

Abstract
Background
The arrhythmogenic effects of hyperthermia have been highlighted in the Brugada Syndrome, but remain largely unexplored in other arrhythmic syndromes. The present study examines the effect of hyperthermia on transmural dispersion of action potential duration (TD-APD), early afterdepolarization (EAD) activity, and Torsade de Pointes (TdP) under long QT conditions.
Methods and Results
Standard and floating glass microelectrodes were used to record action potentials from epicardial, M-cell, and endocardial regions of the arterially-perfused LV wedge, from tissue slices isolated from these regions, as well as from isolated Purkinje fibers. A transmural ECG was simultaneously recorded across the wedge. Under baseline conditions and in the presence of IKs block (chromanol 293B), hyperthermia (39-40°C) abbreviated APD in tissue slices from all three regions. In the presence of IKr block (E-4031), hyperthermia prolonged APD and induced or augmented EADs in M cell and Purkinje preparations at pacing cycle lengths ≥ 800 ms, but abbreviated APD in epicardium and endocardium, resulting in a marked accentuation of TD-APD. Ryanodine prevented the hyperthermia-induced EAD. In perfused wedge preparations, hyperthermia abbreviated APD throughout both in the absence or presence of IKr or IKs block and did not induce EADs or TdP. Combined IKr and IKs block increased TD-APD, and induced EADs (4/12) and spontaneous TdP (3/12) at 36-37°C; hyperthermia (39-40°C) further accentuated TD-APD and facilitated the development of EAD activity (9/12) and TdP (6/12).
Conclusions
Our findings suggest that hyperthermia can be associated with an increased arrhythmic risk when the repolarization reserve of the myocardium is compromised.
Keywords: Arrhythmias, Electrophysiology, Fever, Long QT Syndrome, Triggered activity
INTRODUCTION
Although reduced repolarization reserve of the myocardium is encountered under a number of clinical conditions (congenital and acquired long QT syndromes, heart failure, hypertrophic and dilated cardiomyopathy, hypothyroidism etc.) and hyperthermia (secondary to fever, heat stroke, drugs, or exercise) is a common clinical syndrome, little is known about electrophysiological effects of hyperthermia in hearts with a reduced repolarization reserve. Independently, prolonged repolarization due to reduced repolarization reserve and hyperthermia are known to be capable of inducing or promoting ventricular arrhythmias in man.1-11 A recent clinical study has demonstrated that fever can be a risk factor for the development of life-threatening ventricular arrhythmias in the LQT2 form of congenital long QT syndrome.12 Under normothermic conditions, ventricular arrhythmias associated with prolonged repolarization generally involve the development of early afterdepolarizations (EAD) and augmented spatial dispersion of repolarization.13 The mechanisms responsible for hyperthermia-induced arrhythmias are not well understood in the majority of cases. Nine years ago our group revealed an interesting mechanism by which hyperthermia could unmask or accentuate ST segment elevation and arrhythmogenicity in the Brugada syndrome.14 The aim of the present study was to probe the effects of hyperthermia on transmural dispersion of repolarization (TDR), EAD activity, and the development of spontaneous Torsade de Pointes (TdP) arrhythmias under conditions of prolonged repolarization (IKr and/or IKs blockers) in canine left ventricular isolated tissue, Purkinje fibers, and coronary-perfused wedge preparations.
METHODS
This investigation conforms to the Guide for Care and Use of Laboratory Animals published by the National Institutes of Health (NIH publication No 85-23, Revised 1996) and was approved by the ACUC of the Masonic Medical Research Laboratory.
Isolated tissue slice preparations
Free running Purkinje fiber, epicardial, endocardial, and M cell preparations (strips 1 × 0.5 × 0.1 cm) were isolated from left ventricle (LV) of hearts removed from anesthetized (30 mg/kg sodium pentobarbital) mongrel dogs. The preparations, isolated using a dermatome (Davol Simon Dermatome, Cranston, RI, USA), were placed in a tissue bath (volume 5 ml, flow rate 12 ml/min) and allowed to equilibrate for at least 3 hours while superfused with oxygenated Tyrode’s solution (36.5±0.5°C, pH=7.35) and stimulated at a basic cycle length (BCL) of 500 msec using field or point stimulation (rectangular stimuli 1 - 3- ms duration, 2-3 times diastolic threshold intensity). The composition of the Tyrode’s solution was (in mM): NaCl 129, KCl 4, NaH2PO4 0.9, NaHCO3 20, CaCl2 1.8, MgSO4 0.5, and D-glucose 5.5.
Perfused left ventricular wedge
The methods used for isolation, perfusion, and recording of transmembrane activity from the arterially perfused canine LV wedge preparation, as well as the viability and electrical stability of the preparation, are detailed in previous studies.15, 16 Briefly, transmural wedges with dimensions of approximately 3 × 1.8 × 1.2 cm were dissected from the anterior wall of the canine LV (see above). The wedge preparations were cannulated via a small (diameter ∼100 μm) coronary artery and perfused with cold cardioplegic solution ([K+]0 = 8.5 mM, t0 = 4°C). The total period of time from excision of the heart to cannulation and perfusion of the artery was less than 4 min. Unperfused tissue was carefully removed using a razor blade. The preparations then were placed in a small tissue bath and arterially perfused with Tyrode’s solution of the same composition as tissues. The perfusate was delivered to the artery by a roller pump (Cole Parmer Instrument Co., Niles, IL). Perfusion pressure was monitored with a pressure transducer (World Precision Instruments, Inc., Sarasota, FL) and maintained between 40 and 50 mm Hg by adjustment of the perfusion flow rate. Ventricular wedges were allowed to equilibrate in the tissue bath until electrically stable, usually 1 hour. The preparations were stimulated using bipolar silver electrodes insulated except at the tips and applied to the endocardial surface. A pseudo-electrocardiogram (ECG) was recorded using two electrodes consisting of Ag/AgCl half cells placed in the Tyrode’s solution bathing the preparation, 1.0 to 1.2 cm from the two opposite sides of the atrial or ventricular coronary-perfused preparations.
Action potential recordings
Action potentials were recorded using standard (tissue slices) and floating (wedges) glass microelectrodes filled with 2.7 M KCl (10 to 20 MΩ DC resistance) connected to a high input-impedance amplification system (World Precision Instruments, Sarasota, FL, USA). The signals were displayed on oscilloscopes, amplified, digitized and analyzed (Spike 2, Cambridge Electronic Design, Cambridge, England).
Drugs
A highly selective IKr blocker, E-4031 (gift of Eisai, Co, Ltd, Japan), and sarcoplasmic reticulum (SR) calcium release blocker ryanodine (SIGMA, MO) were dissolved in distilled water to make a 1 mmol/L stock solution. A relatively specific IKs blocker, chromanol 293B (gift of Aventis Pharma Deutschland GmbH, Germany) was dissolved in 100% DMSO to form a stock solution of 10 or 30 mmol/L.
Study protocols
Electrophysiologic and electrocardiographic activity was assessed in the range of temperatures of 36-40°C, first under control conditions and then in the presence of the drug(s), in both cases starting from a steady state at 36-37°C. In the experiments where several levels of temperature were tested (superfused tissues), temperature was increased in a step-wise manner. Each new temperature was kept constant for at least 5 minutes before the start of recording. In some experiments involving M cell preparations, the temperature range was 33-40°C so as to characterize the temperature-dependence of EAD activity over a wider range. The effect of temperature was assessed over a BCL range of 300-5000 ms in tissue and 500 and 2000 ms in wedge preparations. In the wedge preparation, EAD and spontaneous arrhythmias were assessed whenever possible over a BCL range of 500-4000 ms. Transmural dispersion of action potential duration (TD-APD90) was approximated as the difference between the longest and shortest APD90. QT interval was measured as the time from the beginning of the QRS to the intersection of the tangent drawn to the maximum slope of the descending limb of the T wave with the isoelectric line.
Statistics
One-way repeated measure analysis variance (ANOVA) followed by Bonferroni’s test was performed when comparing data obtained at 36°C with those obtained at 38°C and 40°C in superfused tissue preparations. When comparing normal temperature (36-37°C) vs. hyperthermia (39-40°C) in perfused-wedge experiments, where only one temperature step was performed in each given case, paired student’s t-test was used. The differences between no-drug vs. drug conditions at the same temperature were determined using unpaired Student’s t-test.
The authors had full access to the data and take responsibility for its integrity. All authors have read and agree to the manuscript as written.
RESULTS
Isolated tissues
Under baseline conditions, elevation of temperature from 36 to 38-40°C produced an abbreviation of APD in epicardial, M cell, endocardial, and Purkinje fiber preparations in the BCL range of 300 - 5000 ms. Figure 1 shows summary data for a BCL of 2000 ms. Interestingly, even under control conditions hyperthermia accentuated the dispersion of APD among the different isolated tissue preparations, due to a larger abbreviation of APD in epicardial and endocardial, compared to M cell regions (Table 1). At a temperature of 36°C, IKr block with E-4031 (1 μmol/L) produced a significant prolongation of repolarization in all four isolated preparations, more prominently in M cell and Purkinje (Fig.1). In the continued presence of E-4031, a rise in temperature from 36 to 38-40°C led to APD abbreviation in all four ventricular cell types at rapid rates (BCL = 300-500 ms), but to a paradoxical prolongation of APD and induction or accentuation of EAD activity in M cell (10 of 10) and Purkinje fiber (7 of 8) preparations at longer BCLs (≥ 600 ms) (Fig. 1). These opposite effects of hyperthermia on the APD of M cell and epicardial and endocardial tissues at slow rates caused a remarkable exaggeration of TD-APD (Table 1). It is noteworthy that E-4031 produced a significant APD prolongation and was able to induce EAD activity in M cell (in 8/10) and Purkinje fiber (5/8) preparations at 36-37°C (Fig. 2). However, APD prolongation was less pronounced and EAD activity developed only at relatively slow pacing rates (BCLs ≥ 1500 ms) (Fig. 2). At 38-40°C, EADs developed at much shorter BCLs, approaching the physiological range of heart rates (at a BCL as short as 600 ms) (Fig. 2). A similar relationship was observed in Purkinje fibers.
Figure 1.
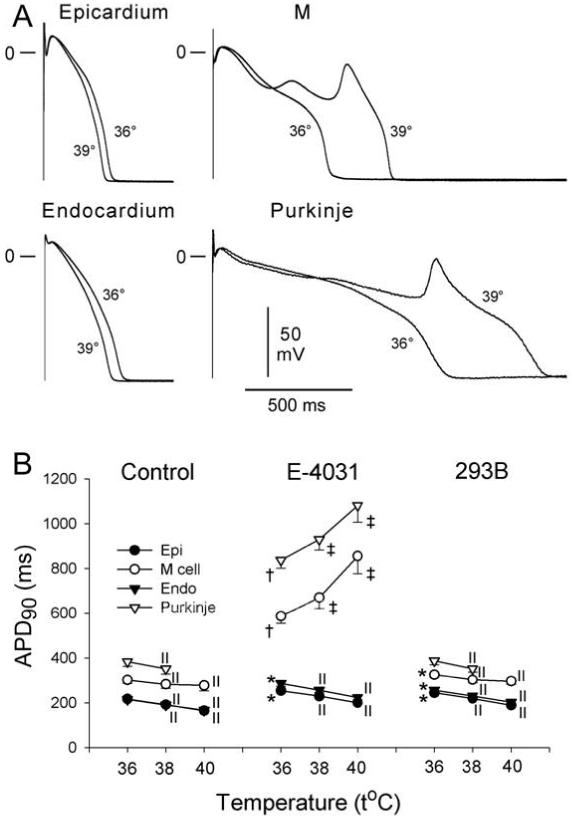
Effect of temperature on action potential duration (APD90) of canine isolated epicardial (Epi), M cell, endocardial (Endo) tissue slices, and Purkinje fiber preparations under baseline conditions (control) and in the presence of E-4031 (1 μmol/L, IKr block) or chromanol 293B (293B, 30 μmol/L, IKs block) (n=8 for each). BCL = 2000 ms.
A: Superimposed action potentials recorded from the four ventricular cell types before and after an increase in ambient temperature of the superfusion solution from 36 to 39°C.
B: Summary data for the effect of hyperthermia on APD90 of Epi, M cell, Endo, and Purkinje preparations under conditions of prolonged repolarization.
Control and chromanol 293B APD data from Purkinje fibers at 40°C are lacking because of the effect of hyperthermia to accelerate spontaneous automaticity. * - p<0.05 and † - p<0.001 vs. control 36°C for each region, ‡ - p<0.05 and ∥- p<0.001 vs. 36°C for each condition. Mean ± SEM.
Table 1.
The effect of temperature on transmural dispersion of action potential duration (APD90) recorded from isolated canine left ventricular tissue slices and arterially-perfused wedge preparations in the absence (control) and presence of E-4031 (1 μmol/L), chromanol 293B (293B, 30 μmol/L), or combination of these 2 agents. BCL = 2000 ms.
| Tissue Slices | Perfused Wedge | ||||
|---|---|---|---|---|---|
| T°C | 36° | 38° | 40° | 36-37° | 39-40° |
| Control | 84±16 | 91±22 | 110±27* | 52±17 | 47±15 |
| E-4031 | 332±63∥ | 451±129†∥ | 654±180‡∥ | 61±22 | 58±27 |
| 293B | 81±18 | 88±19 | 109±23* | 54±15 | 51±14 |
| E-4031+293B | - | - | - | 91±28¶ | 173±61§# |
p<0.05
p<0.001
p<0.0005 vs. 36°C
p<0.05 vs. 36-37°C.
p<0.0005
p<0.05
p<0.001 vs. control for the same temperature. N=6-12. Mean±SD.
Figure 2.
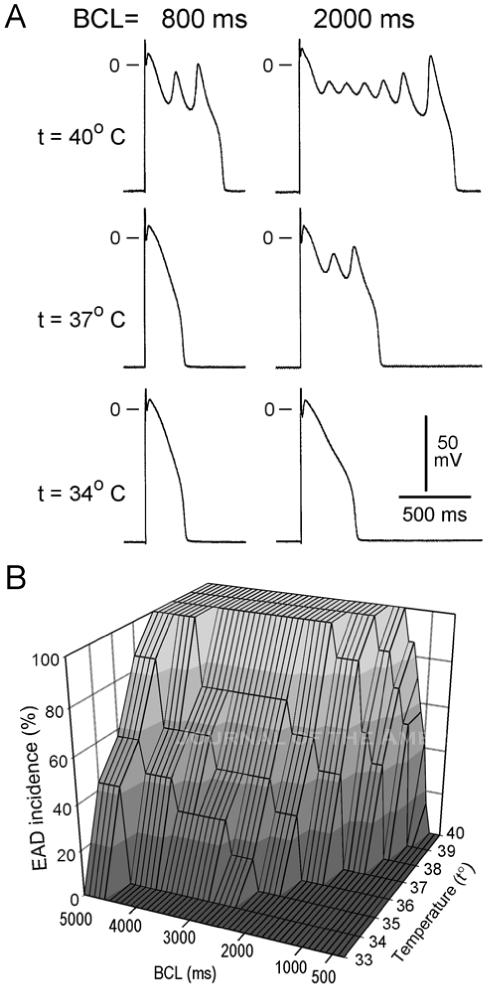
Temperature- and rate- dependence of early afterdepolarization (EAD) activity in M cell preparations pretreated with an IKr blocker, E-4031 (1.0 μmol/L).
A: Transmembrane activity recorded from an M cell preparation at temperatures of 34, 37, and 40°C, recorded at basic cycle lengths (BCL) of 800 and 2000 ms.
B: Plot of temperature- and rate-dependence of EAD incidence in M cell preparations (n=7).
Ryanodine (1 μmol/L) prevented the hyperthermia-induced prolongation of APD and induction of EAD in 4/4 M cell preparations tested (Fig. 3).
Figure 3.
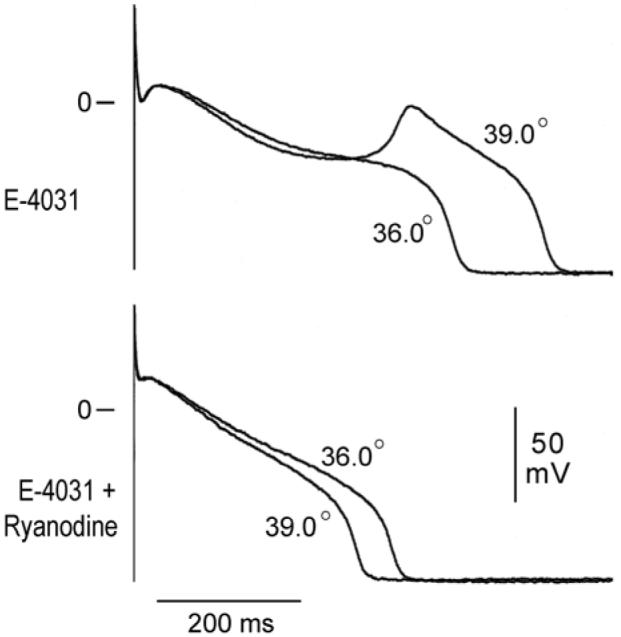
Ryanodine (1 μmol/L) suppresses hyperthermia-induced early afterdepolarization activity in the presence of E-4031 (1 μmol/L). Shown are superimposed transmembrane action potentials recorded from an M region tissue slice preparation at a BCL of 1000 ms.
Chromanol 293B (30 μmol/L, IKs block) produced a relatively homogeneous prolongation of APD in the epicardial, M cell, and endocardial preparations with little effect in Purkinje fibers. Hyperthermia abbreviated APD in all preparations at all pacing rates and did not induced EAD activity. However, there was a modest accentuation of TD-APD at 40°C (Fig. 1 and table 1).
Arterially-perfused wedge preparations
Both E-4031 (1 μmol/L) and chromanol 293B (30 μmol/L) caused a prolongation of repolarization in the coronary-perfused wedge preparations at 36-37°C (Fig. 4). E-4031 had a tendency to increase TD-APD, whereas chromanol 293B produced little effect on TD-APD (Table 1, Fig. 4). Under control conditions, as well as in the presence of E-4031 or chromanol 293B, increasing temperature from 36-37 to 39-40°C produced an abbreviation of repolarization in all regions of the wedge at a BCL of 500 (not shown) and 2000 ms, without accentuation of TD-APD (Table 1 and Fig. 4). Neither EAD nor spontaneous tachyarrhythmia was recorded under these conditions (Table 2).
Figure 4.

Effect of temperature on action potential duration (APD90) and QT interval in the arterially perfused left ventricular wedge preparation. BCL = 2000 ms.
A: Superimposed transmembrane action potentials recorded from subepicardial and subendocardial M cell regions before and after an increase in temperature of the superfusion solution from 37 to 40°C in control (left) and in the presence of E-4031 (1 μmol/L) and chromanol 293B (30 μmol/L)
B: Left panel: Graph plots summary data of the effect of hyperthermia on the APD90 of epicardial (Epi), subepicardial M cell (Subepi M), subendocardial M cell (Subendo M), and endocardial (Endo) regions of the wedge under control conditions and in the presence E-4031 (1 μmol/L), chromanol 293B (30 μmol/L), and the combination of the two agents (n=8 for each). Right panel shows a large variability in the response of QT interval to hyperthermia.
* p<0.05 and † p<0.001 vs. control 36°C for each region; ‡ - p<0.05 and § - p<0.001 vs. 36°C for each condition. n=8 for each. Mean ±SEM.
TABLE 2.
Temperature-dependent incidence of early afterdepolarization activity (EAD) and Torsade de Pointes (TdP) in arterially-perfused wedge
| t°C | EAD activity | TdP | ||||
|---|---|---|---|---|---|---|
| Epi | M Subepi | M Subendo | Endo | |||
| Control | 36-37 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 |
| 39-40 | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | |
| E-4031 (1μ mo/L) | 36-37 | 0/8 | 0/8 | 0/8 | 0/8 | 0/8 |
| 39-40 | 0/8 | 0/8 | 0/8 | 0/8 | 0/8 | |
| 293B (30 μ mol/L) | 36-37 | 0/6 | 0/6 | 0/6 | 0/6 | 0/6 |
| 39-40 | 0/6 | 0/6 | 0/6 | 0/6 | 0/6 | |
| E-4031+293B | 36-37 | 4/12 | 4/12 | 3/12 | 2/12 | 3/12 |
| 39-40 | 8/12 | 9/12 | 4/12 | 1/12 | 6/12 | |
Epi = epicardium; M Subepi = subepicardial M cell region; M Subendo = subendocardial M cell region; Endo = endocardium; 293B = chromanol 293B.
The combination of E-4031 and chromanol 293B produced a prominent prolongation of repolarization and accentuation of TD-APD (Fig. 4, Table 1). Under these conditions, an increase in temperature produced an abbreviation of repolarization in all regions of the wedge at a BCL of 500 ms and highly variable responses at a CL of 2000 ms (Fig. 4). In general, the repolarization in the epicardial and subepicardial regions tended to prolong and in the endocardial and subendocardial regions tended to abbreviate. These regionally dissimilar responses to hyperthermia resulted in a further accentuation of TD-APD (Table 1). The combined IKr and IKs block induced EADs, EAD-induced extrasystoles, and spontaneous Torsade de Pointes (TdP) at 36-37°C (Table 2). Superimposition of hyperthermia significantly increased the incidence of EAD activity and TdP (Fig. 5 and Table 2). Non-sustained monomorphic VT was observed as well (in 3/12 wedges) at 39-40°C. EAD activity originated predominantly in epicardial and subepicardial M regions (Table 2). Spontaneous TdPs were always short (≤ 8 sec). EAD and TdP were bradycardia-dependent; no EAD and TdP were recorded at a BCL of ≤ 800 ms at any temperature tested.
Figure 5.

Hyperthermia-induced EAD-mediated triggered beat and spontaneous TdP in the presence of combined IKr and IKs block (E-4031 and chromanol 293B).
A: Action potentials and ECG recorded from a wedge preparation perfused with 37°C (left) and 40°C (right) Tyrode’s solution and paced at a BCL of 1000 ms.
B: An ECG tracing of a spontaneous TdP observed when the coronary perfusate was warmed to 39°C.
DISCUSSION
The chief finding of the present study is that under conditions of reduced repolarization reserve, hyperthermia can promote EADs, produce an increase of transmural dispersion of repolarization, and precipitate tachyarrhythmias in isolated canine ventricular preparations.
Hyperthermia or a febrile state has been shown to be capable of inducing or promoting ventricular arrhythmias in humans.2-11 This adverse effect of fever is commonly associated with a variety of factors including tricyclic antidepressant poisoning,4 overdose of quinine,8 amphetamines or digitalis,2 long QT intervals,11, 12 long QT and/or acute rheumatic fever,5, 6 the Brugada syndrome,10 malignant hyperthermia induced by anesthesia.3 Fever-induced VF has been reported in humans without detectable structural abnormalities or other factors known to precipitate sudden cardiac death.9 The mechanisms responsible are poorly defined.
In a previous study14 we provided a mechanistic understanding for the development of VT/VF in the Brugada syndrome under hyperthermic conditions. The T1620M mutation of SCN5A linked to the Brugada syndrome was shown to cause premature inactivation of sodium channel current (INa) leaving Ito unopposed. This acceleration of INa decay was found to be a sensitive function of temperature, with inactivation of the current dramatically accelerated under hyperthermic conditions, leading to a loss of function. The increase in net outward current causes repolarization of the right ventricular epicardium at the end of phase 1 of the action potential at some sites, but not others. This epicardial dispersion of repolarization gives rise to phase 2 reentry, which provides a closely coupled extrasystole that captures the vulnerable window created across the right ventricular wall (due to marked abbreviation of the action potential in epicardium, but not endocardium), thus precipitating reentry in the form of VT/VF. We suggested that phase 2 reentry initiates conventional reentry in the setting of Brugada syndrome based on the results of transmembrane action potential recordings.10, 14 A recent study involving optical mapping in a similar Brugada syndrome model provided an elegant experimental confirmation of these mechanisms.17 In patients with Brugada syndrome displaying a missense mutation in SCN5A (F1344S), a temperature-induced a positive shift in the voltage of steady-state activation and a change in slope factor of INa was shown to account for the reduction of INa and the induction of VF by hyperthermia.18
Amin et al. studied the mechanism of fever-induced QT prolongation and ventricular arrhythmias in LQT2 patients by heterologously expressing the A558P HERG missense mutation in HEK-293 cells.12 The current generated by WT + A558P HERG increased in response to hyperthermia to a much lesser degree than that that of WT, thus contributing to fever-induced QT prolongation in these LQT2 patients.
Consistent with the clinical observations,11,12 the present experimental study demonstrates the potential for hyperthermia to lead to arrhythmogenesis under long QT conditions. The prolongation of the QT interval in both congenital and acquired long QT syndromes (LQTS) is associated with the appearance of an atypical polymorphic ventricular tachycardia (PVT), known as Torsade de Pointes (TdP). Both acquired and congenital LQTS are bradycardia-, or pause-dependent,1 although acceleration of heart rate from an initially slow rate has been shown to precede the development of TdP in the clinic19 as well as in experimental models of LQTS.20 These conditions are similar to those under which agents that prolong repolarization induce EAD activity in isolated Purkinje fibers and M cells.21 While initiation of TdP is commonly thought to be caused by an EAD-induced extrasystole, maintenance of TdP is thought to be due to reentry.22 Under some conditions, such as those encountered in experimental models of LQT1 and Timothy syndrome or LQT8, delayed afterdepolarization (DAD)-induced triggered extrasystoles are thought to precipitate TdP.23,24 Increased transmural dispersion of repolarization has been demonstrated in LQTS patients1 as well as in experimental models of LQTS.13, 15
The results of the present study indicate that under conditions of reduced repolarization reserve, an increase in temperature can lead to both induction of EAD activity as well as accentuation of transmural dispersion of repolarization, thus setting the stage for the development of TdP.
The ionic mechanisms responsible for the differential effects of hyperthermia in the four cell types are most likely multifactorial and complex in nature. On a simplistic level, one could hypothesize that the hyperthermia-induced prolongation of APD and induction of EAD is due to augmentation of ICa25 and, as a consequence of calcium loading of the cells, an augmentation of INa-Ca. The temperature-induced prolongation of APD and induction of EAD are not seen in the absence of IKr block because temperature-induced increases of the delayed rectifier currents (IKr and IKs) likely predominate. Temperature elevation is known to increase Ik.25 Note that, in the presence of IKr block, the paradoxical behavior occurs principally in cells with low levels of IKs (M cell26 and Purkinje27), but not those with a prominent IKs (epicardium and endocardium26). In the wedge, in the presence of electrotonic interactions between the surface regions (epicardium and endocardium) with the M cell region,16 hyperthermia-induced abbreviation of APD in epicardial/endocardial regions is likely to abbreviate cells in M region. When the repolarization reserve of the myocardium is further reduced with combined IKr and IKs block, hyperthermia-induced M cell prolongation is further exaggerated and the electrotonic interaction with endocardium and epicardium, while enough to limit its prolongation, is insufficient to permit abbreviation of the M cells. Consequently, with combined IKr and IKs block, Epicardium/Endocardium and M cells show opposite APD responses to hyperthermia (see Table 1).
Evidence in support of the hypothesis that these hyperthermia-induced changes in APD are in part mediated by an increase intracellular calcium activity (Cai) derives from the observation that ryanodine eliminates the hyperthermia-induced APD prolongation and EAD (Fig. 3). Interestingly, an increase of Cai at 37°C by other means, such as acceleration of pacing rate or β adrenergic stimulation, can also prolong APD and induce and/or augment EAD, as well as shift the rates at which EADs develop to shorter CLs.21, 28, 29 It is also noteworthy that manifestation of DADs is also temperature dependent, developing more readily at 37°C than at 33°C.30
The modulatory effect of hyperthermia on the electrophysiological response to pharmacological drugs that prolong cardiac repolarization is an area that is largely unexplored. An increase in temperature from 37°C to 38.5-40°C has been shown to induce EAD activity in quinidine-treated isolated canine Purkinje fibers.31 Hyperthermia exaggerates block of HERG by erythromycin in LTC cells as well as APD prolongation by this drug in mouse isolated ventricular myocytes.32 Erythromycin is an IKr blocker which can induce EAD, long QT syndrome, and TdP.33 It is also of interest that the incidence of spontaneous bradycardia-dependent arrhythmias in isolated rabbit heart pretreated with quinidine is higher at 36°C than at 25-30°C.34
Study limitations
As always, extrapolation of experimental data obtained from canine ventricular tissues and wedge preparations to the clinic must be approached with great caution. The absence of autonomic factors, hormones and other blood-related factors in our Tyrode’s solution-perfused preparations may alter responses from those observed in vivo. Another limitations is that in vivo, hyperthermia-induced increase in sinus rate is expected to antagonize the effects of hyperthermia to prolong APD of the M cell and Purkinje fiber and thus to augment spatial dispersion of repolarization.
Clinical Implications
Apart from the acquired and congenital long QT syndromes, a variety of diseases including heart failure, hypertrophic and dilated cardiomyopathy, and hypothyroidism are associated with a reduced repolarization reserve.35 In the case of heart failure and hypertrophic and dilated cardiomyopathies a coordinated reduction of IKr and IKs has been demonstrated, similar to that employed in our study.
In addition to Class I and III antiarrhythmic drugs, a number of other pharmacological agents including tri- and tetracyclic antidepressants, antibiotics, antihistaminics, diuretics and neuroleptic agents prolong the QT interval and are capable of inducing induce TdP in humans and experimental models.33, 36 Our data raise the possibility that a febrile state may be associated with an increased arrhythmic risk under these conditions of acquired long QT syndrome.
The effect of hyperthermia to accelerate heart rate is expected to prevent ventricular arrhythmias that develop secondary to the bradycardia-dependent mechanisms such as those described in this study. Accordingly, fever-induced ventricular arrhythmias may more readily manifest when in addition to weakened repolarization, pauses or/and slow heart rates are present, for example as a result of AV node block.
CONCLUSIONS
Our data suggest that hyperthermia may promote pro-arrhythmic potential of IKr blockers and other conditions associated with prolonged ventricular repolarization and suggest the inclusion of fever as a potential risk factor in the long QT syndrome.
ACKNOWLEDGMENTS
We gratefully acknowledge the expert technical assistance of Judy Hefferon, Robert Goodrow and Kathy Sullivan.
SOURCES of FUNDING
Supported by grant HL47678 from NHLBI (CA) and grants from the American Heart Association (AB and CA) and NYS and Florida Free and Accepted Masons.
Footnotes
DISCLOSURES
None
REFERENCES
- 1.Roden DM, Lazzara R, Rosen MR, Schwartz PJ, Towbin JA, Vincent GM, The SADS Foundation Task Force on LQTS. Antzelevitch C, Brown AM, Colatsky TJ, Crampton RS, Kass RS, Moss AJ, Sanguinetti MC, Zipes DP. Multiple mechanisms in the long-QT syndrome: Current knowledge, gaps, and future directions. Circulation. 1996;94:1996–2012. doi: 10.1161/01.cir.94.8.1996. [DOI] [PubMed] [Google Scholar]
- 2.Olson KR, Benowitz NL. Environmental and drug-induced hyperthermia. Pathophysiology, recognition, and management. Emergency Medicine Clinics on North America. 1984;2:459–474. [PubMed] [Google Scholar]
- 3.Dunn D. Malignant hyperthermia. AORN J. 1997;65:728–754. doi: 10.1016/s0001-2092(06)62996-7. [DOI] [PubMed] [Google Scholar]
- 4.Pentel P, Benowitz Nl. Tricyclic antidepressant poisoning. Management of arrhythmias. Med Toxicol. 1986;1:101–121. doi: 10.1007/BF03259831. [DOI] [PubMed] [Google Scholar]
- 5.Freed MS, Sacs P, Ellman MH. Ventricular Tachycardia in acute rheumatic fever. Arch Intern Med. 1985;145:1904–1905. [PubMed] [Google Scholar]
- 6.Gimrikh EO, Popov SV, Pekarsky VV. Electrocardiostimulation in therapy of recurring ventricular fibrillation in a patient with the syndrome of a QT elongated interval on the ECG. Ter Arkh. 1985;57(10):45–46. [PubMed] [Google Scholar]
- 7.Hantson P, Benaissa M, Clemessy JL, Baud FJ. Hyperthermia complicating tricyclic antidepressant overdose. Intensive Care Med. 1996;22:453–455. doi: 10.1007/BF01712165. [DOI] [PubMed] [Google Scholar]
- 8.Jacqz-Aigran E, Bennasr S, Desplanques L, Peralma A, Beaufils F. Les risques d’intoxication grave lies a l’administration intraveineuse de quinine. Arch Pediatr. 1994;1:14–19. [PubMed] [Google Scholar]
- 9.Pasquie JL, Sanders P, Hocini M, Hsu LF, Scavee C, Jais P, Takahashi Y, Rotter M, Sacher F, Victor J, Clementy J, Haissaguerre M. Fever as a precipitant of idiopathic ventricular fibrillation in patients with normal hearts. J Cardiovasc Electrophysiol. 2004;15:1271–1276. doi: 10.1046/j.1540-8167.2004.04388.x. [DOI] [PubMed] [Google Scholar]
- 10.Antzelevitch C, Brugada R. Fever and the Brugada Syndrome. PACE. 2002;25:1537–1539. doi: 10.1046/j.1460-9592.2002.01537.x. [DOI] [PubMed] [Google Scholar]
- 11.Weiner I, Rubin DA, Martinez E, Postman J, Herman MV. QT prolongation and paroxysmal ventricular tachycardia occurring during fever following trimethoprim-sulfamethoxazole administration. Mt Sinai J Med. 1981;48:53–55. [PubMed] [Google Scholar]
- 12.Amin AS, Herfst LJ, Delisle BP, Klemens CA, Rook MB, Bezzina CR, Underkofler HAS, Holzem KM, Tan HL, January CT, Wilde AAM. Fever-induced QTc prolongation and ventricular arrhythmias in type 2 congenital long QT. J Clin Invest. 2008 doi: 10.1172/JCI35337. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antzelevitch C, Shimizu W, Yan GX, Sicouri S, Weissenburger J, Nesterenko VV, Burashnikov A, Di Diego JM, Saffitz J, Thomas GP. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J Cardiovasc Electrophysiol. 1999;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- 14.Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, Brugada J, Brugada R, Antzelevitch C. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res. 1999;85:803–809. doi: 10.1161/01.res.85.9.803. [DOI] [PubMed] [Google Scholar]
- 15.Shimizu W, Antzelevitch C. Sodium channel block with mexiletine is effective in reducing dispersion of repolarization and preventing torsade de pointes in LQT2 and LQT3 models of the long-QT syndrome. Circulation. 1997;96:2038–2047. doi: 10.1161/01.cir.96.6.2038. [DOI] [PubMed] [Google Scholar]
- 16.Burashnikov A, Antzelevitch C. Prominent IKs in epicardium and endocardium contributes to development of transmural dispersion of repolarization but protects against development of early afterdepolarizations. J Cardiovasc Electrophysiol. 2002;13:172–177. doi: 10.1046/j.1540-8167.2002.00172.x. [DOI] [PubMed] [Google Scholar]
- 17.Morita H, Zipes DP, Morita ST, Wu J. Temperature modulation of ventricular arrhythmogenicity in a canine tissue model of Brugada syndrome. Heart Rhythm. 2007;4:188–197. doi: 10.1016/j.hrthm.2006.10.015. [DOI] [PubMed] [Google Scholar]
- 18.Keller DI, Huang H, Zhao J, Frank R, Suarez V, Delacretaz E, Brink M, Osswald S, Schwick N, Chahine M. A novel SCN5A mutation, F1344S, identified in a patient with Brugada syndrome and fever-induced ventricular fibrillation. Cardiovasc Res. 2006;70:521–529. doi: 10.1016/j.cardiores.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 19.Locati EH, Maison-Blanche P, Dejode P, Cauchemez B, Coumel P. Spontaneous sequences of onset of torsade de pointes in patients with acquired prolonged repolarization: Quantitative analysis of Holter recordings. J Am Coll Cardiol. 1995;25:1564–1575. doi: 10.1016/0735-1097(95)00100-i. [DOI] [PubMed] [Google Scholar]
- 20.Vos MA, Verduyn SC, Gorgels APM, Lipcsei GC, Wellens HJ. Reproducible induction of early afterdepolarizations and Torsade de Pointes arrhythmias by d-sotalol and pacing in dogs with chronic atrioventricular block. Circulation. 1995;91:864–872. doi: 10.1161/01.cir.91.3.864. [DOI] [PubMed] [Google Scholar]
- 21.Burashnikov A, Antzelevitch C. Acceleration-induced action potential prolongation and early afterdepolarizations. J Cardiovasc Electrophysiol. 1998;9:934–948. doi: 10.1111/j.1540-8167.1998.tb00134.x. [DOI] [PubMed] [Google Scholar]
- 22.Akar FG, Yan GX, Antzelevitch C, Rosenbaum DS. Unique topographical distribution of M cells underlies reentrant mechanism of torsade de pointes in the long-QT syndrome. Circulation. 2002;105:1247–1253. doi: 10.1161/hc1002.105231. [DOI] [PubMed] [Google Scholar]
- 23.Burashnikov A, Antzelevitch C. Block of I(Ks) does not induce early afterdepolarization activity but promotes beta-adrenergic agonist-induced delayed afterdepolarization activity. J Cardiovasc Electrophysiol. 2000;11:458–465. doi: 10.1111/j.1540-8167.2000.tb00342.x. [DOI] [PubMed] [Google Scholar]
- 24.Sicouri S, Timothy KW, Zygmunt AC, Glass A, Goodrow RJ, Belardinelli L, Antzelevitch C. Cellular basis for the electrocardiographic and arrhythmic manifestations of Timothy syndrome: effects of ranolazine. Heart Rhythm. 2007;4:638–647. doi: 10.1016/j.hrthm.2006.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kiyosue T, Arita M, Muramatsu H, Spindler AJ, Noble D. Ionic mechanisms of action potential prolongation at low temperature in guinea-pig ventricular myocytes. J Physiol (Lond) 1993;468:85–106. doi: 10.1113/jphysiol.1993.sp019761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu DW, Antzelevitch C. Characteristics of the delayed rectifier current (IKr and IKs) in canine ventricular epicardial, midmyocardial, and endocardial myocytes. Circ Res. 1995;76:351–365. doi: 10.1161/01.res.76.3.351. [DOI] [PubMed] [Google Scholar]
- 27.Han W, Wang Z, Nattel S. Slow delayed rectifier current and repolarization in canine cardiac Purkinje cells. Am J Physiol Heart Circ Physiol. 2001;280:H1075–H1080. doi: 10.1152/ajpheart.2001.280.3.H1075. [DOI] [PubMed] [Google Scholar]
- 28.Priori SG, Corr PB. Mechanisms underlying early and delayed afterdepolarizations induced by catecholamines. Am J Physiol. 1990;258:H1796–H1805. doi: 10.1152/ajpheart.1990.258.6.H1796. [DOI] [PubMed] [Google Scholar]
- 29.Burashnikov A, Antzelevitch C. Late-Phase 3 EAD. A Unique Mechanism Contributing to Initiation of Atrial Fibrillation. PACE. 2006;29:290–295. doi: 10.1111/j.1540-8159.2006.00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gambassi G, Cerbai E, Pahor M, Capogrossi MC, Carbonin P, Mugelli A. Temperature modulates calcium homeostasis and ventricular arrhythmias in myocardial preparations. Cardiovasc Res. 1994;28:391–399. doi: 10.1093/cvr/28.3.391. [DOI] [PubMed] [Google Scholar]
- 31.Kaseda S, Gilmour RF, Jr., Zipes DP. Depressant effect of magnesium on early afterdepolarizations and triggered activity induced by cesium, quinidine and 4-aminopyridine in canine Purkinje fibers. Am Heart J. 1989;118:458–466. doi: 10.1016/0002-8703(89)90258-5. [DOI] [PubMed] [Google Scholar]
- 32.Guo J, Zhan S, Lees-Miller JP, Teng G, Duff HJ. Exaggerated block of hERG (KCNH2) and prolongation of action potential duration by erythromycin at temperatures between 37 degrees C and 42 degrees C. Heart Rhythm. 2005;2:860–866. doi: 10.1016/j.hrthm.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 33.Antzelevitch C, Sun ZQ, Zhang ZQ, Yan GX. Cellular and ionic mechanisms underlying erythromycin-induced long QT intervals and torsade de pointes. J Am Coll Cardiol. 1996;28:1836–1848. doi: 10.1016/S0735-1097(96)00377-4. [DOI] [PubMed] [Google Scholar]
- 34.Spear JF, Moore EN. Modulation of quinidine-induced arrhythmias by temperature in perfused rabbit heart. Am J Physiol. 1998;274:H817–H828. doi: 10.1152/ajpheart.1998.274.3.H817. [DOI] [PubMed] [Google Scholar]
- 35.Tomaselli GF, Marban E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 36.Haverkamp W, Breithardt G, Camm AJ, Janse MJ, Rosen MR, Antzelevitch C, Escande D, Franz MR, Malik M, Moss A, Shah R. The potential for QT prolongation and proarrhythmia by non- antiarrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Eur Heart J. 2000;21:1216–1231. doi: 10.1053/euhj.2000.2249. [DOI] [PubMed] [Google Scholar]