

Abstract
Potential methods for the generation of the guanidine hemiaminal functionality of palau’mine (1) from a spirocyclic thiohydantoin precursor are examined using a spirobicyclic model system, culminating in the identification of an efficient means of effecting this transformation.
INTRODUCTION
The architecturally intricate marine natural product palau’amine (1) was first isolated from Stylotella aurantium in 1991. i Biochemical assays have since revealed palau’amine to possess exceptional immunosuppressive activity (IC50 <18 ng/mL, mixed lymphocyte reaction), identifying it as an important target for total synthesis. Despite considerable synthetic investigation in the intervening years, ii palau’amine (1) has yet to succumb to total synthesis. During efforts toward the total synthesis of 1, we required methods for constructing the spiro guanidine hemiaminal (2-aminoimidazolin-5-ol) functionality of the palau’amine F-ring. Several methods for the preparation of closely related heterocycles have been reported previously: (1) rearrangement of 2-(hydroxylamino)imidazoles,iiia (2) condensation of guanidine salts with glyoxaliiib or glyoxylate hemiacetals,iv and (3) oxidation of 2-aminoimidazoles in the presence an alcohol.v However, none of these methods appeared readily compatible with our proposed synthetic approach to 1, wherein an S-methylated thiohydantoin (cf., 3) was envisioned as a precursor of the spirobicyclic guanidine hemiaminal functionality. To evaluate alternative approaches for the synthesis the spiro 2-aminoimidazolin-5-ol portion of 1, we examined a range of methods for the conversion of 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one (3), an analog of the D/F-ring system of 1, to the corresponding spirobicyclic guanidine hemiaminal (2) (2-amino-1,3-diazaspiro[4.4]non-1-en-4-ol).
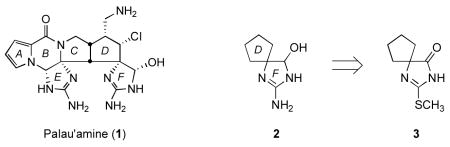
RESULTS AND DISCUSSION
We commenced these studies by preparing S-methylated thiohydantoin (3) from commercially available 1-aminocyclopentane carboxylic acid (4) (Scheme 1). This process was initiated by Fischer of 4, followed by condensation of the resulting α-amino ester with benzoyl isothiocyanate, affording thiourea (5). Exposure of 5 to methanolic potassium carbonate led to cyclization to the corresponding thiohydantoin, followed by cleavage of the N-benzoyl group. S-Methylation of the resulting thiohydantoinvi then furnished diazaspiro[4.4]nonenone (3).
Scheme 1.
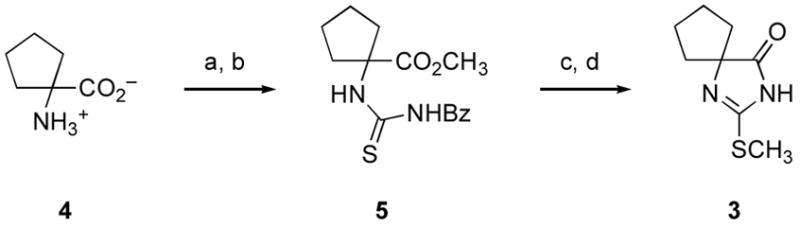
Reagents and conditions: (a) AcCl, CH3OH, 0→40 °C; (b) BzNCS, Et3N, CH2Cl2, 23 °C; (c) K2CO3, CH3OH, 23 °C (85%; 3 steps); (d) CH3I, NaOH, CH3OH, H2O, 23 °C (82%).
Our initial studies toward the conversion of 3 to guanidine hemiaminal (2) were guided by the goal of delaying guanidine installation as long as possible, as such a strategy promised to simplify the chromatographic purification of intermediates in a subsequent synthesis of 1. We therefore initially examined reduction of the carbonyl group of 3 to the corresponding hemiaminal prior to introduction of the final nitrogen. We achieved this goal by adding an N-9-fluorenylmethoxycarbonyl (N-Fmoc) protective group to thiohydantoin derivative (3), thereby potentiating borohydride-mediated reduction of 3 to N-Fmoc 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-ol (6) (Scheme 2).vii In an attempt to generate the parent heterocycle, the N-Fmoc group was then cleaved from 6 by exposure to an excess of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) at room temperature. However, the product formed in this reaction, 7, was found to exist exclusively as the ring-opened tautomer in chloroform, methanol, and dimethyl sulfoxide. Ring-formation could be promoted by incubating aldehyde (7) with benzyl alcohol and 4Å molecular sieves in the presence of 10-camphorsulfonic acid (CSA), allowing for the isolation of 4-benzyloxy-1,3-diaza-2-(methylthio)spiro[4.4]non-1-ene (8). Unfortunately, attempts to convert O-benzyl hemiaminal (8) to the corresponding spirobicyclic guanidine hemiaminal were unsuccessful, as exposure of 8 to ethanolic ammonia at room temperature only resulted in substitution of ammonia or ethanol for the benzyloxy group. Prolonged heating of these substitution products with ethanolic ammonia triggered rearrangement of the products to 2-methylthio-4,5,6,7-tetrahydrobenzimidazole,viii and attempts to facilitate the aminolysis of 8 by converting 8 to the corresponding sulfoneix were likewise unsuccessful and led only to substrate decomposition.
Scheme 2.
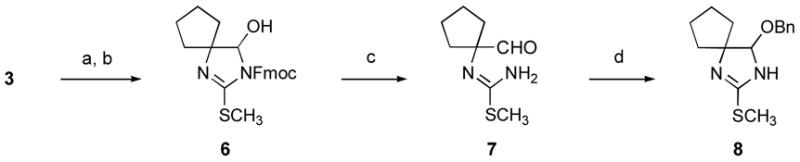
Reagents and conditions: (a) FmocCl, NaH, THF, 0 °C (92%); (b) NaBH4, CH3OH, 0 °C (86%); (c) DBU, CH2Cl2, 23 °C (85%); (d) BnOH, CSA, 4Å mol. sieves, CH2Cl2, 23 °C (55%).
The difficulties encountered in converting O-benzyl hemiaminal (8) to the corresponding guanidine, engendered by the acid- and base-lability of the aminal portion of 8, led us to pursue an alternative strategy for the synthesis of aminodiazaspiro[4.4]nonenol (2) in which generation of the hemiaminal function of 2 would be deferred until after guanidine installation. As the N-substitution of pseudothioureas with electron-withdrawing protective groups has frequently been employed to facilitate pseudothiourea aminolysis, x we next examined the aminolysis of N-2-(trimethylsilyl)ethylsulfonyl-protected (N-SES)xi spirobicyclic (methylthio)imidazolone (9), which was prepared in one step from S-methylated thiohydantoin (3) (Scheme 3). Gratifyingly, exposure of 9 to ammonia-saturated methanol at 23 °C rapidly and cleanly led to the formation of the corresponding spiro glycocyamidine (10).xii Reduction of glycocyamidine (10) to the corresponding hemiaminal proved more difficult. This reduction was ultimately accomplished through the use of excess diisobutylaluminum hydride (DIBAL), affording hemiaminal (11) in moderate yield (47%).xiii,xiv Our inability to cleave the N-SES protective group from this product ultimately proved the undoing of this synthetic route, however, as treatment of 11 with aqueous HF or HF· pyridine resulted in no detectible reaction.xv
Scheme 3.
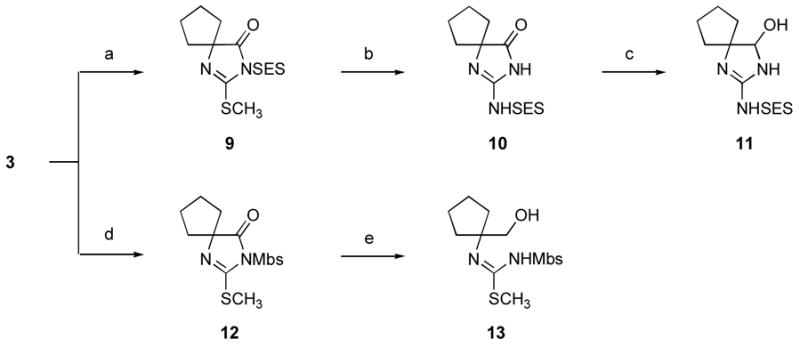
Reagents and conditions: (a) SESCl, NaH, THF, 0 °C (66%); (b) NH3, CH3OH, 0→23 °C (92%); (c) DIBAL, THF, –78→0 °C (47%); (d) MbsCl, NaH, THF, 0 °C (89%); (e) NaBH4, CH3OH, 0 °C (100%). (SES = 2-(trimethylsilyl)ethylsulfonyl; Mbs = 4-methoxybenzenesulfonyl.)
The facility of the aminolysis of N-sulfonyl pseudothiourea (9) prompted renewed investigation of a synthetic sequence involving initial generation of the hemiaminal function of 2, followed by guanidine installation. Reduction of an N-sulfonyl-protected intermediate such as 12 to the corresponding hemiaminal would be expected to proceed under considerably milder conditions than those required for reduction of glycocyamidine (10).xiii It was hoped that aminolysis of the resulting N-sulfonyl-pseudothiourea hemiaminal might, like the aminolysis of N-sulfonyl pseudothiourea (9), prove rapid, allowing efficient access to guanidine hemiaminals related to 11. Unfortunately, facile over-reduction of the Mbs-protected derivative (12) to the ring-opened primary alcohol (13) ultimately thwarted this synthetic plan.
As an N-Fmoc congener of 12 had demonstrated excellent kinetic stability toward over-reduction (Scheme 2), we next considered a related strategy in which an N-acyloxy group would facilitate late-stage conversion of a pseudothiourea fragment to the corresponding guanidine (Scheme 4).xvi To examine this possibility, N-Cbz-protected 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-ol (14) was prepared in high yield from S-methylated thiohydantoin (3) by sequential N-alkoxyacylation (CbzCl, NaH) and reduction (NaBH4, CH3OH, 0 °C).xvii,xviii The products obtained upon subsequent aminolysis of pseudothiourea (14) proved to be remarkably solvent dependant. For example, treatment of 14 with methanolic ammonia at 80 °C in a sealed tube for 22 h led exclusively to cleavage of the carbamate, followed by rearrangement of the resulting hydroxyimidazoline to 2-methylthio-4,5,6,7-tetrahydrobenzimidazole. However, in non-protic solvents (e.g., acetonitrile, N,N-dimethylformamide, or tetrahydrofuran) carbamate cleavage was suppressed, allowing aminolysis to proceed in good yield at 80 °C to generate N-Cbz-protected 2-amino-1,3-diazaspiro[4.4]non-1-en-4-ol (15). Furthermore, in contrast to the results observed with hemiaminal (8), only a limited amount of the C4-amino analog of 15 was detected during this reaction, indicating limited ammonia exchange into the hemiaminal under these conditions. xix Efficient access to guanidine hemiaminal (15) set the stage for a final deprotection reaction: hydrogenolytic cleavage of the N-Cbz protective group of 15 proved straightforward (10% Pd/C, 1 atm H2, AcOH, THF), delivering 2-amino-1,3-diazaspiro[4.4]non-1-en-4-ol (2) in high yield (73%).
Scheme 4.
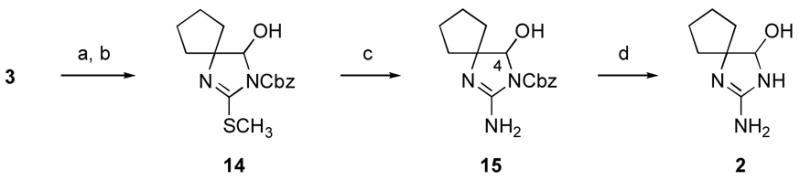
Reagents and conditions: (a) CbzCl, NaH, THF, 0 °C (95%); (b) NaBH4, CH3OH, 0 °C (88%); (c) NH3, THF, 80 °C, sealed tube (68%); (d) 10% Pd/C, H2 (1 atm), THF, AcOH, 23 °C (73%).
In this note, we have reported the identification of a method for the conversion of S-methyl thiohydantoin (3) to the corresponding guanidine hemiaminal (2), establishing a potential method for the generation of the guanidine hemiaminal portion of palau’amine (1) from a thiohydantoin precursor. These studies have additionally identified several key features of the chemistry of 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-ols, including: (1) the influence of N-protecting groups on the position of the tautomeric equilibrium between ring-open and ring-closed forms, (2) the lability of these compounds and their corresponding ether derivatives (e.g., 8) toward acid- and base-mediated exchange with external nucleophiles at C4 and toward rearrangement to 2-methylthio-4,5,6,7-tetrahydrobenzimidazole, and (3) the role of electron-withdrawing N-protective groups in facilitating the aminolysis of pseudothiourea hemiaminals. This knowledge is currently being applied in efforts toward the total synthesis of palau’amine (1).
EXPERIMENTAL
Reactions were performed in oven-dried glassware fitted with rubber septa under a positive pressure of argon. Dichloromethane and tetrahydrofuran were dried by passage through a bed of activated alumina.xx Commercial reagents were used as received. Organic solutions were concentrated by rotary evaporation (1 Torr, 23 °C) unless otherwise noted. Thin-layer chromatography was performed on Merck 60 F254 pre-coated silica gel plates, which were visualized by exposure to ultraviolet light (254 nm) or stained by submersion in aqueous ceric ammonium molybdate solution or ethanolic phosphomolybdic acid solution followed by brief heating on a hot plate (~200 °C, 10–15 s). Flash chromatography was performed as described by Stillxxi (silica gel: 60 Å, 230–400 mesh, Merck KGA). 1H NMR spectra were recorded at 500 or 600 MHz and 13C NMR spectra at 125 MHz or 150 MHz with Brucker Avance spectrometers. Infrared spectra were recorded using an ASI ReactIR™ 1000 spectrometer. Mass spectra were measured with a Micromass LCT spectrometer. Melting points were determined using a Thomas Hoover Unimelt apparatus and are uncorrected.
2-Thioxo-1,3-diazaspiro[4.4]nonan-4-one
An ice-cooled solution of AcCl (3.0 mL, 42 mmol, 1.4 equiv) and MeOH (40 mL) was added to 1-aminocyclopentanecarboxylic acid (4.0 g, 31 mmol, 1 equiv), and the resulting solution was heated at reflux for 2.5 h. Half of the solvent was then removed in vacuo, and Et2O (125 mL) was added to the remaining solution, precipitating methyl 1-aminocyclopentanecarboxylate hydrochloride, which was collected by filtration and dried in vacuo. CH2Cl2 (250 mL), Et3N (33 mL, 240 mmol, 7.7 equiv), and benzoylisothiocyanate (5.9 mL, 36 mmol, 1.2 equiv) were sequentially added to the dry solid, and the resulting solution was stirred at 23 °C for 4 h. The reaction solution was then washed with a 1:1 mixture of saturated aqueous NaHCO3 solution–brine (200 mL). The organic layer was isolated, and the aqueous layer was extracted with CH2Cl2 (2×100 mL). The combined organic fractions were dried over MgSO4 and concentrated in vacuo, affording methyl 1-(3-benzoylthioureido)cyclopentanecarboxylate (5) as a yellow-orange oil. MeOH (250 mL) and K2CO3 (1.5 g, 11 mmol, 0.35 equiv) were subsequently added to 5, and the resulting solution was stirred at 23 °C for 15 h. The reaction solution was then concentrated to 40 mL final volume and partitioned between AcOEt (100 mL) and saturated aqueous NaHCO3 solution (50 mL). The organic layer was isolated, and the aqueous layer was extracted with AcOEt (3×100 mL). The combined extracts were dried over Na2SO4 and concentrated in vacuo, affording a yellow solid. Recrystallization of the solid from hot CH2Cl2 afforded 2-thioxo-1,3-diazaspiro[4.4]nonan-4-one as a colorless solid (2.85 g, 54%, 3 steps):xxii mp 190–193 °C; 1H NMR (500 MHz, CDCl3) δ 8.52 (br s, 1H, CONH), 7.77 (br s, 1H, CSNH), 2.20 (m, 2H, CCH2), 1.94 (m, 4H, CCH2CH2), 1.83 (m, 2H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 180.4, 104.8, 73.0, 37.7, 25.4; FTIR (film) 3301, 3098, 1734, 1518, 1406, 1319, 1269, 1235, 1196, 1148, 1069, 1005, 948, 761 cm− (CI) m/z calcd for C7H10N2OS (M)+: 170.0514, found: 170.0515.
1,3-Diaza-2-(methylthio)spiro[4.4]non-1-en-4-one (3)
Aqueous NaOH (1.0 M, 16.5 mL, 16.5 mmol, 1 equiv) and MeI (1.6 mL, 26 mmol, 1.6 equiv) were sequentially added to a solution of 2-thioxo-1,3-diazaspiro[4.4]nonan-4-one (2.80 g, 16.5 mmol, 1 equiv) in MeOH (164 mL), and the resulting solution was stirred at 23 °C for 18 h. Brine (25 mL) was then added, and the resulting mixture was concentrated in vacuo to a volume of 35 mL. The residual solution was extracted with CHCl3 (6×60 mL), and the combined extracts were dried over Na2SO4 and concentrated in vacuo. Chromatographic purification of the residue (60% AcOEt–hexanes→100% AcOEt) furnished diazaspirononenone (3) as a colorless solid (2.49 g, 82%): mp 129–130 °C; 1H NMR (600 MHz, CDCl3) δ 9.20 (br s, 1H, CONH), 2.60 (s, 3H, SCH3), 1.96 (m, 4H, CCH2), 1.89 (m, 2H, CCH2CH2), 1.81 (m, 2H, CCH2CH2); 13C NMR (CDCl3, 125 MHz) δ 187.6, 156.8, 78.9, 37.7, 26.2, 12.7; FTIR (film) 3032, 2950, 2872, 1709, 1499, 1443, 1414, 1340, 1277, 1159, 1073, 1022, 986 cm−1; HRMS (CI) m/z calcd for C8H12N2OS (M)+: 184.0670, found: 184.0678.
9-Fluorenylmethyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one-3-carboxylate
NaH (60% w/w in mineral oil, 163 mg, 4.08 mmol, 3.0 equiv) was added in one portion to a solution of diazaspirononenone (3) (250 mg, 1.36 mmol, 1 equiv) in THF (27 mL) at 0 °C. The resulting suspension was stirred at 0 °C for 10 min, then 9-fluorenylmethyl chloroformate (369 mg, 1.43 mmol, 1.05 equiv) was added, and the resulting solution was stirred at 0 °C for 5 min. Saturated aqueous NH4Cl solution (10 mL) was carefully added to the reaction solution, and the resulting biphasic mixture was partitioned between AcOEt (90 mL) and half-saturated aqueous NH4Cl solution (10 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (2×30 mL). The combined organic extracts were dried over Na2SO4 and then concentrated in vacuo. Chromatographic purification of the residue (15%→25% AcOEt–hexanes) furnished 9-fluorenylmethyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one-3-carboxylate as a colorless solid (507 mg, 92%): mp 135–136 °C; 1H NMR (500 MHz, CDCl3) δ 7.82 (d, 2H, J = 7.4 Hz, ArH), 7.79 (d, 2H, J = 7.5 Hz, ArH), 7.44 (t, 2H, J = 7.4 Hz, ArH), 7.37 (dt, 2H, J = 1.1, 7.5 Hz, ArH), 4.58 (d, 2H, J = 7.5 Hz, CO2CH2), 4.38 (t, 1H, J = 7.5 Hz, CO2CH2CH), 2.45 (s, 3H, SCH3), 2.15–2.07 (m, 2H, CCH2), 2.07–1.94 (m, 4H, CH2CH2), 1.94–1.85 (m, 2H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 181.0, 156.8, 149.8, 143.3, 141.4, 128.1, 127.4, 125.6, 120.1, 78.4, 70.0, 46.5, 38.7, 26.1, 14.2; FTIR (film) 3066, 3043, 2960, 2873, 1777, 1736, 1574, 1452, 1383, 1298, 1236, 1172, 1118, 1023, 1003, 758, 741 cm−1; HRMS (ESI) m/z calcd for C23H22N2NaO3S (M+Na)+: 429.1249, found: 429.1248.
9-Fluorenylmethyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-ol-3-carboxylate (6)
9-Fluorenylmethyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one-3-carboxylate (224 mg, 551 μmol, 1 equiv) was dissolved in MeOH (30 mL) with gentle heating, and the resulting solution was cooled to 0 °C. NaBH4 (416 mg, 11.0 mmol, 20 equiv) was added in one portion, and the resulting solution was stirred at 0 °C for 10 min. AcOEt (10 mL), saturated aqueous NH4Cl solution (8 mL), and water (2 mL) were sequentially added to the reaction solution, and the resulting biphasic mixture was allowed to warm to 23 °C. The mixture was then partitioned between AcOEt (150 mL) and half-saturated aqueous NH4Cl solution (10 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (2×50 mL). The organic extracts were combined and washed with brine (10 mL), then dried over Na2SO4 and concentrated in vacuo. Chromatographic purification of the residue (25% AcOEt–hexanes) gave N-Fmoc diazaspirononenol (6) as a colorless solid (194 mg, 86%): mp 129–131 °C; 1H NMR (500 MHz, CDCl3) δ 7.79 (dm, 2H, J = 7.5 Hz, ArH), 7.63 (d, 2H, J = 7.4 Hz, ArH), 7.43 (dd, 2H, J = 2.3, 7.4 Hz, ArH), 7.36 (ddt, 2H, J = 1.1, 2.3, 7.5 Hz, ArH), 5.01 (br s, 1H, NCHOH), 4.67 (d, 2H, J = 5.2 Hz, CO2CH2), 4.34 (t, 1H, J = 5.4 Hz, CO2CH2CH), 2.38 (s, 3H, SCH3), 2.07 (m, 1H, CCH2), 1.83 (m, 2H, CCH2), 1.69 (m, 1H, CCH2), 1.64 (m, 1H, CCH2CH2), 1.57 (m, 2H, CCH2CH2), 1.49 (m, 1H, CCH2CH2); 13C NMR (125 MHz, CDCl3; minor rotamer denoted with *) δ 154.5, 151.5, 143.7*, 143.5, 141.5, 141.5*, 128.3*, 128.2, 127.6*, 127.6, 125.0*, 124.9, 120.3*, 120.3, 88.7, 80.3, 68.0, 47.1, 39.3, 31.4, 24.2, 23.7, 14.9; FTIR (film) 3459, 3067, 3040, 2955, 2874, 1724, 1570, 1451, 1396, 1351, 1301, 1245, 1146, 1088, 1024, 758, 741 cm−1; HRMS (ESI) m/z calcd for C23H24N2NaO3S (M+Na)+: 431.1405, found: 431.1415.
1-(1-Formylcyclopentyl)-2-methylisothiourea (7)
A solution of diazaspirononenol (6) (59 mg, 150 μmol, 1 equiv) and 1,8-diazabicyclo[5.4.0]undec-7-ene (24 μL 160 μmol, 1.1 equiv) in CH2Cl2 (7.25 mL) was stirred at 23 °C for 15 min, then directly chromatographically separated (30% AcOEt–hexanes + 2% Et3N→70% AcOEt–hexanes + 2% Et3N), furnishing 2-methylisothiourea (7) as a clear oil (23 mg, 85%): 1H NMR (500 MHz, CDCl3) δ 8.61 (s, 1H, CHO), 2.58 (s, 3H, SCH3), 2.14–2.05 (m, 2H, CCH2), 1.99–1.87 (m, 4H, CCH2CH2), 1.83–1.77 (m, 2H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 191.1, 173.1, 93.5, 32.9, 26.4, 14.5; FTIR (film) 3445, 3078, 2964, 2931, 2872, 1574, 1514, 1437, 1316, 1292, 1215, 1122, 1085, 954, 907, 695 cm−1; HRMS (ESI) m/z calcd for C8H15N2OS (M+H)+: 187.0905, found: 187.0903.
1,3-Diaza-2-methylthio-4-(phenylmethoxy)spiro[4.4]non-1-ene (8)
10-Camphorsulfonic acid (16 mg, 69 μmol, 0.8 equiv) was added in one portion to a suspension of 2-methylisothiourea (7) (16 mg, 86.0 μmol, 1 equiv), benzyl alcohol (13 μL, 130 μmol, 1.5 equiv), and 4Å molecular sieves (4 mg) in CH2Cl2 (2.1 mL) at 23 °C. After 100 min, the resulting solution was partitioned between CH2Cl2 (8 mL) and saturated aqueous NaHCO3 solution (4 mL). The organic layer was separated, and the aqueous layer was extracted with CH2Cl2 (8 mL). The organic extracts were combined, dried over Na2SO4, and concentrated in vacuo. Chromatographic purification of the residue (40% AcOEt–hexanes + 2% Et3N) furnished diazaspirononene (8) as a clear oil (16 mg, 55%): 1H NMR (500 MHz, CDCl3) δ 7.38 (m, 2H, ArH), 7.35 (d, 2H, J = 4.2 Hz, ArH), 7.31 (m, 1H, ArH), 4.98 (br s, 1H, NH), 4.71 (s, 2H, OCH2), 4.56 (m, 1H, NCHO), 2.48 (br s, 3H, SCH3), 2.34–1.98 (m, 2H, CCH2), 1.98–1.73 (m, 2H, CCH2), 1.73–1.43 (m, 4H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 141.2, 128.8, 127.8, 127.7, 127.2, 94.9, 70.1, 65.5, 38.9, 31.4, 24.7, 24.2, 13.8; FTIR (film) 3200, 3066, 3031, 2954, 2931, 2871, 1559, 1457, 1208, 1067, 1025, 735, 699 cm−1; HRMS (ESI) m/z calcd for C15H21N2OS (M+H)+: 277.1375, found: 277.1368.
1,3-Diaza-2-methylthio-3-[2-(trimethylsilyl)ethylsulfonyl]spiro[4.4]non-1-en-4-one (9)
NaH (60% w/w in mineral oil; 65 mg, 1.6 mmol, 3.0 equiv) was added in one portion to a solution of diazaspirononenone (3) (100 mg, 540 μmol, 1 equiv) in THF (10 mL) at 0 °C, and the resulting suspension was stirred for 5 min at 0 °C. 2-(Trimethylsilyl)ethylsulfonyl chloride (98 μL, 570 μmol, 1.0 equiv) was then added, and the resulting solution was stirred at 0 °C. After 5 min, saturated aqueous NH4Cl solution (5 mL) was carefully added, and the resulting biphasic mixture was partitioned between AcOEt (40 mL) and water (5 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (40 mL). The combined organic extracts were dried over Na2SO4 and concentrated in vacuo. Chromatographic purification of the residue (10%→25% AcOEt–hexanes) afforded N-SES-diazaspirononenone (9) as a colorless solid (124 mg, 66%): mp 72–75 °C; 1H NMR (600 MHz, CDCl3) δ 3.40 (m, 2H, SO2CH2), 2.45 (s, 3H, SCH3), 1.98 (m, 4H, CCH2), 1.92 (m, 2H, CCH2CH2), 1.84 (m, 2H, CCH2CH2), 1.00 (m, 2H, CH2TMS), 0.06 (s, 9H, Si(CH3)3); 13C NMR (150 MHz, CDCl3) δ 182.7, 155.2, 78.1, 50.8, 38.6, 26.0, 14.1, 10.0, –1.8; FTIR (film) 2925, 2896, 2873, 1760, 1569, 1370, 1252, 1196, 1154, 1113, 1073, 861, 843 cm−1; HRMS (ESI) m/z calcd for C13H24N2NaO3S2Si (M+Na)+: 371.0895, found: 371.0899.
2-[2-(Trimethylsilyl)ethylsulfonylamino]-1,3-diazaspiro[4.4]non-1-en-4-one (10)
Ammonia was bubbled through a solution of N-SES-diazaspirononenone (9) (124 mg, 357 μmol, 1 equiv) in MeOH (18 mL) at 0 °C in a sealable high-pressure vessel. After 3 min, the vessel was sealed, and the resulting solution was stirred at 23 °C for 1.5 h. The vessel was then cooled to 0 °C and opened, allowing excess ammonia to evaporate upon subsequent warming to 23 °C. Concentration of the reaction solution in vacuo furnished sulfonamide (10) as a colorless solid (104 mg, 92%). Recrystallization from a layered 1:1 AcOEt–Et2O mixture (−20 °C) afforded colorless plates suitable for X-Ray diffraction: mp 170–173 °C; 1H NMR (500 MHz, CDCl3) δ 8.60 (br s, 1H, NH), 7.59 (br s, 1H, NH), 3.04 (m, 2H, SO2CH2), 2.19 (m, 2H, CCH2), 1.93 (m, 4H, CCH2CH2), 1.84 (m, 2H, CCH2CH2), 1.05 (m, 2H, CH2TMS), 0.07 (s, 9H, Si(CH3)3); 13C NMR (125 MHz, CDCl3) δ 176.8, 155.2, 77.4, 51.1, 38.0, 25.5, 10.3, –1.7; FTIR (film) 3296, 2954, 1764, 1638, 1443, 1295, 1250, 1167, 1131, 855, 758, 702 cm−1; HRMS (ESI) m/z calcd for C12H23N3NaO3SSi (M+Na)+: 340.1127, found: 340.1130.
2-[2-(Trimethylsilyl)ethylsulfonylamino]-1,3-diazaspiro[4.4]non-1-en-4-ol (11)
Diisobutylaluminum hydride (1.0M in hexane, 100 μL, 100 μmol, 7.5 equiv) was added to a solution of sulfonamide (10) (4 mg, 13 μmol, 1 equiv) in THF (1.0 mL) at −78 °C. After 10 min, the resulting solution was transferred to a 0 °C bath for 1 h. Saturated aqueous NH4Cl solution (1 mL) and AcOEt (1 mL) were sequentially added to the reaction solution at 0 °C, and the resulting biphasic mixture was allowed to warm to 23 °C. The mixture was then extracted with AcOEt (10 mL), and the organic layer was separated and sequentially washed with water (2×20 mL) and brine (2 mL). All aqueous layers were then combined and extracted with AcOEt (10 mL). All organic extracts were then combined, dried over Na2SO4, and concentrated in vacuo. Chromatographic purification of the residue (3% MeOH–CH2Cl2) provided N-SES diazaspirononenol (11) as a colorless solid (2 mg, 47%): mp 146–149 °C; 1H NMR (500 MHz, CDCl3) δ 7.45 (s, 1H, SO2NH), 6.65 (s, 1H, CHNH), 4.98 (d, 1H, J = 4.8 Hz, CHOH), 4.15 (d, 1H, J = 5.6 Hz, OH), 2.94 (ddd, 2H, J = 3.4, 6.6, 14.6 Hz, SO2CH2), 2.28 (m, 1H, CCH2), 1.87–1.64 (m, 7H, CCH2CH2), 1.03 (ddd, 2H, J = 4.3, 7.2, 15.0 Hz, CH2TMS), 0.05 (s, 9H, Si(CH3)3); 13C NMR (125 MHz, CDCl3) δ 158.6, 84.4, 72.0, 50.9, 38.6, 30.7, 23.5, 23.4, 10.5, –1.7; FTIR (film) 3338, 2955, 2875, 1605, 1497, 1247, 1131, 1077, 1023, 840, 755, 700 cm−1; HRMS (ESI) m/z calcd for C12H25N3NaO3SSi (M+Na)+: 342.1284, found: 342.1283.
1,3-Diaza-2-methylthio-3-(4-methoxybenzenesulfonyl)spiro[4.4]non-1-en-4-one (12)
NaH (27 mg, 680 μmol, 2.5 equiv) was added in one portion to an ice-cooled solution of diazaspirononenone (3) (50.0 mg, 271 μmol, 1 equiv) in THF (10.0 mL). After 10 min, 4-methoxybenzenesulfonyl chloride (140 mg, 680 μmol, 2.5 equiv) was added, and the resulting solution was stirred at 0 °C for 10 min. Saturated aqueous NH4Cl solution (1 mL) was then slowly added at 0 °C to quench excess NaH, and the resulting mixture was partitioned between AcOEt (10 mL) and half-saturated aqueous NH4Cl solution (3 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (2×10 mL). All organic extracts were then combined, dried over Na2SO4, and concentrated in vacuo. Chromatographic purification of the residue (20%→30% AcOEt–hexanes) provided diazaspirononenone (12) as a colorless solid (86 mg, 89%): mp 90–92 °C; 1H NMR (500 MHz, CDCl3) δ 8.03 (d, 2H, J = 8.9 Hz, SO2CCH), 7.01 (d, 2H, J = 8.9 Hz, CHCOCH3), 3.88 (s, 3H, OCH3), 2.44 (s, 3H, SCH3), 1.90–1.81 (m, 6H, CCH2CH2), 1.74–1.71 (m, 2H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 181.5, 164.7, 155.1, 130.8, 129.1, 114.6, 77.8, 56.0, 38.3, 25.9, 14.1; FTIR (film) 3105, 2954, 2873, 1769, 1595, 1567, 1498, 1378, 1266, 1199, 1164, 1091, 1022, 972, 835, 805, 674 cm−1; HRMS (ESI) m/z calcd for C15H18N2NaO4S2 (M+Na)+: 377.0606, found: 377.0611.
1-[1-(Hydroxymethyl)cyclopentyl]-3-(4-methoxybenzenesulfonyl)-2-methylisothiourea (13)
NaBH4 (2.0 mg, 53 μmol, 1.3 equiv) was added in one portion to a solution of diazaspirononenone (12) (15 mg, 42 μmol, 1.0 equiv) in MeOH (0.5 mL) at 0 °C. After 1 h, AcOEt (2 mL) was added to the reaction solution, followed by half-saturated aqueous NH4Cl solution (2 mL). The resulting biphasic mixture was allowed to warm to 23 °C and was then partitioned between AcOEt (15 mL) and water (2 mL). The organic layer was separated, and the aqueous layer was extracted with AcOEt (2×10 mL). The organic extracts were combined, dried over Na2SO4, and concentrated in vacuo. Chromatographic purification of the residue (70% AcOEt–hexanes) provided N-SES pseudothiourea (13) as a clear oil (15 mg, 100%): 1H NMR (600 MHz, CDCl3) δ 8.37 (br s, 1H, NH), 7.86 (dt, 2H, J = 3.0, 8.4 Hz, SO2CCH), 6.95 (dt, 2H, J = 3.0, 9.0 Hz, SO2CCHCH), 3.86 (s, 3H, OCH3), 3.72 (s, 2H, CH2OH), 2.39 (s, 3H, SCH3), 1.96 (m, 2H, CCH2), 1.85 (m, 2H, CCH2), 1.70 (m, 2H, CCH2CH2), 1.63 (m, 2H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 162.6(2), 134.5, 128.6, 114.1, 68.5, 65.7, 55.8, 36.1, 24.5, 15.1; FTIR (film) 3396, 2958, 2873, 1567, 1499, 1256, 1140, 1088, 834, 805, 704 cm−1; HRMS (ESI) m/z calcd for C15H22N2NaO4S2 (M+Na)+: 381.0919, found: 381.0919.
Benzyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one-3-carboxylate
NaH (60% w/w in mineral oil, 270 mg, 6.8 mmol, 2.5 equiv) was added in one portion to a solution of 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one (3) (500 mg, 2.70 mmol, 1.0 equiv) in THF (35 mL) at 0 °C. After 10 min, benzyl chloroformate (410 μL, 2.9 mmol, 1.1 equiv) was added, and the resulting solution was stirred at 0 °C for 5 min. Saturated aqueous NH4Cl solution (8 mL) was then added, and the resulting biphasic mixture was partitioned between AcOEt (120 mL) and half-saturated aqueous NH4Cl solution (20 mL). The organic layer was isolated, and the aqueous layer was extracted with AcOEt (2×100 mL). All organic extracts were then combined, dried over Na2SO4, and concentrated in vacuo. Chromatographic purification of the residue (20%→30% AcOEt–hexanes) provided benzyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one-3-carboxylate as a colorless solid (819 mg, 95%). Recrystallization from Et2O (23 °C) afforded colorless spars suitable for X-Ray diffraction: mp 111–112 °C; 1H NMR (500 MHz, CDCl3) δ 7.49 (d, 2H, J = 6.9 Hz, ArH), 7.41–7.34 (m, 3H, ArH), 5.38 (s, 2H, OCH2Ph), 2.43 (s, 3H, SCH3), 2.05–1.93 (m, 4H, CCH2), 2.93–1.86 (m, 2H, CCH2CH2), 1.84–1.76 (m, 2H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 181.1, 156.7, 149.6, 134.5, 128.9(2), 128.5, 78.4, 69.3, 38.7, 26.1, 14.3; FTIR (film) 3064, 3035, 2958, 2873, 1775, 1737, 1574, 1457, 1382, 1297, 1229, 1171, 1119, 1000, 766, 735, 697 cm−1; HRMS (ESI) m/z calcd for C16H18N2NaO3S (M+Na)+: 341.0936, found: 341.0923.
Benzyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-ol-3-carboxylate (14)
NaBH4 (230 mg, 6.2 mmol, 10 equiv) was added in one portion to a solution of benzyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one-3-carboxylate (197 mg, 619 μmol, 1.0 equiv) in MeOH (8 mL) at 0 °C. The resulting suspension was stirred at 0 °C for 5 min and was then diluted with AcOEt (10 mL). Saturated aqueous NH4Cl solution (8 mL) and water (2 mL) were sequentially added, and the resulting biphasic mixture was allowed to warm to 23 °C. The mixture was then partitioned between AcOEt (30 mL) and water (2 mL). The organic layer was isolated, and the aqueous layer was extracted with AcOEt (2×30 mL). All organic extracts were then combined, dried over Na2SO4, and concentrated in vacuo. Chromatographic purification of the residue (30%→40% AcOEt–hexanes) furnished 14 as a colorless solid (176 mg, 88%): mp 138–139 °C; 1H NMR (500 MHz, CDCl3) δ 7.42–7.34 (m, 5H, ArH), 5.38 (s, 1H, NCHOH), 5.26 (dd, 2H, J = 12.2, 16.7 Hz, CO2CH2), 2.41 (s, 3H, SCH3), 2.15 (m, 1H, CCH2), 1.86 (m, 2H, CCH2), 1.77 (m, 1H, CCH2), 1.60 (m, 4H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 154.4, 151.6, 135.2, 128.9, 128.8, 128.6, 88.7, 80.4, 68.4, 39.2, 31.4, 24.2, 23.7, 15.0; FTIR (film) 3438, 3066, 3035, 2960, 2871, 1719, 1573, 1391, 1351, 1301, 1144, 1082, 1023, 766, 699 cm−1; HRMS (ESI) m/z calcd for C16H21N2O3S (M+H)+: 321.1273, found: 321.1270.
Benzyl 2-amino-1,3-diazaspiro[4.4]non-1-en-4-ol-3-carboxylate (15)
Ammonia (7.0 mL, 280 mmol, 1000 equiv) was condensed into a solution of 14 (90 mg, 280 μmol, 1.0 equiv) in THF (3 mL) in a glass-lined stainless-steel high-pressure vessel at −78 °C. The vessel was then sealed and heated at 80 °C for 1 d. After cooling to −78 °C, the vessel was opened, excess ammonia was allowed to evaporate with warming to 23 °C, and the resulting solution was concentrated in vacuo. Chromatographic purification of the residue (20→40→60% THF–CH2Cl2)xxiii yielded 15 as a colorless solid (55 mg, 68%): mp 154–156 °C; 1H NMR (600 MHz, CDCl3) δ 7.90 (br s, 1H, NH2), 7.67 (br s, 1H, NH2), 7.39 (d, 2H, J = 7.1 Hz, ArH), 7.35 (t, 2H, J = 7.1 Hz, ArH), 7.31 (t, 1H, J = 7.1 Hz, ArH), 5.09 (dd, 2H, J = 12.6, 15.9 Hz, CO2CH2), 4.84 (s, 1H, NCHOH), 2.16 (m, 1H, CCH2), 1.79 (m, 1H, CCH2), 1.72–1.64 (m, 2H, CCH2), 1.64–1.53 (m, 4H, CCH2CH2); 13C NMR (125 MHz, CD3OD) δ 164.9, 163.8, 138.9, 129.5, 128.9(2), 85.8, 72.2, 67.6, 39.4, 31.3, 24.4, 24.2; FTIR (film) 3384, 2954, 2875, 1648, 1615, 1497, 1455, 1383, 1328, 1277, 1237, 1090, 1077, 1028, 801, 743, 699 cm−1; HRMS (ESI) m/z calcd for C15H20N3O3 (M+H)+: 290.1505, found: 290.1498.
2-Amino-1,3-diazaspiro[4.4]non-1-en-4-ol, acetic acid salt (2)
10% Palladium on activated carbon (50% w/w water; 54.0 mg, 25.4 μmol, 0.15 equiv) was added to an argon-sparged solution of 15 (49 mg, 170 μmol, 1.0 equiv) and AcOH (29 μL, 510 μmol, 3.0 equiv) in THF (5.6 mL) at 23 °C. The flask containing the resulting suspension was alternately evacuated (1 torr) and refilled with hydrogen (1 atm) (3×), and the resulting suspension was then stirred under hydrogen atmosphere (1 atm) at 23 °C for 1 h. Filtration of the resulting suspension through a Celite pad (subsequently washed with 50 mL of THF), followed by concentration of the filtrate, afforded 2 as a waxy, colorless solid (33 mg, 73%): 1H NMR (500 MHz, CDCl3) δ 8.22 (br s, 1H, NH), 7.92 (br s, 2H, NH2), 6.51 (br s, 2H, CHOH, CO2H), 4.95 (s, 1H, NCHOH), 2.21 (m, 1H, CCH2), 2.03 (s, 3H, COCH3), 1.81–1.77 (m, 3H, CCH2), 1.77–1.64 (m, 4H, CCH2CH2); 13C NMR (125 MHz, CDCl3) δ 179.0, 158.2, 86.1, 73.3, 38.6, 30.6, 23.5(2), 23.5; FTIR (film) 3174, 2960, 2919, 2850, 1690, 1563, 1409, 1262, 1086, 1021, 799 cm−1; HRMS (ESI) m/z calcd for C7H14N3O (M+H)+: 156.1137, found: 156.1136.
Acknowledgments
Support from the National Heart, Lung, and Blood Institute (HL-25854) and NIH NRSA postdoctoral fellowship support to B.A.L. (GM-71132) are gratefully acknowledged. We also thank Bruce N. Rogers, Fang-Tsao Hong, and Patrick W. Papa for preliminary experiments in this area. Joseph Ziller is acknowledged for assistance with X-Ray crystallography.
Footnotes
In recognition of his many contributions to organic synthesis and heterocyclic chemistry, this contribution is dedicated to a close friend, Steven Weinreb, on the occasion of his 65th birthday.
REFERENCES (AND NOTES)
- i.(a) Kinnel RB, Gehrken HP, Scheuer PJ. J Am Chem Soc. 1993;115:3376. [Google Scholar]; (b) Kinnel RB, Gehrken HP, Swali R, Skoropowski G, Scheuer PJ. J Org Chem. 1998;63:3281. [Google Scholar]
- ii.Jacquot DEN, Lindel T. Curr Org Chem. 2005;9:1551. [Google Scholar]
- iii.(a) McClelland RA, Panicucci R. J Am Chem Soc. 1985;107:1762. [Google Scholar]; (b) McClelland RA, Panicucci R, Rauth AM. J Am Chem Soc. 1987;109:4308. [Google Scholar]
- iv.Schwarz F-TD, Altreiter J. Ger Offen. 1993 December 23; DE 4220069. [Google Scholar]
- v.(a) Olofson A, Yakushijin K, Horne DA. J Org Chem. 1998;63:1248. doi: 10.1021/jo972295d. [DOI] [PubMed] [Google Scholar]; (b) Wiese KJ, Yakushijin K, Horne DA. Tetrahedron Lett. 2002;43:5135. These methods afford O-alkyl guanidine hemiaminals. [Google Scholar]
- vi.An alternative preparation of 2-thioxo-1,3-diazaspiro[4.4]nonan-4-one has previously been reported: Carrington HC, Vasey CH, Waring WS. J Chem Soc. 1953:3105. This compound is also available commercially from Chemstep.
- vii.The position of the N-acyloxy protective group in 6 (and the N-sulfonyl protective groups in 9 and 12) is assigned by analogy with benzyl 1,3-diaza-2-(methylthio)spiro[4.4]non-1-en-4-one-3-carboxylate, whose structure was established by X-Ray crystallography.
- viii.Exposure of 8 to ethanolic ammonium chloride (75 °C, 19 h) exclusively led to the generation of the corresponding O-ethyl hemiaminal.
- ix.Prepared by the oxidation of pseudothiourea (8) with dimethyldioxirane (DMDO). Attempts to convert 8 to the corresponding sulfone using 3-chloroperoxybenzoic acid afforded only recovered starting material.
- x.Cliffe IA. In: Comprehensive Organic Functional Group Transformations. Katritzky AR, Meth–Cohn O, Rees CW, editors. Vol. 6. Elsevier; Oxford: 1995. pp. 639–75. [Google Scholar]
- xi.(a) Weinreb SM, Demko DM, Lessen TA, Demers JP. Tetrahedron Lett. 1986;27:2099. [Google Scholar]; (b) Weinreb SM, Chase CE, Wipf P, Venkatraman S. Org Synth. 1988;75:161. [Google Scholar]
- xii.Aminolysis of 9 initially generates the ring-opened primary amide, which subsequently cyclizes to N-SES glycocyamidine (10). The structure of 10 has been confirmed by X-Ray crystallography.
- xiii.The deoxoguanidine analog of 10 is the major by-product of this reaction.
- xiv.The resistance of glycocyamidines bearing acidic hydrogens to metal hydride-mediated reduction has previously been reported: Stearns JF, Rapoport H. J Org Chem. 1977;42:3608.
- xv.Direct aminolysis of S-methylated thiohydantoin (3) (NH3, EtOH, 80 °C, 24 h) afforded a related, unprotected glycocyamidine; however, subsequent reduction of this product to the corresponding guanidine hemiaminal (PtO2/H2) proved unsuccessful (cf., Wegner MM, Rapoport H. J Org Chem. 1978;43:3840.Cbz-protection of this related glycocyamidine facilitated reduction to the corresponding guanidine hemiaminal (NaBH4); however, low yields for this process and complicated 1H NMR spectra of the protected intermediates (Cbz rotamers) rendered this route to guanidine hemiaminal (2) unattractive.
- xvi.The use of N-Fmoc pseudothiourea (6) in such a strategy was precluded by the base-lability of the N-Fmoc group.
- xvii.A large excess of sodium borohydride was employed in the reduction step to favor reduction over competitive methoxide-mediated Cbz cleavage and imide solvolysis processes.
- xviii.As before, no over-reduction product was observed in this two-step process. The only by-product of this process was S-methylated thiohydantoin (3), which arose from cleavage of the Cbz group from N-Cbz 3 prior to reduction.
- xix.The C4-amino analog of 15 was the sole by-product detected in this reaction.
- xx.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- xxi.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923. [Google Scholar]
- xxii.Alternatively, chromatographic purification of the crude product (35→40→50% AcOEt–hexanes) afforded 2-thioxo-1,3-diazaspiro[4.4]nonan-4-one in 85% yield (3 steps).
- xxiii.Substitution of MeOH for THF as an eluent resulted in partial conversion of 15 to the corresponding O-methyl hemiaminal during chromatography.