

Abstract
RNA interferences in the unicellular green alga, Chlamydomonas reinhardtii, can be silenced. We have used the silencing of a transgene (aadA) that confers resistance to spectinomycin to investigate the mechanisms responsible for silencing by an artificial inverted repeat (IR) of the aadA gene. The IR construct provided strong silencing, but the RNAi efficiency varied among subclones of a single RNAi-transformed strain with successive cell divisions. Northern blot analyses revealed an inverse correlation between the copy number of the hairpin RNA and the spectinomycin resistance of the subclones. There is an inverse correlation between the efficiency of RNAi and the frequency of methylated CpG (*CpG) in the silenced region. No significant methylated cytosine was observed in the target aadA gene, which suggests the absence of RNA-directed DNA methylation in trans. Several experiments suggest the existence of an intrinsic IR sequence-dependent but a transcription-independent DNA methylation system in C. reinhardtii. The correlation between the *CpG levels and the IR transcript implies the existence of IR DNA-dependent DNA methylation. Treatment of RNAi-induced cells with a histone deacetylase inhibitor, Trichostatin A, rapidly increased the amount of the hairpin RNA and suggests that transcription of the silencer construct was repressed by *CpG-related silencing mechanisms.
RNA interference (RNAi) provides a means of silencing genes that protects eukaryotic cells against transposable elements and viruses that produce double-stranded RNAs. In addition, the introduction of transgenes can also elicit RNAi. The major biochemical reactions of RNA interference are shared among various eukaryotes (Cerutti and Casas-Mollano 2006). These reactions include the Piwi and the Dicer family of proteins. However, additional pathways are not identical among eukaryotes. In some organisms that include Caenorhabditis elegans, Schizosaccharomyces pombe, and Arabidopsis thaliana, there is a pathway to convert the target mRNA into dsRNA by siRNA-primed RNA-dependent RNA polymerase (RdRP) (Cerutti 2003; Cerutti and Casas-Mollano 2006). In these organisms, the newly generated dsRNA is also recognized by Dicer to produce a large amount of secondary siRNA, which leads to efficient degradation of the target mRNA (Dalmay et al. 2000). Variation exists in the duration of effective RNAi among organisms. In C. elegans, RNAi can even be passed through the germ line to subsequent generations (Grishok et al. 2000). However, in the unicellular green alga, Chlamydomonas reinhardtii, it is often reported that silencing efficiency decreases over variable numbers of cell divisions without rearrangement of the silencer DNA construct (e.g., Koblenz et al. 2003; Schroda 2006). For example, 106-bp long dsRNA worked successfully to knock down centrin (a protein located in multiple places in the flagellar apparatus) and cells became aflagellate (Koblenz et al. 2003). However, the effect was transient and most transformants restored two flagella within 200 days after transformation when they were grown continuously in liquid medium. The AGG2 and AGG3 genes were silenced within 20–30 days in liquid medium (Iomini et al. 2006).
In some eukaryotes, DNA sequences that are complementary to the siRNA are methylated. The siRNA-dependent de novo methylation is called RNA-directed DNA methylation (RdDM). Generally, cytosine methylation is detected in symmetric CpG sequences, because only this type of methylation is efficiently preserved by maintenance-type methyltransferases and inherited by daughter cells. In land plants, cytosine methylation at CpHpG sites (where H equals A, T, or C) is faithfully inherited by daughter cells in addition to the symmetric CpG sites (Tariq and Paszkowski 2004; Chan et al. 2005). In A. thaliana, >30% of total C's in the genome are methylated (Adams 1990), while in the case of C. reinhardtii (Hattman et al. 1978) and its closely related multicellular relative, Volvox carteri (Babinger et al. 2001), the percentages of cytosine methylation are only 0.7 and 1.1%, respectively. In V. carteri, limited CpG methylation undoubtedly contributes to gene silencing of transgenes and endogenous transposons (Babinger et al. 2001, 2007).
Histones located in heterochromatic domains are distinguished by particular modifications, such as methylation, on specific amino acid residues of the histone tails. In some eukaryotes including C. reinhardtii, monomethylation of lysine 4 in histone H3 (H3K4) and monomethylation of H3K9 are putative epigenetic markers for a transcriptionally silent/inactive chromatin state (Cheung and Lau 2005; Van Dijk et al. 2005; Waterborg et al. 1995; Casas-Mollano et al. 2007; Pfluger and Wagner 2007), and the underlying DNA in heterochromatin is often accompanied by methylation. In contrast, histones located in euchromatic regions usually have different types of modifications, such as acetylation, and the DNA in these regions is usually not highly methylated. Thus, DNA methylation status, histone modifications, and heterochromatin formation are closely related (Loidl 2004; Berger 2007). Moreover, in S. pombe, siRNA is able to induce heterochromatin in the absence of concomitant DNA methylation (Grewal and Elgin 2007). Curiously, the above phenomenon appears to be predominantly a cis-effect that requires transcription of the gene.
In this study, we induced RNAi in C. reinhardtii by expressing hairpin RNA from an inverted repeat (IR) DNA construct, which is designed to target the aadA mRNA.
This mRNA encodes an aminoglycoside 3′-adenyltransferase protein that inactivates an aminocyclitol antibiotic, spectinomycin, by adenylation (Kehrenberg et al. 2005). Therefore, the C. reinhardtii 19-P[1030] strain (Cerutti et al. 1997a), which stably expresses the aadA gene, will reduce the spectinomycin resistance by successful induction of RNAi targeting the aadA mRNA.
We closely analyzed temporal changes in the efficiency of RNAi, which fluctuates with successive mitotic cell divisions.
MATERIALS AND METHODS
Strains and culture conditions:
C. reinhardtii 19-P[1030] used in this study is a derivative of the wild-type strain cc-124 (mt−). This strain carries a single copy of the P-679 plasmid carrying an exogenous aadA gene driven by the C. reinhardtii RbcS2 promoter (Cerutti et al. 1997a). This strain is available from the Chlamydomonas Center (Duke University, Durham, NC). Prior to our experiments, single-colony isolation was carried out on agar plates of Tris–acetate–phosphate (TAP) containing 90 μg/ml spectinomycin. Several fast-growing colonies were picked and mixed to start a liquid culture for further transformation to induce RNAi targeting of the aadA mRNA.
Composition of a silencer DNA construct:
To target the aadA mRNA by RNAi, a construct with five parts was made. It has the RbcS2 promoter (Goldschmidt-Clermont and Rahire 1986), the sense strand of the aadA gene minus the first 15 bp, a 79-bp fragment from the second intron of COX2 (Watanabe and Ohama 2001), the antisense strand of the aadA gene, and the RbcS2 3′-UTR. Four of the five components were prepared by PCR, using appropriate templates and primers. The second intron of COX2 was chemically synthesized. The primers used for these experiments are summarized in supplemental Table 1. DNA segments consisting of the RbcS2 promoter, the sense-strand of aadA, and the second intron of COX2 were integrated into a single fragment by the Megaprimer PCR method (Sambrook and Russell 2001). Subsequently, the PCR product was cloned into the EcoRI/NotI site of pSP124S, which contains the ble gene conferring zeocin resistance (Lumbreras et al. 1998). We named this construct front-silencer half/pSP124S. Segments for antisense aadA and RbcS2 3′-UTR were also fused by the Megaprimer PCR method and cloned into the NotI/SacI site of the front-silencer half/pSP124S. The completed silencer DNA construct is shown schematically in Figure 1A.
Figure 1.—

The silencer DNA construct and analysis of RNAi-induced cell features. (A) Restriction enzyme sites utilized to make this construct are indicated by arrows. A 786-bp coding region for the aadA gene (gray box) with a deletion of the first 15 bases was arranged to make an inverted repeat. A 79-bp fragment containing the second intron of COX2 was located in the middle of the inverted repeat. A ble gene (black box) was used as a transformation marker. This gene carries two copies of the first intron of RbcS2 (dotted line) that provides transcriptional enhancer activity. To regulate transcriptional activity of the silencer construct and the ble marker gene, the C. reinhardtii RbcS2 promoter and RbcS2 terminator (white box) were used. The silencer DNA construct and the marker gene were arranged in opposite transcriptional directions. In, intron; pro, promoter; ter, terminator. (B) Results of the spotting tests for cells transformed with the silencer DNA construct. cc-124, wild-type; 19-P[1030], a cc-124 transformant that stably expresses the aadA mRNA; RNAi-13, -17, -18, and -37, 19-P[1030] transformants of an aadA silencer DNA plasmid linearized by KpnI. TAP, Tris–acetate–phosphate medium; TMM, TAP not containing acetate; zeo, zeocin; spc, spectinomycin. (C) Detection of the aadA IR transcript by Northern blot analysis. Hairpin RNA transcribed from the silencer DNA construct was detected by a DIG-labeled sense-aadA RNA probe. Analysis of the same blot with a ble probe was also carried out. About 10 μg of total RNA was loaded per lane. Ethidium bromide staining of the agarose gel was carried out to confirm that equal amounts of RNA were loaded per lane. (D) Detection of siRNA. A small-sized RNA fraction prepared from 30 μg of total RNA was loaded into an 8-m urea-containing 15% polyacrylamide gel. After RNA was transferred to a membrane, detection was carried out by a sense-aadA RNA probe. Ethidium bromide staining of the polyacrylamide gel was carried out to monitor the amount of the loaded RNA. Positions of molecular weight marker bands are shown on the left of each hybridization photogram.
Culture conditions and transformation procedures:
Unless noted otherwise, cells used for transformation were grown mixotrophically in liquid TAP medium under moderate and constant white fluorescent light (20 μmol m−2 sec−1) at 25° with vigorous shaking. The plasmid containing the silencer construct (see above) was linearized by KpnI and used to transform the 19-P[1030] cells, using the biolistic particle delivery system (PDS-1000/He; Bio-Rad, Hercules, CA). Transformants were directly selected on TAP-agar plates containing 10 μg/ml zeocin. Plates were incubated under white fluorescent light (20 μmol m−2 sec−1) (a 16:8-hr light:dark regimen) at 25° for colony formation.
Spectinomycin resistance assay for RNAi-induced transformants:
The liquid culture of RNAi-induced transformants was spread on TAP-agar plates at low density for single colonies, which were cultured independently in 3 ml of TAP medium until they reached stationary growth phase. For each culture, the level of spectinomycin resistance was assayed by the spotting test (see below).
Spotting test to compare the relative spectinomycin resistance:
Independent transformants were cultured to reach stationary phase in liquid TAP medium and subsequently subjected to 10-fold dilution with TAP medium. Five microliters of the diluted samples were spotted on plates containing various concentrations of spectinomycin and incubated for 7–10 days under white fluorescent light (20 μmol m−2 sec−1) (a 16:8-hr light:dark regimen) at 25°. Relative growth rates were monitored by the greenish color of the spots.
Preparation of genomic DNA and RNA:
To prepare genomic DNA, cells were grown to logarithmic phase, harvested by centrifugation, and suspended in 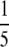 volume of TEN wash buffer [10 mm Tris-HCl (pH 8.0), 10 mm EDTA (pH 8.0), 150 mm NaCl] and subsequently pelleted by centrifugation. Total DNA was isolated from the pellet by the CTAB method (Sambrook and Russell 2001). Total RNA was prepared using TRIzol reagent (Invitrogen, Carlsbad, CA), following the manufacturer's instructions. A mixture of RNaseA and RNaseT1 from the RPAIII Ribonuclease protection kit (Ambion, Austin, TX) was used to digest single-stranded RNA. To isolate small-sized RNAs, high molecular weight RNAs were precipitated with a 5% PEG 8000–0.5 m NaCl mixture (final concentration), and the remaining small-sized RNAs were purified from the supernatant using the QIAGEN (Valencia, CA) RNA/DNA midi kit.
volume of TEN wash buffer [10 mm Tris-HCl (pH 8.0), 10 mm EDTA (pH 8.0), 150 mm NaCl] and subsequently pelleted by centrifugation. Total DNA was isolated from the pellet by the CTAB method (Sambrook and Russell 2001). Total RNA was prepared using TRIzol reagent (Invitrogen, Carlsbad, CA), following the manufacturer's instructions. A mixture of RNaseA and RNaseT1 from the RPAIII Ribonuclease protection kit (Ambion, Austin, TX) was used to digest single-stranded RNA. To isolate small-sized RNAs, high molecular weight RNAs were precipitated with a 5% PEG 8000–0.5 m NaCl mixture (final concentration), and the remaining small-sized RNAs were purified from the supernatant using the QIAGEN (Valencia, CA) RNA/DNA midi kit.
Reverse transcriptase–PCR analysis:
To generate cDNA from aadA and RbcS2 mRNA, total RNA was treated with RNase-free DNase I (Takara, Kyoto, Japan) to remove contaminant DNA. The RNA was subsequently reverse transcribed according to the manufacturer's instructions, using AMV reverse transcriptase (Promega, Madison, WI) and primers specific for each of the genes. The relative amount of cDNA was measured by the real-time PCR method, using the Sequence Detection System 7000 (Applied Biosystems, Foster City, CA) and SYBR green premixed perfect real-time solution (Takara). All data sets were analyzed using ABI Prism SDS software (Applied Biosystems). The aadA cDNA level was normalized by the level of RbcS2 in each sample. Primers used for reverse transcriptase (RT)–PCR are summarized in supplemental Table 2.
DNA digestion with methylcytosine-sensitive restriction enzymes:
To examine the methylation status at the CCGG sites, we used isoschizomeric restriction enzymes, HpaII and MspI. These two enzymes recognize the same target sequence CCGG but differ in their sensitivity to methylcytosine. HpaII does not cleave *CCGG (*C is 5-methylcytosine) and C*CGG, whereas MspI is able to cleave C*CGG but not *CCGG. Sequences flanking the recognition site can affect the cleavage. For example, GGC*CGG is cleaved at an exceptionally slow rate by MspI. Digested samples were subjected to agarose gel electrophoresis to separate the resultant fragments.
Preparation of DNA- and RNA-blotted filters:
To prepare membranes for Southern hybridization, total DNA was completely digested with appropriate restriction enzyme(s), resolved on a 0.8% agarose gel, and then blotted onto positively charged nylon membranes (Hybond N+; GE Healthcare, Little Chalfont, England) by capillary transfer using alkali buffer (0.4 m NaOH, 1 m NaCl). To prepare membranes for Northern hybridization, RNA was separated by a formaldehyde-containing agarose gel and then blotted onto nylon membranes, using a semidry electroblotter (Atto, Tokyo). UV cross-linking and baking affixed the RNA molecules to the filter (for 2 hr at 80°). Small-sized RNAs were separated using a 15% polyacrylamide/8-m urea gel and then transferred to Hybond N+ membrane, using the semidry electroblotter.
Preparation of digoxigenin-labeled DNA probes:
All digoxigenin (DIG)-labeled DNA probes were prepared using a PCR DIG probe synthesis kit (Roche, Basel, Switzerland). We prepared four kinds of DIG-labeled DNA probes (supplemental Table 3) for Southern and Northern hybridization.
Preparation of DIG-labeled RNA probes:
We prepared five kinds of RNA probes for Northern hybridization. DIG-conjugated UTP was transcriptionally incorporated using DIG RNA labeling mix (Roche). As transcriptional templates, some DNA fragments carrying a T3 or a T7 RNA polymerase promoter were prepared by PCR. Alternately, appropriate DNA fragments prepared by PCR were cloned into the pBluescript II SK (−) vector that has T3 and T7 RNA polymerase promoters flanking the multicloning sites. PCR primers used to prepare these DNA fragments are listed in supplemental Table 4.
Hybridization and wash conditions:
Hybridization experiments were carried out using High-SDS hybridization buffer (Roche), anti-DIG-AP fab fragment (Roche), and CDP-star (Roche), following the manufacturer's instructions (DIG Application Manual for Filter Hybridization, Ed. 3). Hybridization with DNA probes and high-stringency washes were carried out at 40°. Hybridization with RNA probes and the subsequent high-stringency washes were carried out at 68°. To detect siRNA, hybridization in the High-SDS hybridization buffer (Roche) and the subsequent high-stringency washes were carried out with 2× SSC (1× SSC contains 0.15 m NaCl and 0.015 m sodium citrate) at 35°. A luminescent image analyzer, LAS-1000 (Fuji Film, Tokyo), was used to detect signals.
Bisulfite genomic DNA PCR:
Genomic DNA was completely digested with 10 restriction enzymes whose restriction sequences are not in the IR region, i.e., SacI, KpnI, BbrPI, BamHI, BglII, XbaI, Eco22TI, PstI, MluI, and XhoI to analyze the positions of methylated C's in the aadA IR region. For analysis of methylated C's in the aadA gene, genome DNA was completely digested with SacI and KpnI. Digested DNA was treated with chemical reagents to convert unmethylated C's to U's while leaving *C unchanged (Liu et al. 2004). Nested PCR was carried out to amplify the region for sequencing. Considering that virtually all *C in CCGG was limited to C*CGG and no *CCGG was detected by DNA digestion with methylcytosine-sensitive restriction enzymes (see text), we designed degenerate PCR primers as follows: Y (Y equals C or T) instead of C was incorporated into the forward primers when C was in the context of CpG in the original sequence and R (R equals A or G) instead of G for the reverse primers when G was in the context of CpG in the original sequence. PCR primers used for the nested PCR are listed in supplemental Table 5. Amplified PCR fragments were cloned into pT7-Blue vector (Novagen, Madison, WI) by TA cloning, and at least eight clones were sequenced for each sample.
Trichostatin A treatment:
Trichostatin A (TSA) (Wako, Osaka, Japan) treatment was carried out for cultures in midlogarithmic growth phase. A TSA/ethanol stock solution (5 mg/ml) was added to the cell culture (final concentration 50 ng/ml) and cells were kept under constant light at 25°.
Chromatin immunoprecipitaion analyses:
To determine modification states of histone H3, chromatin immunoprecipitation (ChIP) was performed on 4 × 107 cells of RNAi-37 subclones. The samples were incubated in 0.75% formaldehyde for 5 min and then in 125 mm glycine for 5 min to quench the cross-link reaction. The cells were washed with PBS (100 mm NaCl, 80 mm Na2HPO4, and 20 mm NaH2PO4) and resuspended in 1.5 ml ChIP lysis buffer [20 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm EDTA (pH 8.0), 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, and 2 μl/ml of Protease inhibitor cocktail for plant cell and tissue extracts (P9599; Sigma, St. Louis)]. Followed the treatment, the cells were disrupted twice in a gas pressure device (Mini-Bom Cell; Kontes, Vineland, NJ) at 10.5 MPa under nitrogen (Shneyour and Avron 1970). Then, the disrupted cells were centrifuged at 3000 × g for 5 min to pellet the nuclei. The pellet was resuspended in 0.75 ml of the ChIP lysis buffer and the chromatin was sheared into tri- to pentanucleosomes size by sonication, using Vibra-Cell (Sonics & Materials, Newtown, CT) with a 3-mm tip (amplitude 35, 10-sec pulses and 1-sec rest cycle, for 12 min), and then centrifuged at 10,000 × g for 10 min. The supernatant containing sheared chromatin was used for immunoprecipitation analyses. Preparation of immunoprecipitants was carried out, using the antibodies against histone H3 (ab1791; Abcam, Cambridge, UK), histone H3K4me1 (Abcam, ab8895), histone H3K9me1 (Abcam, ab8896), and acetyl histone H3 (06-599; Millipore, Bedford, MA). Normal rabbit IgG (PP64B, Millipore) was used as an internal control. The histone–antibody complexes were collected by magnetic dynabeads coupled with protein G (Invitrogen). Fifty microliters of chromatin solution were mixed with the same volume of ChIP lysis buffer, 20 μl of protein G-conjugated beads (previously washed once with the ChIP lysis buffer and resuspended in ChIP lysis buffer), and 2–4 μg of the intended antibody. The mixture was incubated on a shaker for 4 hr at 4°. Subsequently, the beads were serially washed with ChIP lysis buffer, high-salt ChIP lysis buffer [Tris-HCl (pH 8.0), 500 mm NaCl, 1 mm EDTA (pH 8.0), 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate, and 2 μl/ml of the protease inhibitor cocktail for plant cell and tissue extracts (Sigma)], and LiCl wash buffer [10 mm Tris-HCl (pH 8.0), 250 mm LiCl, 1 mm EDTA (pH 8.0), 0.5% NP-40, 0.5% sodium deoxycholate, and 2 μl/ml of Protease inhibitor cocktail for plant cell and tissue extracts (Sigma)]. The washed beads were suspended in ChIP elution buffer (1% SDS, 0.1 m NaHCO3, and 250 mm NaCl) and incubated for 4 hr at 65°. Then, 20 μg of proteinase K were added to the solution and further incubated for 1 hr at 45°. DNA in the immunoprecipitant was purified using a PCR purification kit (Promega). Relative amounts of DNA regions of interest were measured by the real-time PCR method. Primer sets used to amplify specific regions are listed in supplemental Table 6.
RESULTS
Induction of RNAi:
To induce RNAi targeting of the aadA mRNA, we transformed 19-P[1030] cells, which stably express the aadA mRNA and form colonies on TAP agar plates containing 800 μg/ml spectinomycin (spc800/TAP plate) (data not shown), with a linearized silencer/pSP124S plasmid carrying an inverted repeat of aadA, as well as the ble gene as a marker for cotransformation (Figure 1A). Potential cotransformants were selected on medium with zeocin. Transformants in which the aadA mRNA was reduced by the silencer construct were identified by slow growth on spc800/TAP plates.
The growth rate of 59 randomly chosen zeocin-resistant transformants was assayed on various spectinomycin concentrations. When tested on spc250/TAP plates, 40 of 59 transformants showed slower growth than the parental 19-P[1030] strain. All of these 40 transformants survived on spc100/TAP plates on which wild-type cells cannot grow. We selected four transformants that showed slow growth on spectinomycin plates. These strains could not be distinguished from the parent strain under nonselective conditions that included TAP or TAP without acetate (TMM, Tris-minimal-medium) plates. These four strains could not survive on spc400/TAP plates on which parental strain 19-P[1030] grows as well as on TAP plates (Figure 1B). These four transformants were promising candidates for RNAi-induced transformants and we named them RNAi-13, -17, -18, and -37, respectively. Southern hybridization to analyze the copy number of the integrated silencer plasmid and PCR analyses confirmed that the entire silencer construct was integrated in the genome. RNAi-17 and -18 carried three and four copies of the plasmid, respectively. In contrast, RNAi-37 carried a single intact silencer construct and also the ble marker gene (data not shown). Therefore, RNAi-37 was used for further experiments to confirm the existence of siRNA, silencer transcript, and aadA hairpin RNA.
Using a sense aadA RNA probe, we detected a double band (most probably due to incomplete denaturation of the hairpin RNA) from total RNA of RNAi-37. The lower major band was ∼1.6 kb in length (Figure 1C), which is consistent with the expected intact transcript of the silencer construct. We also detected siRNA from the small-sized RNA fraction by the sense-aadA RNA probe (Figure 1D). Therefore, we concluded that reduction of spectinomycin resistance observed for RNAi-37 was due to induction of RNAi that successfully targeted the aadA mRNA.
To determine the genomic locus of the integrated plasmid, we amplified the flanking genomic region of the plasmid by the restriction enzyme site-directed amplification–PCR method (Gonzalez-Ballester et al. 2005). BLAST searches using the sequence obtained from the RESDA–PCR products showed that the plasmid inserted in the proximity of several genes between nucleotide positions 1,422,072 and 1,422,078 of Scaffold_3 (JGI website: http://genome.jgi-psf.org/Chlre3/Chlre3.home.html; Merchant et al. 2007). Therefore, the silencer DNA construct most likely integrated into a euchromatic region of the genome.
Fluctuation of spectinomycin resistance during successive mitotic cell divisions:
We noted that colonies arising from liquid cultures of RNAi-37 and their subclones spread on spc100/TAP plates varied in size (Figure 2A). This observation was remarkably different from that of the parental strain 19-P[1030], which formed colonies of fairly uniform size on similar plates (Figure 2A).
Figure 2.—

Spotting test procedures for fluctuating spectinomycin resistance of subclones originated from a single RNAi-37 colony. (A) Growth of 19-P[1030] and RNAi-37 cells on spectinomycin-containing agar plates. Cells were spread on 100 μg/ml spectinomycin-containing agar plates and incubated for 10 days. (B) Flow chart of clonal analysis. After >100 mitotic cell divisions, RNAi-37 was spread on a TAP plate, and then six colonies were isolated and their spectinomycin resistance was analyzed using plates containing various concentrations of spectinomycin (phase-I spotting). Each RNAi-37 clone was subjected to additional cell divisions (>20 times) and then spread on TAP plates. Ten randomly chosen subclones for each of the 6 RNAi-37 clones (total of 60 subclones) were spotted on various spectinomycin plates (phase II spotting). Tenfold dilution of the cell culture was used for spotting. (C) Results of phase-I and -II spotting tests for RNAi-37 subclones. Tenfold dilution of the cell culture was used for spotting. (D) Results of phase-II spotting test for 19-P[1030]. Ten randomly chosen subclones were used.
To examine whether various levels of growth appear specifically on spectinomycin-containing plates and the observed growth rate is genetically stable, we randomly picked six RNAi-37 single colonies grown on nonselective TAP-plates. Phase I of the spotting test was carried out as follows (Figure 2B): each of the six clones was grown to stationary phase in nonselective liquid TAP-medium independently, and then a 10-fold dilution (5 μl) of each culture was spotted on spc50/TAP, spc100/TAP, spc200/TAP, spc300/TAP, TAP plates, and TMM plates and incubated for 10 days. Various greenish colors were observed for the spots on spc50/TAP, spc100/TAP, and spc200/TAP-plates, while their growth showed no prominent difference on the TAP and TMM plates (Figure 2C). They were arranged by their sensitivity to spectinomycin, as clones 1–6, from the highest to the lowest on the phase-I spotting plate. Then, each remaining sample for the phase-I spotting test was subjected to an approximate total of 20 mitotic cell divisions in liquid TAP medium by successive transfers at midlogarithmic growth phase (Figure 2B). Afterward, a second round of colony isolation was carried out by spreading a small portion of the culture on TAP plates. After 10 days, 10 colonies for each clone were randomly picked and cultured in 2 ml of TAP medium until reaching stationary phase. Then, a total of 60 subclones (10 subclones from each of the six types of RNAi-37 clones) were subjected to the spotting test, using plates containing various concentrations of spectinomycin (spc50/TAP, spc100/TAP, spc200/TAP, and spc300/TAP).
As expected, none of the 60 subclones showed substantial growth difference on TAP, zeo50/TAP, zeo200/TAP, and TMM plates (data not shown). Growth differences among the 10 subclones were observed in the presence of spectinomycin from each starting colony. Some of the subclones (clone 2) showed variation on lower concentrations (spc100/TAP) while others showed variation among the clones on higher concentrations (spc200/TAP for clone 5) (phase-II spotting test, Figure 2C). Subclones could become more resistant or more sensitive than the starting parental clone. Subclones of the parental strain, 19-P[1030], which were subjected to treatments similar to those for RNAi-37, showed no detectable difference among growth rates tested on any concentration of spectinomycin (e.g., Figure 2D).
The analysis shows that spectinomycin resistance of the subclones fluctuated multiple times throughout mitotic cell divisions. Southern hybridization analysis was carried out, and we confirmed, for all of the 60 subclones, that no extensive rearrangements occurred inside or in regions bordering the aadA gene or the silencer construct (supplemental Figure 1). This suggested that the fluctuating spectinomycin resistance was due to epigenetic effects.
In addition to the epigenetic change in spectinomycin sensitivity, genetic changes leading to recovery of spectinomycin resistance were also detected, albeit rarely. Cells bearing such changes were easily distinguishable from those exhibiting epigenetic effects, because these genetic revertants showed the spectinomycin resistance to the level of the parental cell 19-P[1030], i.e., 800 μg/ml spectinomycin resistance. Four clones grew on spc800/TAP plates, following successive liquid cultures in TAP medium. Southern hybridization analysis following digestion with EcoT14I showed that one of these clones had acquired new EcoT14I restriction sites and consequently additional DNA fragments originating from the silencer construct while the others had completely lost the silencer region (supplemental Figure 1).
To correlate various growth rates of RNAi-37 subclones with the amount of remaining aadA mRNA, we carried out quantitative analyses of aadA mRNA levels by real-time RT–PCR using aadA-specific primers, one of which specifically hybridizes to the boundary region between the RbcS2 promoter and the aadA ORF coded in the P-679 plasmid (Cerutti et al. 1997a). This experiment showed that the relative level of aadA mRNA decreased to ∼38% of 19-P[1030] for a strong spectinomycin-resistant RNAi-37 subclone, while it decreased to as low as ∼9% of 19-P[1030] for a weak spectinomycin-resistant RNAi-37 subclone and it was ∼25% for an intermediate-resistance subclone (supplemental Figure 2). This supports our idea that the RNAi efficiency is reflected by the growth rates of the transformants on spc/TAP plates.
Inverse correlation between the level of spectinomycin resistance and the amount of accumulated hairpin RNA and siRNA:
We performed Northern blot analyses to compare the relative amounts of hairpin RNA and siRNA for three newly reisolated RNAi-37 subclones of low spectinomycin resistance (nos. 1–3 in Figure 3A) and three of high spectinomycin resistance (nos. 4–6 in Figure 3A), all of which are different from clones shown in Figure 2. Using the DIG-labeled sense aadA RNA as a probe, we detected the expected IR transcript from all of the subclones at the position corresponding to ∼1.6 kb as a double band, presumably due to incomplete denaturation of the hairpin structure. It was noteworthy that the 1.6-kb signal detected from the highly resistant subclones (nos. 4–6) was consistently weaker than that from the less resistant ones (nos. 1–3) (Figure 3B). By treating the samples with RNase A/T1 mixture to digest single-stranded RNA molecules or single-stranded regions in the dsRNA, the 1.6-kb band shifted to the position corresponding to half of the original molecular weight (data not shown). This demonstrated that the 1.6-kb RNA in the cytosol indeed had formed a hairpin structure with a short loop in the middle. The small-sized RNA fraction was separated on a 15% polyacrylamide gel and a sense-aadA RNA probe was used to detect the presence of the siRNA. A much stronger signal for the siRNA was detected from the weaker resistant subclones (nos. 1–3) than from the stronger resistant subclones (nos. 4–6) (Figure 3B).
Figure 3.—

Northern blot analyses to compare the relative amount of aadA hairpin RNA and siRNA for RNAi-37 subclones with various levels of spectinomycin resistance. (A) Evaluation of spectinomycin resistance for six RNAi-37 clones. Three strongly silenced subclones (nos. 1–3) and weakly silenced subclones (nos. 4–6) were spotted on various kinds of plates after serial dilutions of 10−1, 10−2, and 10−3. (B) Northern blot analyses to detect aadA hairpin RNA and siRNA. A sense-aadA RNA probe was used to detect the hairpin RNA and siRNA. Total RNA was loaded on a 1% agarose/formaldehyde gel and a small-sized RNA fraction was resolved on a 15% polyacrylamide gel containing 8 m urea. Ethidium bromide staining and detection of the RbcS2 mRNA were performed to confirm that the amount of RNA loaded was almost equal. Hybridization using the RbcS2 RNA probe was carried out last, since complete stripping of the probe was difficult.
On the basis of the fact that the amount of siRNA and the amount of the hairpin RNA are positively correlated, it suggests that the primary limiting factor for RNAi efficiency is the amount of silencer hairpin RNA in the cytoplasm.
Analysis of 5-methylcytosine in the inverted repeat DNA region:
Our observation suggests that the fluctuating RNAi effect among subclones is primarily due to the varied amounts of the silencer hairpin RNA, which most likely depends on the transcriptional frequency of the IR construct in the cytoplasm. Since it is known that DNA methylation affects the transcriptional efficiency (Rountree and Selker 1997; Lorincz et al. 2004), we performed Southern blot analyses and genomic bisulfite sequencing (Liu et al. 2004) to analyze the frequency of 5-methylcytosine (*C) in the strongly silenced and weakly silenced subclones, therefore to determine the correlation between the frequency of *C and the amount of the IR transcript.
To assess the *C frequency in the CCGG sequences located in the silencer DNA construct, we prepared total DNA from two strongly silenced and two weakly silenced subclones, which were newly isolated from the RNAi-37 strain. First, we completely cleaved the genomic DNA with EcoT14I to generate DNA fragments of appropriate sizes. Complete digestion of the genomic DNA by this enzyme was confirmed by Southern hybridization, using a full-length aadA probe (supplemental Figure 1). Subsequently, the DNA sample was divided into two equal volumes. One sample was further digested with HpaII and the other with MspI. Both enzymes recognize CCGG, with HpaII inhibited by methylation of either cytosine (C*CG and *CCGG) and MspI inhibited by methylation only of the outer cytosine (*CCGG). The existence of *CpG in CCGG sites of the RbcS2 promoter regions, which were located upstream of the ble and also the aadA genes, was assayed by the P probe (Figure 4A). Alternatively, the C probe was utilized to detect *CpG in the CCGG sites located within the aadA gene and the aadA IR region (Figure 4A).
Figure 4.—

Analysis of methylated CpG frequency by Southern hybridization with methylation-sensitive restriction enzymes. (A) Schematic restriction maps for the ble marker gene and the inverted repeat silencer construct. The aadA gene is represented by shaded boxes and the ble gene is represented by solid boxes. The first intron of RbcS2 inserted into the ble gene is indicated by dotted lines. Open boxes indicate the RbcS2 promoter and the RbcS2 terminator. The positions where P and C probes hybridize are shown by horizontal bars. Horizontal lines correspond to the detected signals in B and C. ×5 demonstrates five closely located restriction sites for HpaII/MspI at the arrowhead position. The asterisk shows the position of the GGCCGG sequence. (B) Result of hybridization by the P probe. Weak, the weakly silenced RNAi-37 clones; strong, the strongly silenced clones. Gathered fragments, unresolved small DNA fragments; *, unknown band. M, MspI; H, HpaII. (C) Result of hybridization by the C probe. Labels are the same as in B.
According to the DNA sequence, seven DNA fragments, with lengths of 482, 116, 84, 82, 74, 73, and 62 bp, respectively, should be detected by the P probe, when all CCGG sites in the RbcS2 promoters and the aadA IR are completely digested. If *CCGG or C*CGG sites were accumulated abundantly in the RbcS2 promoter region, HpaII digestion should result in larger fragments with combined lengths of the neighboring fragments. However, we could not detect these bands by the P probe. Only the 482-bp fragment, which corresponds to the completely digested RbcS2 promoter in the aadA IR construct, was observed irrespective of the strength of RNAi (Figure 4B). This indicated that existence of *CCGG and C*CGG sites, if present, was undetectable in the RbcS2 promoter region in the IR construct by our method. On the other hand, irrespective of the RNAi efficiency, the 482-bp signal detected in MspI-digested samples was consistently stronger than that in HpaII-digested samples. This indicated C*CGG accumulated slightly in the promoter region, but the frequency of this modification appeared to occur similarly in clones irrespective of the silencing strength.
We carried out analysis of *C accumulation at the restriction sites in the aadA gene, using the C probe (Figure 4A). According to the DNA sequence, a 520-bp fragment should be detectable when the aadA gene is digested completely by HpaII or MspI. In all of the samples, the fact that the only fragment detected was 520 bp in length demonstrated that very few, if any, C*CGG or *CCGG sites were present in the gene. In addition, the strength of the signal had no detectable difference regardless of the restriction enzymes used or the origin of the genomic DNA, i.e., DNA prepared from the strongly silenced clones or the weakly silenced ones (Figure 4C). Taken together, these data proved there was no detectable accumulation of *C in the CCGG sites of the targeted aadA gene.
According to the DNA sequence, a 372-bp fragment should be detected by the C probe when the aadA IR DNA is completely digested by HpaII or MspI. For two strongly silenced clones, the density of the signal corresponding to the 372-bp fragment was the same irrespective of the restriction enzymes used. However, for two weakly silenced clones, the signal was notably stronger in MspI-digested samples than in HpaII-digested ones (Figure 4C). This suggested significant and specific accumulation of C*CGG in the weakly silenced clones. Moreover, the significant accumulation of C*CGG in the aadA IR region of the weakly silenced subclones resulted in a signal that corresponds to two unresolved 553- and 605-bp bands that were specifically detected in HpaII-digested samples (Figure 4C).
A 424-bp band was also detected specifically in the weakly silenced clones. Given the DNA sequence of the aadA IR region and the result of the bisulfite genomic sequencing (see below), the 424-bp band was likely caused by slow digestion by MspI caused by the GGC*CGG sequence, but not by the existence of *CCGG. Altogether, Southern blot analyses showed very slight accumulation of C*CGG in the RbcS2 promoter region and significant accumulation of C*CGG in the aadA IR silencer region.
Detection of methylated cytosine by the bisulfite genomic sequencing method:
We analyzed the frequency and location of the *C in the upper strand of the first half of the aadA IR region by the bisulfite genomic sequencing method (Figure 5A). Methylated cytosines were detected within the context of CpG sequences. Of the 59 analyzed CpG sites in the first half of the aadA IR region, the *CpG frequency was 15.3% for the strongly silenced clones but 53.4% for weakly silenced clones on average (Figure 5, B and C). Clear inverse correlation was detected between the *CpG frequency in the aadA IR region and the amount of accumulated hairpin RNA; however, it is not clear whether *CpG accumulation is the cause or the effect of repressive transcription of the IR region. Further investigation is essential to address this issue.
Figure 5.—

Analyses of methylated cytosine in the inverted repeat region of the silencer. (A) A schematic map of the silencer DNA construct. Annealing sites for the degenerate primers are indicated by arrows. (B and C) The CpG methylation patterns of a strongly silenced subclone and a weakly silenced subclone, respectively. Methylated cytosines were detected by the bisulfite sequencing method. Promoter region, −23 to −1; 5′-truncated aadA ORF region, +1 to +786; loop constructing intron, +787 to +800. Annealing sites for nested primers (see supplemental Table 5) are underlined. CpG dinucleotide positions are marked by stacked circles on the sequence. Each horizontal set of circles represents the methylation pattern of a cloned PCR product. Ten PCR clones obtained from a strongly silenced RNAi-37 subclone and 8 PCR clones obtained from a weakly silenced RNAi-37 subclone were sequenced. Solid circles indicate methylated cytosines and open circles indicate unmethylated cytosines. Each horizontal set of circles represents the methylation patterns of a single cloned PCR product.
We applied the same method to the equivalent region of the aadA gene that encodes the target mRNA. The genomic DNA from six weakly silenced RNAi-37 clones and four strongly silenced RNAi-37 clones was analyzed. The original sequence of the PCR amplicon contained 96 non-CpG sites and 35 CpG sites. In total, we detected 1308 C to T converted sites and two nonconverted C sites. Moreover, these two nonconverted C sites were located in non-CpG sequence. However, we cannot exclude the possibility that these nonconverted C sites are due to incomplete chemical reactions in the bisulfite method. Therefore, the aadA gene may contain *C in the non-CpG sequence with extremely low frequency (<0.2%) or it may not contain any *C in the non-CpG sequence at all. No or very rare *C in the target aadA gene implies that C. reinhardtii probably does not have a siRNA-mediated trans-acting cytosine methylation pathway that targets the complementary DNA region. It is notable that distribution of *CpG was strongly enriched in the region close to the RbcS2 promoter and had the tendency to decrease gradually in the direction of its transcription (Figure 5, B and C).
Relationship between the accumulation of *CpG and aadA IR transcription:
To determine whether accumulation of *CpG in the aadA IR region depends on transcription, we made an aadA IR DNA construct bearing no promoter and no terminator, while retaining the other regions as they were in the original silencer plasmid. This construct was introduced into 19-P[1030]. We selected two transformants, both carrying a single intact promoterless aadA IR construct. Northern hybridization analysis showed that transcription of the promoterless aadA IR was below detection level, which suggests that the construct was not driven by neighboring endogenous promoters at the insertion sites in these transformants (data not shown). The level of aadA mRNA in these transformants was not significantly different from that of the recipient strain 19-P[1030], as determined by the real-time RT–PCR analyses (data not shown). In addition, these two transformants showed no significant growth difference on spc400/TAP plates as compared to that of the recipient strain 19-P[1030]. These observations reinforce the idea that the aadA IR construct is not transcribed at significant levels from a neighboring endogenous promoter.
In these transformants, the frequencies of *C in the CpG sites of the promoterless aadA IR were 20.1 and 18.7%, respectively. These frequencies were comparable to the average frequency of *CpG in the strongly silenced RNAi-37 clones (15.3%). This indicated that a negligible amount of the IR transcript is enough to cause *CpG accumulation or the IR transcript is not necessary at all. Instead, the intrinsic IR structure may be the primary determinant of the CpG methylation in the aadA IR region.
In the C. reinhardtii genome, there are many endogenous microRNA coding genes that bear long perfect inverted repeat sequences (Molnár et al. 2007). To determine whether cytosines within the sequences were methylated, we analyzed regions of six microRNA coding genes, which are coded in the clusters numbered cr.01795, cr.01848, cr.02116, cr.02386, cr.02466, and cr.02798 (Molnár et al. 2007), by the bisulfite method. In spite of the fact that each of these genes has a >150-bp-long perfect inverted repeat, no methylated cytosine was detected in the analyzed regions (data not shown). Moreover, HpaII/MspI analysis showed the 5S rRNA gene cluster of C. reinhardtii has no prominent *CpG (data not shown). Therefore, it is possible that, due to unknown mechanisms, endogenous IR structures can escape DNA methylation in C. reinhardtii. Further investigation is essential to uncover the conditions required for induction of DNA methylation in the IR, e.g., minimum length of the IR and specific location of the IR within the genome.
Analyses of histone H3 N-terminal modifications:
Considering the recent reports that nucleosomes unfold completely in a transcriptionally active promoter in Saccharomyces cerevisiae (Boeger et al. 2003; Reinke and Horz 2003), we first investigated the nucleosome occupancy at several regions to assess the H3 modification levels of interest in C. reinhardtii. These regions include the long terminal repeat (LTR) of TOC1 retrotransposon, the promoter region of active endogenous gene ribosomal protein S3 (RPS3), the IR promoter, and the aadA IR. Relative nucleosome occupancy at a specific region was estimated through analyses of the histone H3 levels using a modification-insensitive anti-H3 antibody (Figure 6A).
Figure 6.—

ChIP assays using antibodies against histone H3, monomethylated H3K4, monomethylated H3K9, and acetylated H3K9/14. Solid bars and Shaded bars indicate relative amounts of the PCR fragments amplified from immunoprecipitated chromatins of strongly silenced RNAi-37 subclones and weakly silenced subclones, respectively. (A) The PCR-amplified regions of the inverted repeat silencer are indicated as horizontal bars under the illustration. Specific primers are listed in supplemental Table 6. (B) H3 occupancy levels normalized with respect to the input DNA level in the TOC1 LTR, the RPS3 promoter, the IR promoter, and the aadA IR regions were determined by immunoprecipitation with a modification-insensitive anti-histone H3 antibody. (C) H3K9me1 levels normalized with respect to histone H3 levels in the same regions were determined by immunoprecipitation with an anti-H3K9me1 antibody. (D) H3K4me1 levels normalized with respect to histone H3 levels in the same regions were determined by immunoprecipitation with an anti-H3K4me1 antibody. (E) Acetyl histone H3 levels normalized with respect to histone H3 levels in the same regions were determined by immunoprecipitation with an anti-acetyl histone H3 antibody.
The histone H3 occupancy rate is most likely to reflect the transcription frequency. Compared to the transcriptionally inactive TOC1 LTR, the RPS3 promoter had about only half of the histone H3 occupancy, irrespective of the difference in aadA silencing efficiency (Figure 6B). On the other hand, a prominent occupancy difference was observed between the strongly and weakly silenced clones at the IR promoter and the aadA IR regions. In a weakly silenced clone, H3 occupancy rate at the IR promoter was almost the same as that at the inactive TOC1 LTR; while in a strongly silenced clone, the rate at the same region was almost equal to that at the active RPS3 promoter. In a weakly silenced clone, H3 occupancy rate at the aadA IR was about twice of that at the TOC1 LTR; while in a strongly silenced clone, the rate was only slightly higher than that at the TOC1 LTR (Figure 6B). Therefore, to estimate the H3 modification frequency in these regions, normalization with histone H3 levels was carried out.
Differences in the H3 modification pattern were most prominent at the IR promoter between the strongly and weakly silenced RNAi-37 subclones. In a weakly silenced subclone, H3K9me1 was extremely (Figure 6C) enriched and H3K4me1 was moderately (Figure 6D) increased in this region. In contrast, the acetylation level of H3 was extremely high in a strongly silenced subclone in the same region (Figure 6E). Furthermore, in the aadA IR region, a similar tendency was observed with moderate bias between the two types of RNAi-37 subclones. Altogether, modifications of H3 suggested that aadA IR in a weakly silenced subclone is less actively transcribed than in a strongly silenced one.
Effect of TSA treatment on the accumulation of hairpin RNA and siRNA:
It has been reported that *CpG induces heterochromatin formation and deacetylated histones in various kinds of eukaryotes (for a review, see Klose and Bird 2006). Since there is a clear inverse correlation between the frequency of *CpG and the amount of accumulated hairpin RNA, we explored the relationship between the transcriptional activity for the aadA IR construct and histone modification using TSA, a histone deacetylase inhibitor. One strongly silenced and one weakly silenced RNAi-37 subclone were treated with TSA (50 ng/ml) during the logarithmic growth phase in liquid culture. Equal amounts of liquid culture were withdrawn periodically during the treatment, and the changing quantities of hairpin RNA and siRNA in the samples were monitored by Northern blot analysis.
After 20–40 min of TSA treatment, a distinct increase in the amount of hairpin RNA was observed in both subclones (Figure 7A). Upregulation of the hairpin RNA reached the maximum after incubating with TSA for 40 min in the strongly silenced subclone. However, in the weakly silenced subclone, the increase of the hairpin RNA was very slow and it reached the maximum after 120–180 min incubation (data not shown).
Figure 7.—

Effects of TSA treatment on the accumulation of hairpin RNA and siRNA. (A) Time course of hairpin RNA accumulation after TSA treatment. Samples were isolated at various time points after TSA inoculation and Northern blot analyses were carried out using a DIG-labeled sense-aadA RNA probe. Detection of the RbcS2 mRNA was also carried out to confirm equal loading of RNA. (B) Time course of siRNA accumulation after TSA treatment. A DIG-labeled sense-aadA RNA probe was used to detect the siRNA from small-sized RNA fractions isolated at different time points after TSA treatment. Ethidium bromide staining of RNA was used to confirm equal loading. +180: cells were first inoculated in TSA–TAP medium for 180 min, followed by a recovery period of 180 min in fresh TAP medium.
Contrasting with the prominent effect on the increased aadA IR transcript, no obvious effect of TSA was detected for the transcript of the endogenous RbcS2 gene (Figure 7A). Therefore, the increased aadA IR transcript was most likely the result of specifically enhanced transcriptional activity that had been repressed by histone deacetylation-related mechanisms.
We also examined the effect of TSA on the production of siRNA. Total RNA was prepared from the cells cultured in the TSA-containing TAP medium after a treatment period of 1.5 hr (1.5 hr TSA) and 3 hr (3 hr TSA) and also from the cells that were cultured for 3 hr in the TSA-containing TAP medium and subsequently washed and recultured for an additional 3 hr in TAP medium without TSA (3 hr TSA/3 hr TAP). A continuous increase of siRNA was observed for both types of subclones; however, it was much greater for strongly silenced cells (Figure 7B). The amount of siRNA detected from 3 hr TSA/3 hr TAP-treated cells greatly exceeded that detected from 1.5 hr TSA and 3 hr TSA-treated ones for both types of subclones (Figure 7B). Furthermore, the increase of siRNA had an ∼1-hr delay compared to that of the hairpin RNA.
Effect of TSA treatment on the accumulation of *CpG in the silencer construct:
We examined the *CpG frequency change for a strongly silenced RNAi-37 subclone and a weakly silenced one by the TSA treatment: both types of subclones were cultured for 2 hr in the TSA-containing TAP medium and subsequently washed and recultured for an additional 14 hr in TAP medium (i.e., enough time to experience a mitotic cell division) without TSA (2 hr TSA/14 hr TAP cell). Then *CpG frequency was estimated by the HpaII/MspI-Southern hybridization method described above, using the C probe and the P probe. No significant *CpG frequency changes were detected in the promoter and the IR region of the silencer construct for both types of subclones (data not shown). This suggests that the increased IR transcript by TSA treatment is not due to the change of *CpG frequency in the IR region but rather to conformational change of the aadA IR-containing chromatin region, which is likely induced by the generation of hyperacetylated nucleosomes.
We also treated RNAi-37 subclones with a DNA methylation inhibitor 5-aza-deoxycytidine to ask whether *CpG contributes to repression of IR transcription in C. reinhardtii. 19-P[1030] cells and RNAi-37 subclones were treated with a DNA methylation inhibitor 5-aza-deoxycytidine (5adc). Previous experiments suggest that 50 μm 5adc is fully effective for blocking DNA methylation in the C. reinhardtii chloroplast (Umen and Goodenough 2001). Cells were grown on TAP agar plates containing 200 μm of 5adc for 5 days. Subsequently, we transferred cells to TAP liquid medium containing a 200-μm concentration of 5adc and cultured them for 10 days. During the period of liquid cultivation, TAP medium containing 5adc was renewed every 48 hr. As a control, the same process was carried out using TAP medium not containing 5adc. No prominent change in the *CpG pattern or the genetic composition of the aadA IR region was observed for the 5adc-treated subclones (data not shown). As expected, there was no significant difference in spectinomycin resistance or the amount of the hairpin RNA between the 5adc-treated cells and untreated cells (data not shown).
Effect of TSA on the elongation step of IR transcription:
Two nonmutually exclusive mechanisms are plausible for the aadA IR repression: (i) initiation of transcription is repressed or (ii) the elongation step of transcription is repressed. To assess the extent of the latter effect, we carried out Northern hybridization using sense and antisense aadA RNA probes to detect complete transcripts and prematurely terminated transcripts. Northern hybridization analyses were carried out for five newly isolated RNAi-37 subclones of which RNAi efficiencies were widely diverse (Figure 8A).
Figure 8.—

Analyses of complete and incomplete silencer transcripts. (A) Five RNAi-37 clones (nos. 1–5), which show distinctive silencing efficiencies, were chosen. A spotting test was carried out using plates containing various concentrations of spectinomycin. (B) Detection of the silencer transcripts. In Northern blot analyses, a membrane was first hybridized with the 5′-terminal sense-aadA RNA probe, which recognized the almost completely transcribed products (top). The probe was stripped and a second hybridization with the 3′-terminal antisense-aadA RNA probe, which hybridized with complete and incomplete transcripts of the silencer DNA, was carried out (middle). Finally, the same membrane was hybridized with a ble probe to ensure equal loading of total RNA (bottom).
When hybridization was carried out using a sense-aadA RNA probe (750 bases long), which hybridizes to most of the second half of the hairpin transcript, a weak smeared signal corresponding to 1.0- to 1.5-kb-long incomplete transcripts was detected in addition to the 1.6-kb-long complete hairpin RNA (Figure 8B). Using the same membrane, another hybridization was carried out with an antisense-aadA RNA probe (620 bases long), which hybridizes to most of the first half of the hairpin transcript and 3′ region of the target aadA mRNA. A strong smeared signal corresponding to 0.5–1.5 kb long was detected (Figure 8B). The signal intensity for the complete 1.6-kb transcript was apparently the same, irrespective of which one of the two RNA probes was used, while the smeared signals were much more strongly detected by the antisense-aadA probe than by the sense-aadA probe (Figure 8B). In 19-P[1030], in spite of the use of an aadA ORF 3′-region-specific RNA probe for hybridization, the aadA mRNA (full intact transcript is ∼0.8 kb long) was detected as smeared signals <0.8 kb (Figure 8). This is most probably due to the fact that transcription of the aadA gene, which originated from Escherichia coli, generates aberrant mRNA in C. reinhardtii (Cerutti et al. 1997b).
Therefore, we consider that the smeared 0.8- to 1.5-kb-long signals contain only incomplete aadA IR transcripts not the aberrant aadA transcripts. Northern blot analyses using the two aadA RNA probes showed an inverse correlation between the ratio of incomplete aadA IR transcripts to the complete aadA IR transcripts and the effectiveness of RNAi; i.e., weakly silenced RNAi-37 subclones contained 3′-truncated IR transcripts at a higher level.
Close analysis of the TSA effect using aadA shorter probes:
We further investigated the effect of TSA on accumulation of the complete length transcripts in the cell, using much shorter probes to distinguish the incomplete aadA IR transcripts (ICP) from the complete aadA IR transcripts (CP): a 280-nt aadA 5′-terminal sense probe, which hybridizes to the 3′ end of the second half of the aadA IR transcript, and a 200-nt aadA 3′-terminal antisense probe, which hybridizes to the 3′ end of the first half of the aadA IR transcript, were utilized. Therefore, the 5′-terminal sense-aadA probe was expected to detect only the nearly complete transcript (∼1.6 kb) while the 3′-terminal antisense-aadA probe could detect both complete and incomplete transcripts. Northern analysis indicated that 1.5 hr of TSA treatment had a very strong effect for increased accumulation of the complete transcripts for both the weakly and the strongly silenced subclones (Figure 9). The ICP/CP ratio decreased to about one-third of that observed before treatment for both types of subclones (Figure 9). The above observation shows two nonmutually exclusive mechanisms are possible for the observed TSA effect: (i) increased efficiency of transcription elongation and (ii) decreased efficiency of transcript degradation.
Figure 9.—

Effect of TSA treatment on the silencer transcripts. Total RNA was prepared from equal numbers of cells before and after 1.5 hr incubation with Trichostatin A (TSA). The 5′-terminal sense-aadA RNA probe and the 3′-terminal antisense-aadA RNA probe were used to distinguish the complete transcripts from the incomplete ones. The positions of the probes are indicated on the right. The ratios between the complete transcripts (CP) and the incomplete high molecular weight transcripts (ICP) were calculated using ImageJ (Abramoff et al. 2004; see text for details) and are shown below each lane.
DISCUSSION
By clonal analysis of RNAi-37, we found that genetically identical subclones show varied spectinomycin resistance (Figure 2). We showed that the epigenetic variability of the sensitivity is at least partially due to CpG-methylation/histone modification-related transcriptional repression of the IR region.
Characteristics of IR DNA methylation in Chlamydomonas:
In A. thaliana, siRNA-directed DNA methylation occurs in the target DNA region even if the target DNA is a promoter (Chan et al. 2005; Herr et al. 2005; Kanno et al. 2005a,b; Onodera et al. 2005). In contrast to these observations, we detected no obvious methylation in the targeted aadA gene in C. reinhardtii (Figure 4). Therefore, C. reinhardtii may have no siRNA-directed DNA methylation at the trans locus. Absence of this key reaction is reinforced by the lack of evidence for genes encoding RNA-dependent DNA methylases of the domains-rearranged methyltransferase (DRM) class (Cao and Jacobsen 2002; Gendrel and Colot 2005) and genes encoding RNA polymerase IV (Luo and Hall 2007).
In this study, an obvious inverse correlation was observed between the accumulation of hairpin RNA and the frequency of *CpG in the IR region (Figures 4 and 5). Unlike the nuclear DNA methylation in A. thaliana (Gong et al. 2002) and Hela cells (Banine et al. 2005), our observations imply that 5adc penetrates into the C. reinhardtii nucleus with very limited efficiency or the nuclear DNA methyltransferase is much less sensitive to this inhibitor.
Maintenance of *CpG:
Close analysis of cytosine methylation in A. thaliana showed that *CpG in the genome is a result of de novo C methylation and the maintenance of *CpG among *CpN during DNA replication. The successful *CpG maintenance ratio was ∼96% per DNA replication in Arabidopsis (Bird 2002). In C. reinhardtii, the number of *CpG sites in the IR seems to fluctuate during cell division more than in A. thaliana. Therefore, *CpG fluctuation in C. reinhardtii is probably the result of rarely occurring de novo methylation and less reliable maintenance of *CpG than in A. thaliana. In addition, the chromatin structure within the aadA IR region in C. reinhardtii seems to shuffle between transcriptionally repressive and permissive states in response to the frequency of *CpG. One should be aware that fluctuation of the *CpG frequency during successive cell divisions might not be limited to C. reinhardtii. Indeed, in Petunia hybrida, induction of antisense chalcone synthase genes caused highly variable flower pigmentation on the same branch of a plant (Van Der Krol et al. 1989; Stam et al. 2000). It is very likely a target gene not allowing for such visual scoring may have a similarly variable expression in different parts of the same organ within an individual.
The possible relationship between *CpG accumulation and IR transcription:
In the aadA IR region, we observed a clear gradient in the frequency of *CpG, which was higher near the promoter and lower in the middle of the coding region (Figure 5). This bias was obvious, irrespective of the efficiency of RNAi. Since various 3′-truncated aadA IR transcripts had accumulated (Figures 8 and 9), the *CpG frequency bias might correlate with the transcription frequency of the region. Moreover, we showed that the *CpG frequency is much lower in the promoterless IR construct (∼20%, data not shown) than that in the promoter-bearing IR silencer (∼50%, Figure 5). In addition, distribution of the *CpG had no prominent bias in the promoterless IR (data not shown). This is likely due to transcription-independent methylation of the region. This also indicates that the process of transcription may improve the efficiency of de novo methylation and/or maintenance of the *CpG. In this study, we detect methylated cytosines within the CpG context only in the aadA IR region. Moreover, in Volvox carteri, epigenetic silencing-related modification of foreign DNA is also restricted to *CpG (Babinger et al. 2001). While in the transcriptionally active retrotransposon REM1 in C. reinhardtii, it has been suggested that *C occurs in non-CpG sequences (*CCGG) and very limited methylation of A is present within GATC (MboI/Sau3AI site) sequences (Pérez-Alegre et al. 2005). This implies the existence of several sophisticated DNA modification systems in C. reinhardtii.
Using a 106-bp hairpin RNA transcript, Koblenz et al. (2003) observed depletion of centrin in C. reinhardtii. However, the depletion effect was repressed after continuous mitotic cell divisions. Using an inducible promoter bearing the IR construct to knock down centrin, Koblenz and Lechtreck (2005) showed deterioration of the silencing effect occurs concomitantly with transcription. A strong RNAi effect was observed when the transformants were shifted to inducible conditions even after long periods of successive cultivation under noninducible conditions. Recently, another experiment showed frequent transit of RNA polymerase II induces efficient de novo and/or maintenance of CpG methylation in C. reinhardtii: increase of *CpG was observed within tandemly repeated transgenes, when transcriptional silencing of them is released by RNAi-mediated suppression of SET3 (encoding histone H3 monomethyltransferase on methylation of H3 lysine 9) (Casas-Mollano et al. 2007). Therefore, it is very likely that RNA polymerase II-associated factors and remodeling factors, in addition to methyltransferases (Jeddeloh et al. 1999) and *C recognition proteins (e.g., MeCP2 and MBDs), are involved in the maintenance of *CpG (Jones et al. 1998; Nan et al. 1998).
We failed to recover any hairpin RNA from total RNA using poly(T) RNA conjugated beads (data not shown). This suggests that our hairpin RNA is most likely deficient of a poly(A) tail and the RNA might not be efficiently incorporated into the mRNA transport system. This inefficiency may be the reason a substantial amount of hairpin RNA was consistently detected from RNAi-37, irrespective of the RNAi efficiency in the cells (Figures 1C and 3B).
Characteristics of histone H3 modifications in Chlamydomonas:
In many eukaryotes, CpG methylation induces heterochromatin formation by recruiting proteins that specifically bind to *CpG (for a review see Craig 2005). Moreover, accumulated *CpG has the potential to bind to diverse types of histone modulators at the N terminus. In Neurospora crassa, *CpG in the coding region hampers the elongation step of the PolII RNA polymerase (Rountree and Selker 1997), and in mammals, *CpG positioned in the downstream region of promoters impedes the elongation step by stimulating the formation of repressive chromatin structures (Lorincz et al. 2004). However, in Chlamydomonas, it is possible that formation of a repressive chromatin structure occurs first, followed by DNA methylation of the silenced region. To better understand the role of *CpG, experiments using cells being depleted of a maintenance DNA methyltransferase will be informative.
We investigated the distribution of H3K4me1, H3K9me1, and H3Ac along with the silencer construct to investigate the relationship between these histone modifications and transcriptional silencing. In C. reinhardtii, it has been reported that transcription of a dispersed retrotransposon TOC1 is repressed by heterochromatin formation in which Mut11, a subunit of H3K4 methyltransferase complexes, and a novel serine/threonine kinase participate in this process (Jeong et al. 2002; Van Dijk et al. 2005; Casas-Mollano et al. 2008). Consistent with the H3K4me1-related transcriptional repression for IR region-containing transgenes, this histone modification was particularly enriched in the promoter region of the silencer in the weakly silenced clone of RNAi-37 (Figure 6D).
In the silencer region, H3K9me1 was highly enriched in the weakly silenced clone (Figure 6C). Moreover, Casas-Mollano et al. (2007) showed that SET3, which monomethylates H3K9, is required for silencing of tandemly repeated aadA genes in C. reinhardtii. However, RNAi-mediated repression of SET3 does not release the silencing of a single-copy transgene or dispersed transposable elements, such as TOC1. Therefore, it is intriguing whether monomethyl H3K9 acts as an epigenetic marker of repression for exogenous single inverted repeat constructs.
H3Ac was also enriched in the silencer promoter region in the strongly silenced clone of RNAi-37. On the other hand, its level was as low as in the TOC1 LTR when detected in the weakly silenced clone (Figure 6E), irrespective of the fact that both types of RNAi-37 subclones have limited *CpG in the region (Figure 4B). This suggests that the H3Ac level is not directly controlled by the *CpG level. It has been reported that in S. cerevisiae, repeated transcription induces specific histone modifications to maintain open chromatin (Hampsey and Reinberg 2003; Krogan et al. 2003; Kizer et al. 2005). The above implies that different acetylation levels in the aadA IR promoter region might be attributed to the transcriptional frequency of the aadA IR region. It is noteworthy that TSA treatment not only increases the ratio of completely transcribed aadA IR, but also prominently increases the total amount of the IR transcripts (Figure 7A). The amount of accumulated RbcS2 mRNA increased only slightly with the same treatment, suggesting no drastic change of mRNA stability in the cells (Figure 7B). TAS treatment might have secondary effects on various histone modifications besides deacetylation, which might further enhance full transcription of the hairpin RNA.
Acknowledgments
We thank Manabu Matsuura, Huawen Lin, and Jamie Vernon for many valuable discussions. We specially thank M. Takeuchi for her encouragement. This work was supported by a grant from the Research Fellowship of the Society for the Promotion of Science for Young Scientists and by a Sasagawa Scientific Research grant.
References
- Abramoff, M. D., P. J. Magelhaes and S. J. Ram, 2004. Image processing with ImageJ. Biophotonics Int. 11 36–42. [Google Scholar]
- Adams, R. L., 1990. DNA methylation. The effect of minor bases on DNA-protein interactions. Biochem. J. 265 309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babinger, P., I. Kobl, W. Mages and R. Schmitt, 2001. A link between DNA methylation and epigenetic silencing in transgenic Volvox carteri. Nucleic Acids Res. 29 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babinger, P., R. Völkl, I. Cakstina, A. Maftei and R. Schmitt, 2007. Maintenance DNA methyltransferase (Met1) and silencing of CpG-methylated foreign DNA in Volvox carteri. Plant Mol. Biol. 63 325–336. [DOI] [PubMed] [Google Scholar]
- Banine, F., C. Bartlett, R. Gunawardena, C. Muchardt, M. Yaniv et al., 2005. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Res. 65 3542–3547. [DOI] [PubMed] [Google Scholar]
- Berger, S. L., 2007. The complex language of chromatin regulation during transcription. Nature 447 407–412. [DOI] [PubMed] [Google Scholar]
- Bird, A., 2002. DNA methylation patterns and epigenetic memory. Genes Dev. 16 6–21. [DOI] [PubMed] [Google Scholar]
- Boeger, H., J. Griesenbeck, J. S. Stratton and R. D. Kornberg, 2003. Nucleosomes unfold completely at a transcriptionally active promoter. Mol. Cell 11 1587–1598. [DOI] [PubMed] [Google Scholar]
- Cao, X., and S. E. Jacobsen, 2002. Role of the Arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr. Biol. 12 1138–1144. [DOI] [PubMed] [Google Scholar]
- Casas-Mollano, J. A., K. van Dijk, J. Eisenhart and H. Cerutti, 2007. SET3p mono-methylates histone H3 on lysine 9 and is required for the silencing of tandemly repeated transgenes in Chlamydomonas. Nucleic Acids Res. 35 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Mollano, J. A., B. R. Jeong, J. Xu, H. Moriyama and H. Cerutti, 2008. The MUT9p kinase phosphorylates histone H3 threonine 3 and is necessary for heritable epigenetic silencing in Chlamydomonas. Proc. Natl. Acad. Sci. USA 105 6486–6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti, H., 2003. RNA interference: Traveling in the cell and gaining functions? Trends Genet. 19 39–46. [DOI] [PubMed] [Google Scholar]
- Cerutti, H., and J. A. Casas-Mollano, 2006. On the origin and functions of RNA-mediated silencing: from protists to man. Curr. Genet. 50 81–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti, H., A. M. Johnson, N. W. Gillham and J. E. Boynton, 1997. a A eubacterial gene conferring spectinomycin resistance on Chlamydomonas reinhardtii: integration into the nuclear genome and gene expression. Genetics 145 97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti, H., A. M. Johnson, N. W. Gillham and J. E. Boynton, 1997. b Epigenetic silencing of a foreign gene in nuclear transformants of Chlamydomonas. Plant Cell 9 925–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, S. W., I. R. Henderson and S. E. Jacobsen, 2005. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 6 351–360. [DOI] [PubMed] [Google Scholar]
- Cheung, P., and P. Lau, 2005. Epigenetic regulation by histone methylation and histone variants. Mol. Endocrinol. 19 563–573. [DOI] [PubMed] [Google Scholar]
- Craig, J. M., 2005. Heterochromatin—many flavours, common themes. BioEssays 27 17–28. [DOI] [PubMed] [Google Scholar]
- Dalmay, T., A. Hamilton, S. Rudd, S. Angell and D. C. Baulcombe, 2000. An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus. Cell 101 543–553. [DOI] [PubMed] [Google Scholar]
- Gendrel, A. V., and V. Colot, 2005. Arabidopsis epigenetics: when RNA meets chromatin. Curr. Opin. Plant Biol. 8 142–147. [DOI] [PubMed] [Google Scholar]
- Goldschmidt-Clermont, M., and M. Rahire, 1986. Sequence, evolution and differential expression of the two genes encoding variant small subunits of ribulose bisphosphate carboxylase/oxygenase in Chlamydomonas reinhardtii. J. Mol. Biol. 191 421–432. [DOI] [PubMed] [Google Scholar]
- Gong, Z., T. Morales-Ruiz, R. R. Ariza, T. Roldán-Arjona, L. David et al., 2002. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell 111 803–814. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Ballester, D., A. de Montaigu, A. Galvan and E. Fernandez, 2005. Restriction enzyme site-directed amplification PCR: a tool to identify regions flanking a marker DNA. Anal. Biochem. 340 330–335. [DOI] [PubMed] [Google Scholar]
- Grewal, S. I., and S. C. Elgin, 2007. Transcription and RNA interference in the formation of heterochromatin. Nature 447 399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishok, A., H. Tabara and C. C. Mello, 2000. Genetic requirements for inheritance of RNAi in C. elegans. Science 287 2494–2497. [DOI] [PubMed] [Google Scholar]
- Hampsey, M., and D. Reinberg, 2003. Tails of intrigue: phosphorylation of RNA polymerase II mediates histone methylation. Cell 113 429–432. [DOI] [PubMed] [Google Scholar]
- Hattman, S., C. Kenny, L. Berger and K. Pratt, 1978. Comparative study of DNA methylation in three unicellular eukaryotes. J. Bacteriol. 135 1156–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herr, A. J., M. B. Jensen, T. Dalmay and D. C. Baulcombe, 2005. RNA polymerase IV directs silencing of endogenous DNA. Science 308 118–120. [DOI] [PubMed] [Google Scholar]
- Iomini, C., L. Li, W. Mo, S. K. Dutcher and G. Pipern, 2006. Two flagellar genes, AGG2 and AGG3, mediate orientation to light in Chlamydomonas. Curr. Biol. 16 1147–1153. [DOI] [PubMed] [Google Scholar]
- Jeddeloh, J. A., T. L. Stokes and E. J. Richards, 1999. Maintenance of genomic methylation requires a SWI2/SNF2-like protein. Nat. Genet. 22 94–97. [DOI] [PubMed] [Google Scholar]
- Jeong, B.-r., D. Wu-Scharf, C. Zhang and H. Cerutti, 2002. Suppressors of transcriptional transgenic silencing in Chlamydomonas are sensitive to DNA-damaging agents and reactivate transposable elements. Proc. Natl. Acad. Sci. USA 99 1076–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. L., G. J. Veenstra, P. A. Wade, D. Vermaak, S. U. Kass et al., 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19 187–191. [DOI] [PubMed] [Google Scholar]
- Kanno, T., B. Huettel, M. F. Mette, W. Aufsatz, E. Jaligot et al., 2005. a Atypical RNA polymerase subunits required for RNA-directed DNA methylation. Nat. Genet. 37 761–765. [DOI] [PubMed] [Google Scholar]
- Kanno, T., W. Aufsatz, E. Jaligot, M. F. Mette, M. Matzke et al., 2005. b A SNF2-like protein facilitates dynamic control of DNA methylation. EMBO Rep. 6 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehrenberg, C., B. Catry, F. Haesebrouck, A. de Kruif and S. Schwarz, 2005. Novel spectinomycin/streptomycin resistance gene, aadA14, from Pasteurella multocida. Antimicrob. Agents Chemother. 49 3046–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizer, K. O., H. P. Phatnani, Y. Shibata, H. Hall, A. L. Greenleaf et al., 2005. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3K36 methylation with transcript elongation. Mol. Cell. Biol. 25 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klose, R. J., and A. P. Bird, 2006. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31 89–97. [DOI] [PubMed] [Google Scholar]
- Koblenz, B., and K. F. Lechtreck, 2005. The NIT1 promoter allows inducible and reversible silencing of centrin in Chlamydomonas reinhardtii. Eucaryot. Cell 4 1959–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblenz, B., J. Schoppmeier, A. Grunow and K. F. Lechtreck, 2003. Centrin deficiency in Chlamydomonas causes defects in basal body replication, segregation and maturation. J. Cell Sci. 116 2635–2646. [DOI] [PubMed] [Google Scholar]
- Krogan, N. J., M. Kim, A. Tong, A. Golshani, G. Cagney et al., 2003. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 23 4207–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L., R. Wylie, N. Hansen, L. G. Andrews and T. O. Tollefsbol, 2004. Profiling DNA methylation by bisulfite genomic sequencing, pp. 169–179 in Methods in Molecular Biology, edited by T. O. Tollefsbol. Humana Press, Clifton, NJ/Totowa, NY. [DOI] [PubMed]
- Loidl, P., 2004. A plant dialect of the histone language. Trends Plant Sci. 9 84–90. [DOI] [PubMed] [Google Scholar]
- Lorincz, M. C., D. R. Dickerson, M. Schmitt and M. Groudine, 2004. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Mol. Biol. 11 1068–1075. [DOI] [PubMed] [Google Scholar]
- Lumbreras, V., D. R. Stivens and S. Purton, 1998. Efficient foreign gene expression in Chlamydomonas reinhardtii mediated by an endogenous intron. Plant J. 14 441–447. [Google Scholar]
- Luo, J., and B. D. Hall, 2007. A multistep process gave rise to RNA polymerase IV of land plants. J. Mol. Evol. 64 101–112. [DOI] [PubMed] [Google Scholar]
- Merchant, S. S., S. E. Prochnik, O. Vallon, E. H. Harris, S. J. Karpowicz et al., 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár, A., F. Schwach, D. J. Studholme, E. C. Thuenemann and D. C. Baulcombe, 2007. miRNAs control gene expression in the single-cell alga Chlamydomonas reinhardtii. Nature 447 1126–1129. [DOI] [PubMed] [Google Scholar]
- Nan, X., H. H. Ng, C. A. Johnson, C. D. Laherty, B. M. Turner et al., 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393 386–389. [DOI] [PubMed] [Google Scholar]
- Onodera, Y., J. R. Haag, T. Ream, P. C. Nunes, O. Pontes et al., 2005. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell 120 613–622. [DOI] [PubMed] [Google Scholar]
- Pérez-Alegre, M., A. Dubus and E. Fernández, 2005. REM1, a new type of long terminal repeat retrotransposon in Chlamydomonas reinhardtii. Mol. Cell. Biol. 25 10628–10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfluger, J., and D. Wagner, 2007. Histone modifications and dynamic regulation of genome accessibility in plants. Curr. Opin. Plant Biol. 10 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke, H., and W. Horz, 2003. Histones are first hyperacetylated and then lose contact with the activated PHO5 promoter. Mol. Cell 11 1599–1607. [DOI] [PubMed] [Google Scholar]
- Rountree, M. R., and E. U. Selker, 1997. DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa. Genes Dev. 11 2383–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J., and D. W. Russell, 2001. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- Schroda, M., 2006. RNA silencing in Chlamydomonas: mechanisms and tools. Curr. Genet. 49 69–84. [DOI] [PubMed] [Google Scholar]
- Shneyour, A., and M. Avron, 1970. High biological activity in chloroplasts from Euglena gracilis prepared with a new gas pressure device. FEBS Lett. 8 164–166. [DOI] [PubMed] [Google Scholar]
- Stam, M., R. de Bruin, R. van Blokland, R. A. van der Hoorn, J. N. Mol et al., 2000. Distinct features of post-transcriptional gene silencing by antisense transgenes in single copy and inverted T-DNA repeat loci. Plant J. 21 27–42. [DOI] [PubMed] [Google Scholar]
- Tariq, M., and J. Paszkowski, 2004. DNA and histone methylation in plants. Trends Genet. 20 244–251. [DOI] [PubMed] [Google Scholar]
- Umen, J. G., and U. W. Goodenough, 2001. Chloroplast DNA methylation and inheritance in Chlamydomonas. Genes Dev. 15 2585–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Krol, A. R., P. A. de Lange, G. M. Gerats, J. N. M. Mol and A. R. Stuitje, 1989. Antisense chalcone synthase genes in Petunia: visualization of variable transgene expression. Gen. Genet. 220 204–212. [Google Scholar]
- Van Dijk, K., K. E. Marley, B. R. Jeong, J. Xu, J. Hesson et al., 2005. Monomethyl histone H3 lysine 4 as an epigenetic mark for silenced euchromatin in Chlamydomonas. Plant Cell 17 2439–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, K. I., and T. Ohama, 2001. Regular spliceosomal introns are invasive in Chlamydomonas reinhardtii: 15 introns in the recently relocated mitochondrial cox2 and cox3 genes. J. Mol. Evol. 53 333–339. [DOI] [PubMed] [Google Scholar]
- Waterborg, J. H., A. J. Robertson, D. L. Tatar, C. M. Borza and J. R. Davie, 1995. Histones of Chlamydomonas reinhardtii. Synthesis, acetylation, and methylation. Plant Physiol. 109 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]