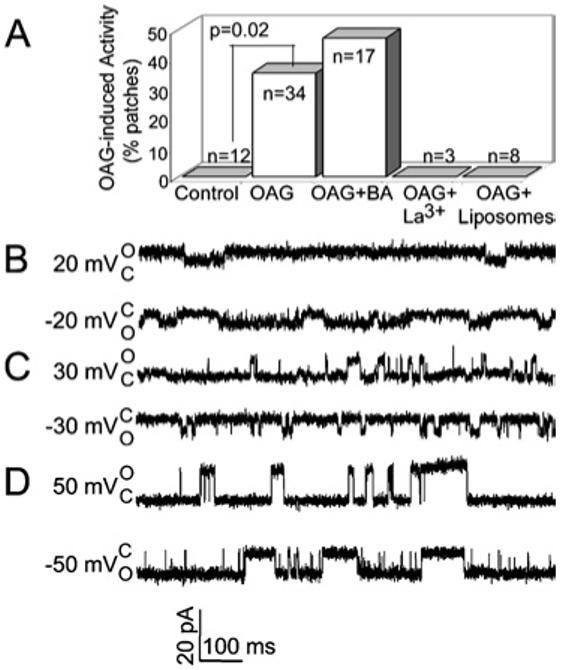
OAG-induced mitoplast channel activity. (A): The frequency of detecting OAG-induced channel activity in rat brain mitoplasts is shown in the absence (Control) and presence (OAG) of 100 μM OAG, and presence of 100 μM OAG plus 20 μM BKA (OAG BA). The frequency is statistically different in the absence and presence of OAG ( p = 0.02) and is not statistically different in the presence and absence of BKA ( p = 0.4, Fisher’s test). The frequency of detecting OAG-induced activity in patches from giant liposomes is also shown (OAG liposomes). “n” indicates the number of independent patches. (B-D). Current traces from three different excised patches from mitoplasts show 202 ± 33 pS (SD) transitions in the presence of 100 μM OAG at indicated voltages. O and C indicate open and closed current levels.
OAG Induces Cationic Channel Activity in Rat Brain Mitoplasts
In the absence of OAG, no channel activity was recorded from mitoplasts in 9 patches and conductances characteristic of PTP were observed in 3 other patches. Seventeen out of 34 patches showed no channel activity of any kind in the presence of OAG. Multiple conductance levels were observed in patches that were slightly cationselective in the presence of OAG in 12 of 34 patches. These patches were scored positive for the presence of OAG-induced activity in the bar graph (Fig. 4(A), p < 0.05). While other transition sizes were observed, current traces recorded in the presence of OAG typically exhibited transitions of 202 ± 33 pS; (n = 8 patches, Fig. 4(B)-(D)). PTP and Tim23 channels with conductances of ∼1200 and 750 pS, respectively, were recorded from 5 patches, ∼ which may have obscured the smaller OAG-induced activity (not shown). While the frequency of observing the OAG-induced activity increased slightly to 8 of 17 patches in the presence of the PTP inhibitor BKA, this increase was not statistically significant ( p = 0.4). Unlike BKA, perfusing the excised patches that exhibited OAG-induced activity with 1 mM LaCl3 led to a blockade of this channel activity (n = 3 patches). We did not observe any current upon exposure of OAG to patches excised from liposomes (Type IV, phosphatidylcholine, prepared as in (Guo et al., 2004), Fig. 4(A)).
DAGs Release Mitochondrial Ca2+ Through a Mechanism Independent of DAG Metabolism or Activation of PKC
1,2-diacylglycerols other than OAG, release Ca2+ from Ca2+-loaded mitochondria, (Fig. 5(A), traces b-f), while 1,3-diacylglycerols are inactive (Fig. 6(A)). Inhibition of PKC using five different PKC inhibitors (Fig. 5(B)) potentiated the OAG-induced initial Ca2+ efflux. No inhibitor exhibited statistically significant respiratory inhibition and/or uncoupling, except Myristoylated Protein Kinase C Inhibitor 20-28 (15% increase in state 4 respiration, not shown). The DAG lipase inhibitor RHC 80267 (Fig. 5(C), trace b) or the DAG kinase inhibitor II (Fig. 5(C), trace c) didn’t influence significantly the initial OAG-induced Ca2+ efflux; RHC 80267 only slightly potentiated the initial OAG-induced Ca2+ efflux while the DAG kinase inhibitor II potentiated the delayed Ca2+ rise. Neither inhibitor exhibited respiratory inhibition or uncoupling (not shown). Addition of OAG to mitochondria prior to loading with CaCl2 did not inhibit subsequent Ca2+ uptake; however, the time elapsedbefore the addition of OAG to the point of Ca2+ challenge influenced the maximal Ca2+ uptake capacity of mitochondria (Fig. 5(D), traces a-d). Various PLC inhibitors (U-73122, Edelfosine, D609, neomycin) were tested for their effects on maximal Ca2+ uptake capacity with the exception of neomycin with these drugs inhibited uptake due to the finding that they are potent respiratory uncouplers (not shown). Inclusion of phospholipids(phosphatidylethanolamine [PE] vs. phosphatidylserine [PS]) in the medium enhanced the potency of the second pulse of OAG-induced Ca2+ efflux from Ca2+-loaded mitochondria (Fig. 5(E)). Addition of OAG to mitoplasts caused release of sequestered Ca2+ (Fig. 5(F), trace a). The presence of RHC 80267 did not alter the initial OAG-induced Ca2+ efflux. The effect of OAG was further potentiated in the presence of Ru360 (Fig. 5(F), trace e). Pretreatment of Ca2+-loaded mitoplasts with 1 mM LaCl3 prior to addition of OAG blocked the effect of the diacylglycerol on releasing stored Ca2+ (Fig. 5(F), trace c).
Fig. 5.
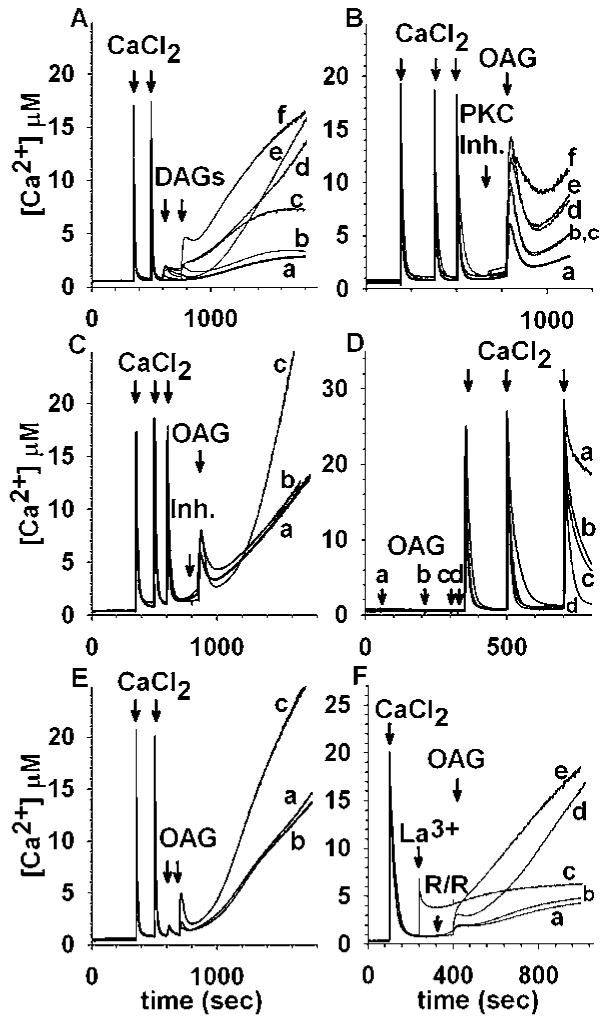
Diacylglycerol-induced release of sequestered Ca2+ in rat brain mitochondria (A-E) and mitoplasts (F): Relationship to putative targets and catabolites. (A): Mitochondria are loaded with two pulses of CaCl2 (400 nmol each at 350 and 500 s) followed by addition of the following DAG at 600 and 750 s (50 μM each pulse): trace a, vehicle (2 μl 96% ethanol [or DMSO, not shown]); trace b, DOL; trace c, DDC; trace d, SAG; trace e, DOG; trace f, HDAG. (B): PKC inhibitors potentiate the OAG-induced mitochondrial Ca2+ release: Mitochondria are loaded with three pulses of 400 nmol CaCl2 (350, 500 and 600 s) and prior to addition of 100 μM OAG (850 s) the following PKC inhibitor is added at 800 s: trace a, vehicle (2 μl 96% ethanol); trace b, Ro-32-0432 (200 nM); trace c, Gö 6976 (100 nM); trace d, myristoylated PKC Inhibitor 20-28 (10 microM); trace e, chelerythrine chloride (1 microM) trace f, Bisindolylmaleimide I (30 nM). (C): Inhibition of OAG catabolism does not mitigate OAG-induced Ca2+ release: Mitochondria are loaded with three pulses of 400 nmol CaCl2 (350, 500, and 600 s) and prior to addition of 100 μM OAG (850 s), inhibitors of DAG lipase and DAG lipase are added at 800 s. In trace a, no inhibitor is present, in trace b the DAG lipase inhibitor RHC 80267 (10 μM) is present; in trace c, the DAG kinase inhibitor “DAG kinase inhibitor II” (1 μM) was added at 800 s. (D): Addition of OAG to mitochondria that have not been loaded with Ca2+ does not abolish subsequent Ca2+ uptake. OAG (100 μM) was added at different time points at 50 s (trace a), 200 s (trace b), 300 s (trace c), 325 s (trace d), prior to challenging mitochondria with three pulses of CaCl2 (at 350, 500, and 700 s, 400 nmol each). (E): Effect of phospholipids on the OAG-induced Ca2+ release: Mitochondria are loaded with two pulses of CaCl2 (400 nmol each at 350 and 500 s) followed by addition of 25 μM OAG at 600 and 700 s in the presence of PS (10 μg/mg mitochondrial protein, trace b) or PE (10 μg/mg mitochondrial protein, trace c). In trace a, ethanol was added instead of a phospholipid. (F): OAG releases sequestered Ca2+ from rat brain mitoplasts: In all traces, mitoplasts are loaded with a single pulse of 400 nmol CaCl2 (100 s), and OAG (100 μM unless otherwise indicated) is added at 400 s. In trace a, no further additions were made. In trace b, RHC 80267 (10 μM) was added at 320 s; in trace c,1mM LaCl3 was added at 240 s; in trace d, 200 μMOAGwas added at 400 s; in trace e, 165 nM Ru360 was added at 320 s, followed by 200 μM OAG at 400 s. “R/R” signifies RHC 80267 or Ru360.
Fig. 6.
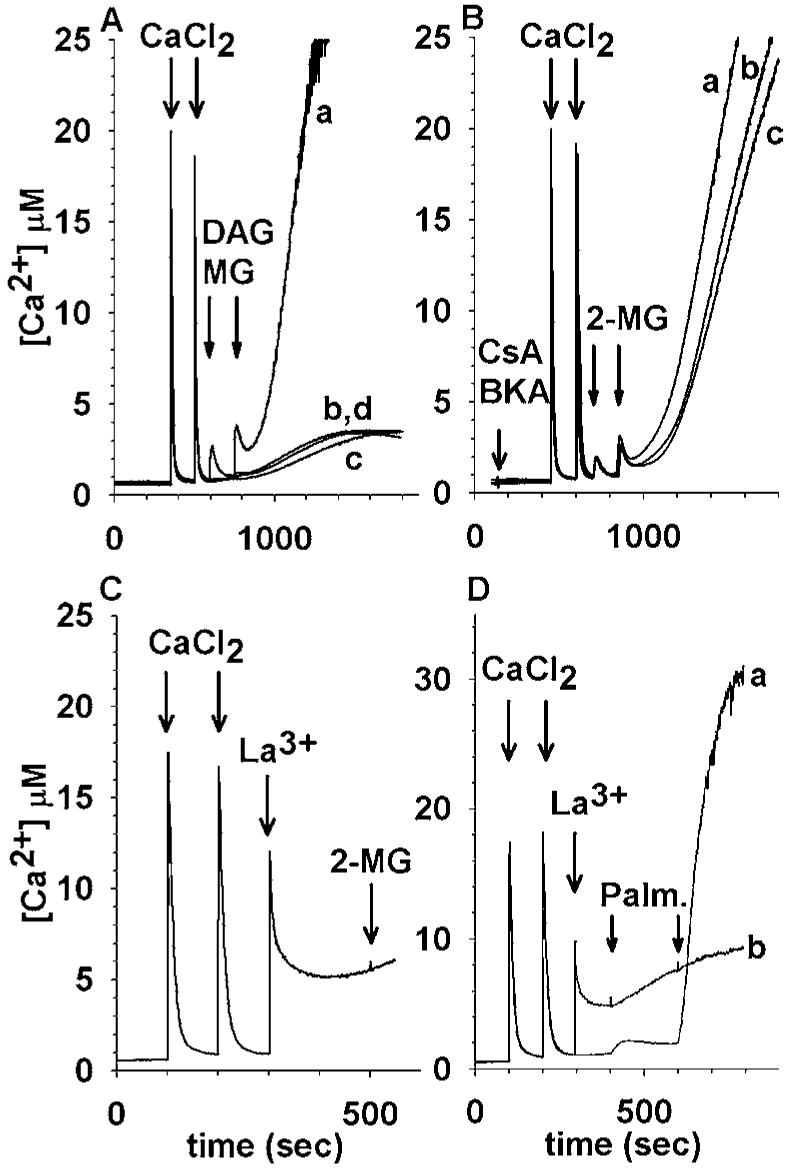
Di- and Monoacylglycerol induced mitochondrial Ca2+ release: Effects of permeability transition inhibition and La3+. (A): Mitochondria are loaded with two pulses of CaCl2 (400 nmol each at 350 and 500 s) followed by addition of the following DAG or MG at 600 and 750 s (100 μM each pulse): trace a, 2-Monooleoylglycerol (C18:1, [cis]-9); trace b, 1,3-Di-([cis]-9-octadecenoyl)-rac-glycerol; trace c, vehicle; trace d, 1-Monooleoyl-rac-glycerol (C18:1, [cis]-9). (B): Mitochondria are pretreated with a PTP inhibitor (at 50 s) and challenged with two pulses of CaCl2 (400 nmol each at 350 and 500 s) followed by addition of 2-Monooleoylglycerol at 600 and 750 s (50 μM each pulse); trace a,no PTP inhibitor; trace b,2 μM Cys A; trace c,20 μM BKA. (C): LaCl3 inhibits the 2-MG induced Ca2+ efflux: Mitochondria are loaded with two pulses of CaCl2 (400 nmol each at 100 and 200 s) followed by addition of 1 mM LaCl3 at 300 s; subsequently, 100 μM 2-Monooleoylglycerol is added at 500 s. (D): LaCl3 inhibits the palmitic acidinduced Ca2+ efflux: In trace a, mitochondria are loaded with two pulses of CaCl2 (400 nmol each at 100 and 200 s); subsequently, 10 μM palmitic acid is added at 400 and 600 s; in trace b, mitochondria are loaded with two pulses of CaCl2 (400 nmol each at 100 and 200 s) followed by addition of 1 mM LaCl3 at 300 s; subsequently, 10 μM palmitic acid is added at 400 and 600 s.
Additional Characteristics of DAG-Induced Release of Mitochondrial Ca2+
We compared the effect of OAG to 1,3-diolein (1,3-Di-([cis]-9-octadecenoyl)-rac-glycerol, (Fig. 6(A), trace b), 2-MG (2-Monooleoylglycerol (C18:1, [cis]-9) (trace a) and 1-MG (1-Monooleoyl-rac-glycerol (C18:1, [cis]-9), (trace d). Only 2-MG induced an initial Ca2+ efflux from Ca2+-loaded rat brain mitochondria, followed by a robust delayed Ca2+ rise (Fig. 6(A), trace a). Neither the initial 2-MG-induced Ca2+ release nor the delayed Ca2+ efflux was sensitive to PTP inhibitors (Fig. 6(B), traces b and c). The 2-MG-induced Ca2+ release was, however, completely abolished by 1 mM LaCl3 (Fig. 6(C)). 1 mM LaCl3 also inhibited release of sequestered Ca2+ from rat brain mitochondria induced by palmitic acid (Fig. 6(D)). Moreover, LaCl3 mitigated the uncoupling effect of this fatty acid (not shown). 2-MG exhibited both significant uncoupling and inhibition of state 3 respiration (Table I). Among the DAGs, DOG exhibited significant uncoupling. The other tested DAGs, including OAG, exhibited little or no inhibition of state 3 respiration.
Table I.
Effects of Di- and Monoacylglycerols and Farnesyl Thiotriazole (FTT) on Rat Brain Mitochondrial Oxygen Consumption
| State 3 | State 4 (effector added before oligomycin) | State 4 (effector added after oligomycin) | |
|---|---|---|---|
| Vehicle, (n = 12) | 131 ± 3 | 16 ± 1 | 16 ± 1 |
| OAG (n = 6) | 102 ± 4* | 16 ± 1ns | 16 ± 1ns |
| 2-MG (n = 5) | 110 ± 9* | 29 ± 2* | 15 ± 1ns |
| DDC (n = 3) | 125 ± 6n.s. | 15 ± 1ns | 14 ± 1ns |
| DOL (n = 3) | 113 ± 6* | 15 ± 1ns | 14 ± 1ns |
| DOG (n = 3) | 130 ± 9n.s. | 21 ± 2* | 17 ± 1ns |
| FTT (n = 3) | 48 ± 4* | 34 ± 2* | 29 ± 2* |
| HDAG (n = 3) | 96 ± 5* | 14 ± 1ns | 15 ± 1ns |
| SAG (n = 3) | 126 ± 7n.s. | 15 ± 1ns | 14 ± 1ns |
Note. All di- and monoacylglycerols and FTT were tested at 100 μM concentration. n: number of independent experiments. Statistics: Mann-Whitney rank sum test
significant compared to control (vehicle), p < 0.001
not significant compared to control. Values are given as nmol/min/mg protein and expressed as standard errors of the mean, rounded to the nearest integer.
DISCUSSION
The novel observations of the current study point to a potential mode of Ca2+ release in mitochondria that can be synchronized with physiological events inducing elevation of cytosolic [Ca2+] either throughout the cell or in microdomains. Our primary finding is that OAG releases Ca2+ sequestered by mitochondria isolated from rodent tissues but not from sweet potato. Plants do possess multiple pathways that ultimately result in DAGs production (Wang, 2004). The importance of the lack of effect of OAG on sweet potato mitochondria is twofold: (i) it demonstrates biological diversity and (ii) it weakens the possibility of an artifact of OAG on lipid membranes, implicated previously (Allan et al., 1978; Leikin et al., 1996; Szule et al., 2002).
Additional lines of evidence argue against a Ca2+-ionophoretic activity of the diacylglycerol or an unspecific effect on bilayers leading to ion permeability: (i) following Ca2+ release upon exposure to OAG in mammalian mitochondria, extramitochondrial [Ca2+] returns to baseline values; an ionophore would lead to the establishment of a new steady-state [Ca2+]; (ii) OAG does not cause any swelling during the initial Ca2+ release, excluding the possibility of an unspecific hole-forming effect or activation of the PTP; (iii) several effective 1,2-DAGs do not exhibit any uncoupling properties that would be obvious if they nonselectively permeabilize the mitochondrial inner membrane; (iv) application of OAG to the patchesdoes not always result in channel activity, arguing against a nonspecific membrane stressing effect; (v) no currents are observed with patched liposomes upon exposure to the diacylglycerol.
To test the hypothesis that OAG induces Ca2+ efflux through previously described mitochondrial flux pathways, we blocked independently the uniporter, the Na+/Ca2+ exchanger, and the PTP. Blocking the uniporter with Ru360 preventing bidirectional Ca2+ transport by this pathway (Matlib et al., 1998) does not inhibit and, as expected, actually potentiates the OAG-induced Ca2+ release from mitochondria or mitoplasts; the amplitude of the release is greater in the presence of Ru360 due to the inability of the uniporter to reaccumulate Ca2+, hence also the lack of post-OAG extramitochondrial [Ca2+] decay. This observation also argues against an uncoupling mode of action of OAG since uncoupler-induced calcium efflux occurs via reversal of the mitochondrial uniporter (Bernardi et al., 1984). In addition, the uniporter is relatively impermeable to K+ while our electrophysiological recordings were made using symmetrical KCl solutions. Moreover, single uniporter channels have multiple subconductance states between 2.6 pS and 5.2 pS at -160 mV, but the OAG-induced conductances are ∼200 pS. Therefore, it is clear that the OAG-induced channel is not the uniporter characterized recently (Kirichok et al., 2004). La3+ inhibits completely the OAG response, while in our hands, La3+(1 mM) does not eliminate the release of Ca2+ from mitochondria induced by respiratory inhibition or uncoupling. Blockade of the Na+/Ca2+ exchanger with CGP-37157 does not antagonize the OAG effect; moreover, the fast kinetics of the initial OAG-induced Ca2+ efflux argue against the involvement of a slow exchanger. In addition, the absence of Na+ in the medium eliminates the Na+/Ca2+ exchanger as the mechanism.
We furthermore conclude that the initial rapid Ca2+ efflux induced by OAG is not due to PTP opening because: (i) it is insensitive to any PTP inhibitor tested; (ii) OAG-induced channel activity exhibits conductances ∼6 times lower than those characteristics of PTP (Loupatatzis et al., 2002); (iii) OAG induces Sr2+ release from Sr2+-loaded mitochondria, whereas Sr2+ does not promote PTP formation (Hunter et al., 1976); (iv) OAG does not induce any large-amplitude swelling during the initial Ca2+ release and, in accordance with our observations, DAGs were shown earlier not to induce swelling of liver mitochondria (Pastorino et al., 1999). In contrast, the delayed Ca2+ rise exhibits robust evidence of PTP, including sensitivity to PTP inhibitors and large-amplitude mitochondrial swelling. At least one component leading to the delayed formation of PTP is the intense Ca2+ cycling substantiated by the OAG-induced Ca2+ channel(s) and the uniporter; however, in rat liver mitochondria, the presence of Cys A did not extend significantly the threshold for PTP induction in the presence of OAG, in spite of the immensely protecting effect of the immunophilin against high-Ca2+ mitochondrial loading in the absence of the diacylglycerol. Since DAGs activate PLA2 which is also found in mitochondria (Nachbaur and Vignais, 1968), the OAG induced secondary phase of Ca2+ release could be due to activation of this enzyme, which is known to contribute to induction and/or maintenance of PTP (Broekemeier and Pfeiffer, 1989; Rustenbeck et al., 1996); however, neither aristolochic acid, nor bromoenol lactone (both inhibitors of PLA2) inhibited mitochondrial Ca2+-induced Ca2+ release (mCICR) under the exact conditions in the absence of OAG (Chinopoulos et al., 2003).
Phorbol esters can substitute for diacylglycerol in activating both protein kinase C (Nishizuka, 1995) and non-PKCs (Brose et al., 2004). Substitution of OAG for PMA fails to induce Ca2+ efflux. Moreover, inclusion of five different PKC inhibitors does not ameliorate the OAG effect and even potentiates it. Such a scenario has been reported for DAG-sensitive TRP channels on the plasma membrane (Venkatachalam et al., 2003). However, the possibility of TRP channels being present in mitochondria has been previously excluded (Chinopoulos et al., 2003). A caveat here is that contemporary pharmacological PKC antagonists targeting the C1 domain bind with similar affinities to non-PKCs possessing the same domain activated by DAG/phorbol esters (Yang and Kazanietz, 2003). The question arises, as to why would there be PKC in our isolated mitochondria preparations, since it translocates to the organelles as a reaction to cell stress; probably, the mitochondrial isolation procedure is damaging enough (decapitation of the animal, transient hypoxia of the tissue) to cause PKC translocation.
To characterize further the mode of action of DAGs on mitochondria, we tested di- and monoacylglycerols by varying the position of the acyl groups. Only 1,2-sn-DAGs induce release of Ca2+, while 1,3-sn-DAGs are inactive. Among monoacylglycerols, 1-MG is not effective, as opposed to 2-MG that releases Ca2+ vigorously. As a comparison, PKC is not activated by 1,3-DAGs nor monoacylglycerols, but is activated by 1,2-DAGs (Nishizuka, 1995). 2-MG induces powerful uncoupling in addition to causing Ca2+ efflux that is insensitive to PTP inhibitors. At this juncture, it is not known if 2-MGs and 1,2-DAGs share the same target(s) on mitochondria or if 2-MG has an alternative mode of action. La3+ inhibits the 2-MG-induced Ca2+ release completely; however, La3+ also inhibits palmitic acid-induced PTP (Sultan and Sokolove, 2001) and Ca2+ flux through the uniporter. The universal ability of La3+ to block all forms of mitochondrial Ca2+ release therefore limits its usefulness in delineating the individual mechanism involved in OAG-induced release.
In addition to regulation by diacylglycerol or phorbol esters, DAG targets require Ca2+ and PS (Bell and Burns, 1991). Inclusion of PS to the suspension does not have an effect on the OAG-induced Ca2+ efflux; however, PE potentiates the OAG response. At this juncture, we cannot attribute the PE potentiating effect on a biologically relevant mechanism or on the possibility that PE is a better vehicle for OAG in solution. Concerning the requirement of matrix Ca2+, our experiments demonstrate that OAG causes release of Ca2+ from Ca2+-loaded mitochondria but does not allow Ca2+ influx in mitochondria treated with Ru360. It is therefore possible that the target molecule(s) of OAG on the inner mitochondrial membrane possess binding site(s) for Ca2+ located at the inner leaflet of the membrane. This is a very similar scenario to the one involving PKC: Ca2+ increases the affinity of conventional PKCs for negatively charged lipids and this increase varies linearly with Ca2+ concentration in the low μM to submillimolar range (Mosior and Epand, 1994). OAG also releases sequestered strontium; however, due to the almost identical molecular radius of Sr2+ to Ca2+, Sr2+ may substitute for Ca2+ for whatever means. The requirement for accumulated intramitochondrial Ca2+ for DAGs-induced Ca2+ release can be envisioned as a mechanism preventing mitochondria from buffering all the Ca2+ coming from other sources during PIP2 hydrolysis. The necessity of an obligatory metabolite (DAG) sets the mechanism in synchrony.
It is clear that mitochondria host DAG targets (see Introduction), though the two major DAG clearance pathways, DAG kinase and DAG lipase, have not been unequivocally identified these organelles (Bothmer et al., 1992); nevertheless, 1,2-diacyl-sn-glycerol is a reactant or product in 23 reactions, spanning among four different biochemical pathways (for a complete description of the involvement of DAGs in biochemical pathways, see: http://www.genome.jp/dbget-bin/wwwbget?compound±C00641), leaving multiple candidates for DAG clearance; parts from the overall reactions in all four pathways are hosted by mitochondrial membranes or take place in the matrix (Murray, 2003).
In summary, DAGs release sequestered Ca2+ from isolated mitochondria, most likely mediated through novel channels located on the inner mitochondrial membrane, or a single channel with multiple substates. It remains to be determined if DAGs are the physiological inducers for this channel activity during signal transduction in cells associated with activation of phospholipase C. This is an exciting possibility linking the triad of signal transduction, intracellular Ca2+ homeostasis, and mitochondrial bioenergetics.
ACKNOWLEDGMENTS
We thank Dr. Alicia J. Kowaltowski for help with the sweet potato mitochondrial isolation and Prof.Miklós Tóth and Dr. György Báthori for comments during the preparation of the manuscript. This work was supported by NIH grant GM57249 and NSF grant MCB-0235834 to K.W.K. and NIH grant NS34152 and USAMRMC grant DAMD 17-99-1-9483 to G.F.
Abbreviations used
- 1,2-DGs
1,2-Diacylglycerols
- 1,3-DGs
1,3-Diacylglycerols
- 2-MGs
2-Monoacylglycerols
- 1-MGs
1-Monoacylglycerols
- ALM
Alamethicin
- BKA
Bongkrekic acid
- Cys A
Cyclosporin A
- PTP
Permeability Transition Pore
- OAG
1-oleoyl-acetyl-sn-glycerol
- DOL
1,2-Dioleoylglycerol (18:1)
- DDC
1,2-Didecanoylglycerol (10:0)
- SAG
1-stearoyl-2-arachidonoyl-sn-glycerol
- DOG
1,2-Dioctanoyl-sn-glycerol (8:0)
- HDAG
1-O-Hexadecyl-2-arachidonoyl-sn-glycerol.
REFERENCES
- Allan D, Thomas P, Michell RH. Nature. 1978;276:289–290. doi: 10.1038/276289a0. [DOI] [PubMed] [Google Scholar]
- Baines CP, Song CX, Zheng YT, Wang GW, Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM, Ping P. Circ. Res. 2003;92:873–880. doi: 10.1161/01.RES.0000069215.36389.8D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RM, Burns DJ. J. Biol. Chem. 1991;266:4661–4664. [PubMed] [Google Scholar]
- Bernardi P, Paradisi V, Pozzan T, Azzone GF. Biochemistry. 1984;23:1645–1651. doi: 10.1021/bi00303a010. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Vassanelli S, Veronese P, Colonna R, Szabo I, Zoratti M. J. Biol. Chem. 1992;267:2934–2939. [PubMed] [Google Scholar]
- Bothmer J, Markerink M, Jolles J. Biochem. Biophys. Res. Commun. 1992;187:1077–1082. doi: 10.1016/0006-291x(92)91307-c. [DOI] [PubMed] [Google Scholar]
- Broekemeier KM, Pfeiffer DR. Biochem. Biophys. Res. Commun. 1989;163:561–566. doi: 10.1016/0006-291x(89)92174-8. [DOI] [PubMed] [Google Scholar]
- Brose N, Betz A, Wegmeyer H. Curr. Opin. Neurobiol. 2004;14:328–340. doi: 10.1016/j.conb.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Brose N, Rosenmund C. J. Cell Sci. 2002;115:4399–4411. doi: 10.1242/jcs.00122. [DOI] [PubMed] [Google Scholar]
- Chen CH, Lehninger AL. Arch. Biochem. Biophys. 1973;157:183–196. doi: 10.1016/0003-9861(73)90404-9. [DOI] [PubMed] [Google Scholar]
- Chinopoulos C, Starkov AA, Fiskum G. J. Biol. Chem. 2003;278:27382–27389. doi: 10.1074/jbc.M303808200. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Clapham DE, Montell C, Schultz G, Julius D. Pharmacol. Rev. 2003;55:591–596. doi: 10.1124/pr.55.4.6. [DOI] [PubMed] [Google Scholar]
- Florin-Christensen J, Florin-Christensen M, Delfino JM, Stegmann T, Rasmussen H. J. Biol. Chem. 1992;267:14783–14789. [PubMed] [Google Scholar]
- Ford DA, Gross RW. J. Biol. Chem. 1990;265:12280–12286. [PubMed] [Google Scholar]
- Freeman M, Mangiapane EH. Biochem. J. 1989;263:589–595. doi: 10.1042/bj2630589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudermann T, Hofmann T, Schnitzler M, Dietrich A. Novartis Found. Symp. 2004;258:103–118. [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Am.J.Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Guo L, Pietkiewicz D, Pavlov EV, Grigoriev SM, Kasianowicz JJ, Dejean LM, Korsmeyer SJ, Antonsson B, Kinnally KW. Am. J. Physiol. Cell Physiol. 2004;286:C1109–C1117. doi: 10.1152/ajpcell.00183.2003. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA, Southard JH. J. Biol. Chem. 1976;251:5069–5077. [PubMed] [Google Scholar]
- Irvine RF. Sci. STKE. 2002:RE13. doi: 10.1126/stke.2002.150.re13. 2002. [DOI] [PubMed] [Google Scholar]
- Kazanietz MG. Mol. Pharmacol. 2002;61:759–767. doi: 10.1124/mol.61.4.759. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Knox CD, Belous AE, Pierce JM, Wakata A, Nicoud IB, Anderson CD, Pinson CW, Chari RS. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G533–G540. doi: 10.1152/ajpgi.00050.2004. [DOI] [PubMed] [Google Scholar]
- Korge P, Honda HM, Weiss JN. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3312–3317. doi: 10.1073/pnas.052713199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Fisher SK, Agranoff BW, Hajra AK. J. Biol. Chem. 1991;266:22837–22846. [PubMed] [Google Scholar]
- Leikin S, Kozlov MM, Fuller NL, Rand RP. Biophys. J. 1996;71:2623–2632. doi: 10.1016/S0006-3495(96)79454-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lorenzo PS, Bogi K, Blumberg PM, Yuspa SH. Mol. Cell Biol. 1999;19:8547–8558. doi: 10.1128/mcb.19.12.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liscovitch M, Czarny M, Fiucci G, Lavie Y, Tang X. Biochim. Biophys. Acta. 1999;1439:245–263. doi: 10.1016/s1388-1981(99)00098-0. [DOI] [PubMed] [Google Scholar]
- Loupatatzis C, Seitz G, Schonfeld P, Lang F, Siemen D. Cell. Physiol. Biochem. 2002;12:269–278. doi: 10.1159/000067897. [DOI] [PubMed] [Google Scholar]
- Majumder PK, Pandey P, Sun X, Cheng K, Datta R, Saxena S, Kharbanda S, Kufe D. J. Biol. Chem. 2000;275:21793–21796. doi: 10.1074/jbc.C000048200. [DOI] [PubMed] [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM. J. Biol. Chem. 1998;273:10223–10231. doi: 10.1074/jbc.273.17.10223. [DOI] [PubMed] [Google Scholar]
- Mikoshiba K, Hattori M. Sci. STKE. 2000:E1. doi: 10.1126/stke.2000.51.pe1. 2000. [DOI] [PubMed] [Google Scholar]
- Mosior M, Epand RM. J. Biol. Chem. 1994;269:13798–13805. [PubMed] [Google Scholar]
- Murray RK. Harper’s Illustrated Biochemistry. Lange Medical Books/McGraw-Hill; New York: 2003. [Google Scholar]
- Murriel CL, Churchill E, Inagaki K, Szweda LI, Mochly-Rosen D. J. Biol. Chem. 2004;279:47985–47991. doi: 10.1074/jbc.M405071200. [DOI] [PubMed] [Google Scholar]
- Nachbaur J, Vignais PM. Biochem. Biophys. Res. Commun. 1968;33:315–320. doi: 10.1016/0006-291x(68)90786-9. [DOI] [PubMed] [Google Scholar]
- Nilius B. Sci. STKE. 2004:e36. doi: 10.1126/stke.2432004pe36. 2004. [DOI] [PubMed] [Google Scholar]
- Nishihira J, Ishibashi T. Lipids. 1986;21:780–785. doi: 10.1007/BF02535412. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- Okada T, Inoue R, Yamazaki K, Maeda A, Kurosaki T, Yamakuni T, Tanaka I, Shimizu S, Ikenaka K, Imoto K, Mori Y. J. Biol. Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- Panagia V, Ou C, Taira Y, Dai J, Dhalla NS. Biochim. Biophys. Acta. 1991;1064:242–250. doi: 10.1016/0005-2736(91)90308-u. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Tafani M, Rothman RJ, Marcinkeviciute A, Hoek JB, Farber JL, Marcineviciute A. J. Biol. Chem. 1999;274:31734–31739. doi: 10.1074/jbc.274.44.31734. [DOI] [PubMed] [Google Scholar]
- Penner R, Fleig A. Sci. STKE. 2004:e38. doi: 10.1126/stke.2432004pe38. 2004. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Rajdev S, Reynolds IJ. Neurosci. Lett. 1993;162:149–152. doi: 10.1016/0304-3940(93)90582-6. [DOI] [PubMed] [Google Scholar]
- Rebecchi MJ, Pentyala SN. Physiol. Rev. 2000;80:1291–1335. doi: 10.1152/physrev.2000.80.4.1291. [DOI] [PubMed] [Google Scholar]
- Reed KC, Bygrave FL. Biochem. J. 1974;140:143–155. doi: 10.1042/bj1400143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustenbeck I, Munster W, Lenzen S. Biochim. Biophys. Acta. 1996;1304:129–138. doi: 10.1016/s0005-2760(96)00113-0. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, Carr BK, May WS. J. Biol. Chem. 1998;273:25436–25442. doi: 10.1074/jbc.273.39.25436. [DOI] [PubMed] [Google Scholar]
- Sato T, O’Rourke B, Marban E. Circ. Res. 1998;83:110–114. doi: 10.1161/01.res.83.1.110. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Wachter E, Sebald W, Neupert W. Eur. J. Biochem. 1984;144:581–588. doi: 10.1111/j.1432-1033.1984.tb08505.x. [DOI] [PubMed] [Google Scholar]
- Schnaitman C, Erwin VG, Greenawalt JW. J. Cell Biol. 1967;32:719–735. doi: 10.1083/jcb.32.3.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G. Biochem. Biophys. Res. Commun. 2001;281:645–650. doi: 10.1006/bbrc.2001.4409. [DOI] [PubMed] [Google Scholar]
- Storz P, Hausser A, Link G, Dedio J, Ghebrehiwet B, Pfizenmaier K, Johannes FJ. J. Biol. Chem. 2000;275:24601–24607. doi: 10.1074/jbc.M002964200. [DOI] [PubMed] [Google Scholar]
- Sultan A, Sokolove PM. Arch. Biochem. Biophys. 2001;386:52–61. doi: 10.1006/abbi.2000.2195. [DOI] [PubMed] [Google Scholar]
- Szule JA, Fuller NL, Rand RP. Biophys. J. 2002;83:977–984. doi: 10.1016/s0006-3495(02)75223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaena de AS, Okamoto Y, Hannun YA. J. Biol. Chem. 2004;279:11537–11545. doi: 10.1074/jbc.M309586200. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, van Rossum DB, Patterson RL, Ma HT, Gill DL. Nat. Cell Biol. 2002;4:E263–E272. doi: 10.1038/ncb1102-e263. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Zheng F, Gill DL. J. Biol. Chem. 2003;278:29031–29040. doi: 10.1074/jbc.M302751200. [DOI] [PubMed] [Google Scholar]
- Wang X. Curr. Opin. Plant Biol. 2004;7:329–336. doi: 10.1016/j.pbi.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM. Biochem. J. 2002;363:657–666. doi: 10.1042/0264-6021:3630657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Kazanietz MG. Trends Pharmacol. Sci. 2003;24:602–608. doi: 10.1016/j.tips.2003.09.003. [DOI] [PubMed] [Google Scholar]