

Abstract
Neutrality tests in quantitative genetics provide a statistical framework for the detection of selection on polygenic traits in wild populations. However, the existing method based on comparisons of divergence at neutral markers and quantitative traits (Qst–Fst) suffers from several limitations that hinder a clear interpretation of the results with typical empirical designs. In this article, we propose a multivariate extension of this neutrality test based on empirical estimates of the among-populations (D) and within-populations (G) covariance matrices by MANOVA. A simple pattern is expected under neutrality: D = 2Fst/(1 − Fst)G, so that neutrality implies both proportionality of the two matrices and a specific value of the proportionality coefficient. This pattern is tested using Flury's framework for matrix comparison [common principal-component (CPC) analysis], a well-known tool in G matrix evolution studies. We show the importance of using a Bartlett adjustment of the test for the small sample sizes typically found in empirical studies. We propose a dual test: (i) that the proportionality coefficient is not different from its neutral expectation [2Fst/(1 − Fst)] and (ii) that the MANOVA estimates of mean square matrices between and among populations are proportional. These two tests combined provide a more stringent test for neutrality than the classic Qst–Fst comparison and avoid several statistical problems. Extensive simulations of realistic empirical designs suggest that these tests correctly detect the expected pattern under neutrality and have enough power to efficiently detect mild to strong selection (homogeneous, heterogeneous, or mixed) when it is occurring on a set of traits. This method also provides a rigorous and quantitative framework for disentangling the effects of different selection regimes and of drift on the evolution of the G matrix. We discuss practical requirements for the proper application of our test in empirical studies and potential extensions.
THE comparison of genetic differentiation at neutral markers and at quantitative traits is a commonly used method to estimate the relative impacts of drift and selection on polygenic traits in the wild. Typically, a set of populations is sampled, from which the differentiation among populations is estimated for a set of molecular markers (Fst) and is compared to the same measure of differentiation at a single or a set of quantitative traits (Qst). Under pure neutrality, and if the traits are additive, Qst = Fst for any trait (Spitze 1993). Departures from this neutral expectation are considered evidence of selection acting on the quantitative trait under study. Qst < Fst is evidence of homogeneous selection for the trait among populations, i.e., selection for the same optimal value of the trait in all populations, while Qst > Fst is evidence of heterogeneous selection for the trait, i.e., selection for different optima among populations (Merila and Crnokrak 2001).
However, proper empirical detection of selection requires being able to detect a statistically significant departure from the neutral expectation (Qst = Fst) and therefore depends on the confidence intervals (C.I.'s) of both Qst and Fst estimates. When studying single traits, confidence intervals on Qst are very large (Merila and Crnokrak 2001; Latta 2004; O'Hara and Merila 2005; Goudet and Buchi 2006), often spanning >50% of their total possible range [0, 1], even in the most recent studies with a large sampling effort (Porcher et al. 2006). Furthermore, the methods employed to estimate the C.I. are not always statistically efficient (O'Hara and Merila 2005). Overall, the power of the test with single traits Qst is very low with the sampling designs typically possible in empirical studies (O'Hara and Merila 2005), so that rejection of the neutral expectation is unlikely, even when fairly strong selection is in fact occurring (Latta 2004). Consequently, most Qst–Fst comparisons use mean Qst values among a set of quantitative traits, which are compared to Fst estimates from several marker loci (Chapuis et al. 2007). In doing so, the C.I. for Qst is reduced (and the power of the test increased) at the cost of losing information on individual traits. However, even with mean Qst values, the C.I.'s obtained are often still large (see, e.g., Merila and Crnokrak 2001). Furthermore, and maybe more importantly, the method used to compute the C.I. for the mean Qst often implicitly assumes that the quantitative traits are mutually independent. This has two drawbacks: (i) in general the traits under scrutiny show some level of covariance within populations so that the resulting C.I. estimates may be unreliable, and (ii) the information contained in these covariances is not used in the analysis. Recent methods can partly correct for covariance between traits, but often not completely, and they are rarely applied in practice, maybe because of their statistical complexity. In addition, even when correctly estimated, the C.I.'s for Qst remain large (O'Hara and Merila 2005). However, Kremer et al. (1997) proposed a measure of Qst on several traits. Although this method does not really use the information contained in covariances between traits, it does correct for these covariances to provide a (still univariate) measure of Qst (called CQst by the authors). As expected, when used by Waldmann and Andersson (1999) in subdivided populations of two plant species, it provided smaller (and more reliable) C.I.'s in the measure of Qst than previously observed on individual traits.
On the other hand, the study of multivariate phenotypic distributions in the wild has led to a flourishing literature on the evolution of genetic covariances between traits, summarized into the matrix of genetic covariances: the G matrix. Since Lande (1979) introduced a multivariate framework to predict the evolution of a set of polygenic traits under selection and drift, the importance of genetic covariances in constraining adaptive evolution has been a major focus of evolutionary biology (see a special issue of The Journal of Evolutionary Biology, Blows 2007). Numerous studies have sought to estimate the G matrix in different species or in different populations of the same species, to test to what extent G matrices could evolve under the influence of various evolutionary forces and how they could constrain the evolutionary trajectory of natural populations (reviewed in Steppan et al. 2002; McGuigan 2006).
However, as for the case of single-quantitative-trait studies, disentangling the effects of selection and drift on multivariate covariances has proved difficult empirically (Steppan et al. 2002). For this purpose, alternative predictions on the pattern of multivariate phenotypic distributions among populations or species must be made according to whether drift or selection is the main driving force of the pattern. It was initially suggested that G matrices in distinct populations undergoing only drift should be proportional to each other, while this was not expected under selection (Roff 2000). However, this prediction is in fact theoretically incorrect (Phillips et al. 2001): G matrices from individual populations can differ largely even under the action of drift alone. It is only the average G among many drifting populations that is expected to be proportional to G in the ancestral population from which they are derived, and this ancestral population is rarely available to the experimenter. This result was demonstrated empirically by Phillips et al. (2001).
In an influential study of morphological traits in stickleback fishes, Schluter (1996) compared the within-populations covariances (G) with the among-populations covariances (the divergence matrix D). He showed that the leading phenotypic axis of within-population variance (the main eigenvector of G) pointed in a similar direction to the main axis of population divergence (the leading eigenvector of D). As this similarity tended to decay with the divergence time between species, he concluded that this pattern was evidence of the action of divergent selection between stickleback species. More recently, McGuigan et al. (2005) extended their approach to comparisons of whole matrices (G vs. D) instead of only leading eigenvectors. Unfortunately, as was pointed out by the authors, similarity between G and D can be generated by selection as well as by drift (Lande 1979) and therefore detecting this pattern alone does not allow disentangling the two effects. In addition, tests of qualitative similarity between matrices may have low power (Steppan et al. 2002), and, to our knowledge, their statistical behavior has never been studied precisely.
More generally the problem that observable patterns in G can be due to both selective and neutral processes has led to criticisms of the whole research program that seeks to use G matrix estimates to get insights into the constraints imposed by evolutionary forces on phenotypic evolution (Pigliucci 2006). Given the already large designs required for evolutionary quantitative genetic studies, it seems that they are more likely to be improved through more appropriate statistical approaches than by increased empirical efforts. In this article, we attempt to overcome these difficulties by devising a neutrality test on the basis of alternative expectations from neutrality vs. selection and by explicitly checking its statistical properties by simulations. Our approach is to extend the classic Qst–Fst comparison to multivariate phenotype distributions, by using the information from neutral markers in comparisons of the G and D matrices.
Rogers and Harpending (1983) proposed a neutrality test that is in fact a multivariate method to compare Qst and Fst, although it was not described in those terms. The method accounts for nonindependence between traits and de facto provides a clear quantitative expectation for the evolution of the G matrix under neutrality. Similar to a previous suggestion of Lande (1979), they suggested to compare G and D and showed that the expected relationship between these two matrices under neutrality was D = 2Fst/(1 − Fst)G (Equation 16 of Rogers and Harpending 1983). In spite of its great potential interest for empirical tests in multivariate evolution, this suggestion has been somewhat overlooked in the empirical literature (but see Merila 1997). This may stem from the fact that their results were obtained in the limiting context of traits encoded by nonpleiotropic and diallelic loci and that the statistical behavior of the test they proposed was not studied in detail. Furthermore, the test compares only the value of the proportionality constant with its expectation (2Fst/(1 − Fst)) but does not test for proportionality of G and D itself. Finally, no explicit statistical approach was proposed by the authors to implement this multivariate test.
The aims of this article are (i) to generalize the prediction on the relationship between G and D under neutrality using results from coalescent theory, (ii) to use this prediction as a basis for a new neutrality test, and (iii) to check the statistical efficiency of our test by simulations of realistic empirical designs. An application of the test to empirical data is provided in an accompanying article (Chapuis et al. 2008). Our statistical test uses the general framework of sample covariance matrix comparisons (Flury 1988), also known as common principal component (CPC) analysis. This method, which has been introduced in evolutionary biology by Phillips and Arnold (1999), allows determining the level of shared structure among an arbitrary number of covariance matrices of arbitrary dimension. These matrices may be equal, be proportional, or have a number of principal components in common. To test our neutral expectation (further detailed below) requires testing for proportionality only between two matrices. The simplicity of this test (one of the simplest subcases of CPC analysis) provides a straightforward testing approach, whose statistical efficiency is evaluated through individual-based simulations.
METHODS
Multivariate neutrality test for additive traits:
Expected pattern under neutrality:
Consider a set of populations for which a set of quantitative traits undergoes only mutation and genetic drift, but no selection. The quantitative characters under study are assumed to be additive, and the loci underlying these characters are assumed to be at linkage equilibrium. The mean value of each trait is expected to diverge randomly across populations because of drift and mutation. Consider that the set of subpopulations diverged from a given common ancestral population. Using coalescent theory, Whitlock (1999) showed that the expected genetic variance of any additive trait both within and among subpopulations can be expressed as a function of (i) the total mutational variance for that trait (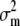 ), (ii) the effective size (Ne), and (iii) Wright's index of population differentiation (Fst) in the metapopulation. Although originally expressed in terms of variances, the argument is also valid in the more general case of covariances between any pair of traits (z1 and z2), simply by replacing
), (ii) the effective size (Ne), and (iii) Wright's index of population differentiation (Fst) in the metapopulation. Although originally expressed in terms of variances, the argument is also valid in the more general case of covariances between any pair of traits (z1 and z2), simply by replacing 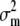 by the mutational covariance between z1 and z2 due to pleiotropy (c12). The expected covariances within and among populations (covw and covb, respectively) can thus be expressed as
by the mutational covariance between z1 and z2 due to pleiotropy (c12). The expected covariances within and among populations (covw and covb, respectively) can thus be expressed as
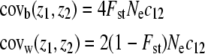 |
(1) |
(from Equations 12 and 13 of Whitlock 1999). Because they are proportional to the same quantity Nec12, a simple relation between the two covariances is deduced from Equation 1: covb(z1, z2) = 2Fst/(1 − Fst)covw(z1, z2) for any pair (z1, z2). This relation can be expressed in matrix form for a set of traits that are all neutral and additive (but not necessarily independent). If E(D) and E(G) are the expected among-populations and within-populations covariance matrices in the metapopulation, we expect
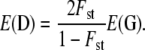 |
(2) |
For a haploid or a completely inbred diploid species, the factor 2 is dropped from the right-hand side of the equation. Furthermore, from Equation 4 of Whitlock (1999), it appears that for a species with a significant level of inbreeding, the right-hand side of Equation 2 above should be divided by (1 + Fis), where Fis, the inbreeding coefficient within populations, is also estimated from the neutral markers.
Equation 2 is similar to Equation 16 of Rogers and Harpending (1983), which was obtained in the limiting case of a polygenic trait encoded by nonpleiotropic diallelic loci and developed in the context of the island model. In fact, the derivation of Equation 2 from coalescent theory shows that this result is valid whatever the forces shaping the neutral divergence among populations (e.g., isolation by distance, extinction–colonization, migration, etc.) and the number of alleles encoding the quantitative traits under focus. Therefore, it should be valid for any metapopulation undergoing neutral divergence and mutation, provided that the determinism of the traits is additive (as for the classic Qst = Fst expectation). The main limitations inherent in the coalescent approach used here are that linkage disequilibrium between quantitative trait loci should be small and that mutational variance per generation on each trait should be independent of the current state of the population (Whitlock 1999). Note that under the specific scenario of population divergence without migration, the proportionality coefficient 2Fst/(1 − Fst) is a direct measure of the scaled time since divergence between the populations τ/2N, where N is the population size assumed to be constant across populations (Slatkin 1995).
Equation 2 suggests that a neutrality test can be performed on the basis of estimates of G, D, and Fst. This test boils down to two tests in fact: testing for proportionality between G and D (and as previously suggested under more restricted cases, Lande 1979; Rogers and Harpending 1983) and testing whether the proportionality coefficient is significantly different from 2Fst/(1 − Fst). In the following, we show how to implement these tests on the basis of empirical estimates of G and D, using statistical approaches for matrix comparisons. We then study the statistical behavior of the proposed test by simulations.
Proportionality between two matrices:
For our multivariate neutrality test, we start from estimates of the covariance matrices among and within populations and of the neutral divergence among populations (Fst) from neutral markers, and we wish to test whether Equation 2 holds. Therefore what is needed here is one of the simplest special cases in CPC analysis: the test for proportionality between two matrices. Fortunately, in this case, the maximum-likelihood estimates (MLEs) of the covariance matrices and the proportionality coefficient between them have a simple close form (Guttman et al. 1985), so that maximization of the log-likelihood need not be performed numerically, contrary to the other tests in CPC analysis. In the following, we recall the close form of these MLEs, and then we adapt the test to metapopulation studies in which a between- and a within-population covariance matrix have been estimated by MANOVA.
Consider two samples (of size n1 and n2) drawn from two multivariate Gaussian distributions with covariance matrices Σ1 and Σ2 of dimension p × p. From these samples, two sample covariance matrices (S1 and S2) can be estimated. Because the samples are drawn into Gaussian distributions, n1S1 and n2S2 are distributed as Wishart deviates W(n1, Σ1) and W(n2, Σ2), respectively (Flury 1988). Using the probability density function of the Wishart, the likelihood of the set (Σ1, Σ2) given that its estimate from the two samples is (S1, S2) can be computed. Under the assumption of proportionality between the covariance matrices (Σ1, Σ2), a positive real number ρ exists such that Σ1 = ρ Σ2, and the MLEs of ρ, Σ1 and Σ2, are the values 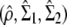 that maximize the likelihood of the observed covariance matrices S1 and S2 given that Σ1 and Σ2 are proportional. Denoting n = n1 + n2 as the total sample size and r1 = n1/n and r2 = n2/n as the relative sample sizes of the two samples, the MLEs are given by
that maximize the likelihood of the observed covariance matrices S1 and S2 given that Σ1 and Σ2 are proportional. Denoting n = n1 + n2 as the total sample size and r1 = n1/n and r2 = n2/n as the relative sample sizes of the two samples, the MLEs are given by
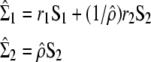 |
(3) |
(Equations 1.1 and 1.2 of Flury 1988, p. 102), where 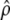 is the estimated coefficient of proportionality that is the unique positive number verifying
is the estimated coefficient of proportionality that is the unique positive number verifying
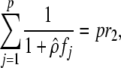 |
(4) |
where the {fj}j∈[1,p] are the eigenvalues of ( (the −1 denotes matrix inverse). Furthermore, the sample distribution of
(the −1 denotes matrix inverse). Furthermore, the sample distribution of  is a Gaussian with mean ρ and variance
is a Gaussian with mean ρ and variance 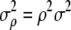 , where
, where 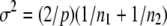 (Equation 4.13 of Flury 1988, p. 119), so that a confidence interval (to the level α) for ρ, given a sample estimate
(Equation 4.13 of Flury 1988, p. 119), so that a confidence interval (to the level α) for ρ, given a sample estimate 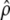 , is
, is
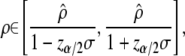 |
(5) |
where zα/2 is the quantile of level α/2 of the standard normal N(0, 1). Note that these results assume nonsphericity, meaning that all eigenvalues of S1 and S2 are distinct. In the case of sphericity (some eigenvalues are equal within both S1 and S2), slight changes have to be made (Flury 1988, p. 106). In empirical estimates, there is little reason to expect sphericity in S1 or S2, unless some eigenvalues are zero (positive semidefinite matrices), but in this case, a correction must anyhow be made to change the matrices to positive definite.
The null hypothesis H0 that Σ1 and Σ2 are proportional can be tested against the alternative that Σ1 and Σ2 are arbitrary. The log-likelihood-ratio statistic to be used is
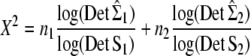 |
(6) |
(Equation 1.1 of Flury 1988, p. 150). Under the hypothesis H0 of proportionality, X2 follows a chi-square distribution with  p(p + 1) − 1 d.f.:
p(p + 1) − 1 d.f.:
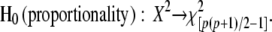 |
(7) |
However, this predicted distribution is valid only asymptotically (i.e., if both n1 and n2 are large), but can be fairly erroneous otherwise (Eriksen 1987). When one or both sample sizes are small, a correction based on the Bartlett adjustment of likelihood ratios gives more accurate results (Eriksen 1987). The likelihood-ratio X2 (Equation 6) is multiplied by a correction factor B1 so that the corrected quantity B1X2 follows the predicted χ2-distribution in Equation 7 to order O(n−3/2). In all our analyses, we used this corrected likelihood ratio B1X2, instead of X2 above, where B1 was implemented as in Theorem 6.1. (i) of Eriksen (1987).
Comparison of among- vs. within-population covariances:
The test presented above can be used to test for the neutral divergence among several populations on a set of traits (i.e., the expected pattern given in Equation 2). The expected covariances among and within populations (D and G) can be estimated by a MANOVA with subpopulations taken as a factor. If SSb and SSw are the matrices of the sum of squares corresponding to each level (among and within subpopulations) in the MANOVA table, then MSb = SSb/nb and MSw = SSw/nw are the mean squares matrices, with nb and nw the corresponding degrees of freedom. The sample distribution of the sum-of-squares matrices is a Wishart (Hill and Thompson 1978) so that the mean square matrices follow exactly the same sample distribution as that of sample covariance matrices S1 and S2 assumed in the Flury proportionality test described above,
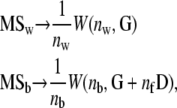 |
(8) |
where nf is the number of families sampled per subpopulation, in a balanced design. In an unbalanced design where distinct sample sizes, nf[i], have been taken in each subpopulation i, a corrected value of nf should be taken,
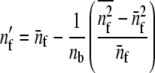 |
(9) |
(Sokal and Rolfh 1981), where bars denote average values across groups (i.e., here, populations). The estimates of the within- and among-populations covariance matrices, D and G, are 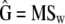 and
and 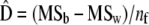 .
.
From these estimates, testing for neutrality on the set of traits considered should reduce to two tests. First, one should test whether the proportionality coefficient 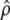 between G and D departs significantly from 2Fst/(1 − Fst), i.e., whether there is an overlap between the confidence interval of
between G and D departs significantly from 2Fst/(1 − Fst), i.e., whether there is an overlap between the confidence interval of  (Equation 5) and that of 2Fst/(1 − Fst) estimated from several molecular markers. Second, proportionality between D and G based on their estimates
(Equation 5) and that of 2Fst/(1 − Fst) estimated from several molecular markers. Second, proportionality between D and G based on their estimates 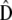 and
and 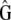 should be tested following the method described in Equations 6 and 7. However, applying this test as such is a priori inexact as
should be tested following the method described in Equations 6 and 7. However, applying this test as such is a priori inexact as 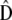 does not follow the distribution assumed in the test, i.e., that of (1/nb)W(nb, D). Note that the problem is relevant only for the among-population covariance matrix as
does not follow the distribution assumed in the test, i.e., that of (1/nb)W(nb, D). Note that the problem is relevant only for the among-population covariance matrix as 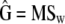 is distributed as (1/nw)W(nw, G). For correspondence with the test described above (Equations 6 and 7), the test of proportionality should be performed on the mean square matrices MSb and MSw, which do follow the assumed distribution (Equation 8). The proportionality test described in the previous section is then performed by setting n1 = nw, n2 = nb, S1 = MSw, S2 = MSb, Σ1 = G, and Σ2 = G + nfD. On the basis of this correspondence and Equation 2, the expected relationship between mean square matrices, under neutrality, is
is distributed as (1/nw)W(nw, G). For correspondence with the test described above (Equations 6 and 7), the test of proportionality should be performed on the mean square matrices MSb and MSw, which do follow the assumed distribution (Equation 8). The proportionality test described in the previous section is then performed by setting n1 = nw, n2 = nb, S1 = MSw, S2 = MSb, Σ1 = G, and Σ2 = G + nfD. On the basis of this correspondence and Equation 2, the expected relationship between mean square matrices, under neutrality, is
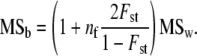 |
(10) |
Equation 10 summarizes the tests to be performed for testing the neutrality of a set of traits, with among-populations and within-populations covariances estimated by MANOVA. Two successive tests have to be performed: (i) testing whether the estimated proportionality coefficient between MS matrices 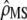 departs significantly from 1 + nf(2Fst/(1 − Fst)) and (ii) testing for proportionality between MSb and MSw, using the likelihood-ratio test proposed in Equations 6 and 7. The proportionality coefficient
departs significantly from 1 + nf(2Fst/(1 − Fst)) and (ii) testing for proportionality between MSb and MSw, using the likelihood-ratio test proposed in Equations 6 and 7. The proportionality coefficient 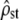 between D and G can be expressed from
between D and G can be expressed from 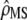 and nf as
and nf as 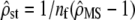 . This transformation from
. This transformation from 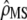 to
to 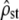 is linear so it does not change the (Gaussian) distributional properties of the estimator of ρ. Therefore, while test ii has to be performed on MS matrices rather than on G vs. D, test i can be performed directly on
is linear so it does not change the (Gaussian) distributional properties of the estimator of ρ. Therefore, while test ii has to be performed on MS matrices rather than on G vs. D, test i can be performed directly on 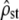 instead of
instead of  and the neutrality assumption to be tested is
and the neutrality assumption to be tested is
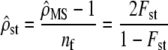 |
(11) |
(from Equation 2). This assumption is tested by checking for an overlap between the empirical C.I. of  and that of 2Fst/(1 − Fst). The C.I. for
and that of 2Fst/(1 − Fst). The C.I. for 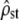 is given by Equation 5, replacing ρ by its estimate
is given by Equation 5, replacing ρ by its estimate  and n1 and n2 by the degrees of freedom of the MANOVA, nw and nb, respectively. The value of 2Fst/(1 − Fst) can be estimated directly from polymorphism data at several neutral markers, by (for diploids)
and n1 and n2 by the degrees of freedom of the MANOVA, nw and nb, respectively. The value of 2Fst/(1 − Fst) can be estimated directly from polymorphism data at several neutral markers, by (for diploids)
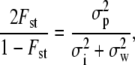 |
(12) |
where 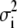 ,
, 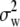 , and
, and 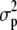 are the components of variance of marker allele frequencies, within individuals, between individuals within populations, and between populations, respectively, and are estimated from a classical Fst analysis. The corresponding C.I. of the ratio in Equation 12 can be obtained, e.g., by bootstrap over the marker loci.
are the components of variance of marker allele frequencies, within individuals, between individuals within populations, and between populations, respectively, and are estimated from a classical Fst analysis. The corresponding C.I. of the ratio in Equation 12 can be obtained, e.g., by bootstrap over the marker loci.
Note that in the case of an unbalanced design these tests are only approximate because of the use of a corrected nf′ (Equation 9). In any case, because a limited number of populations can be studied empirically, the test will always be approximate due to the small number of degrees of freedom for the between-population level (usually 5 < nb < 20). This exemplifies the importance of using the Bartlett adjustment of the χ2-test in Equation 7. These two simple statistical tests were implemented with the software R (Ihaka and Gentleman 1996), and the code is available at http://www.isem.cnrs.fr/spip.php?article934.
Simulations:
We checked the accuracy of our two tests by simulations. We studied whether the tests behaved as expected under the null hypothesis H0 (proportionality), to estimate the importance of type I errors (Figures 1 and 2). We then studied the power of the tests (importance of type II error) by checking whether the tests correctly rejected nonneutrality (Figure 3). These simulations, performed with R (Ihaka and Gentleman 1996), are detailed below.
Figure 1.—


Effect of the number of populations sampled on the accuracy of the tests: application of tests i and ii on 3000 simulated phenotype distributions with proportional G and D matrices, D = 1.5 G (see methods). Each of nb + 1 populations consisted of N = 100 individuals with multivariate normal distributions of phenotypes (five traits), and MS matrices were estimated by MANOVA on samples of nf = 20 individuals per population. The number of sampled populations was varied from 7 to 20 to study the effect of sampling effort on the tests. (a) Test i: the distribution of  -estimates from replicate simulations. Dashed lines give the theoretical C.I. (Equation 5 inverted, see methods), solid lines give the estimated C.I. from simulations, and circles give the mean
-estimates from replicate simulations. Dashed lines give the theoretical C.I. (Equation 5 inverted, see methods), solid lines give the estimated C.I. from simulations, and circles give the mean  from simulations compared to its expected mean ρ = 1.5 (straight line). For all values of nb, the theoretical distribution is accurate. (b) Test ii: the distribution of P-values of the proportionality test between MSb and MSw, using the asymptotic χ2-test for the likelihood-ratio X2 (Equations 6 and 7) (left) or for the Bartlett-adjusted likelihood ratio (B1X2, Eriksen 1987). The solid line gives the density of the uniform over [0, 1], which is the expected distribution of P-values in our simulations (G and D proportional, H0 is true). The Bartlett adjustment makes the test accurate even for small numbers of sampled populations (nb < 10, right).
from simulations compared to its expected mean ρ = 1.5 (straight line). For all values of nb, the theoretical distribution is accurate. (b) Test ii: the distribution of P-values of the proportionality test between MSb and MSw, using the asymptotic χ2-test for the likelihood-ratio X2 (Equations 6 and 7) (left) or for the Bartlett-adjusted likelihood ratio (B1X2, Eriksen 1987). The solid line gives the density of the uniform over [0, 1], which is the expected distribution of P-values in our simulations (G and D proportional, H0 is true). The Bartlett adjustment makes the test accurate even for small numbers of sampled populations (nb < 10, right).
Figure 2.—

Accuracy of the tests under neutral divergence. Among- and within-population MS matrices were estimated by MANOVA for 100 replicate simulations of neutral evolution (see methods), at 20-generations time intervals (corresponding to increasing Fst in the x-axis). Each of 10 isolated subpopulations of size N = 100 haploid individuals with five additive traits underwent mutation, drift, and recombination. For each time point and replicate simulation, 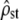 was estimated according to Equation 11 and the proportionality of MS matrices was tested (Bartlett-adjusted test). (Left) Test i: box plot of the estimated
was estimated according to Equation 11 and the proportionality of MS matrices was tested (Bartlett-adjusted test). (Left) Test i: box plot of the estimated 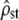 . The estimated 95% C.I.'s of
. The estimated 95% C.I.'s of 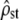 (bars) correspond to the predicted C.I.'s (dashed lines) under neutrality, and the estimated means (solid squares) are in perfect agreement with their predicted value [Fst/(1 − Fst), solid line] at all levels of population divergence (Fst x-axis). (Right) Test ii: distribution of the P-values (pooled from all time intervals) of the Bartlett-adjusted test on MS matrices. The distribution does not differ from the expected uniform [0, 1] (type I error and P-value for the one-sided KS test of comparison with the uniform given on the graph).
(bars) correspond to the predicted C.I.'s (dashed lines) under neutrality, and the estimated means (solid squares) are in perfect agreement with their predicted value [Fst/(1 − Fst), solid line] at all levels of population divergence (Fst x-axis). (Right) Test ii: distribution of the P-values (pooled from all time intervals) of the Bartlett-adjusted test on MS matrices. The distribution does not differ from the expected uniform [0, 1] (type I error and P-value for the one-sided KS test of comparison with the uniform given on the graph).
Figure 3.—



Power to detect nonneutral divergence: the same as Figure 2 but with individuals undergoing selection in addition to drift, mutation, and recombination. In addition to its expected range under neutrality [Equation 5 inverted with ρ = Fst/(1 − Fst), solid dashed lines], the theoretical C.I. of 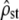 predicted from its observed value (Equation 5 inverted with ρ = mean
predicted from its observed value (Equation 5 inverted with ρ = mean 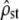 ) was also reported (shaded dashed lines), to check the robustness of Equation 5 to nonproportionality between MS matrices. (a) Homogeneous selection for the same optimum:
) was also reported (shaded dashed lines), to check the robustness of Equation 5 to nonproportionality between MS matrices. (a) Homogeneous selection for the same optimum: 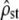 is significantly lower than the neutral expectation Fst/(1 − Fst) (estimated and predicted C.I.'s do not overlap, left) and the P-value distribution of the Bartlett test is significantly different from uniform [0, 1] (right). However, in a large proportion of simulations, proportionality is not rejected, although false (high type II error risk, indicated on the graph). (b) Heterogeneous selection for distinct optima in each population:
is significantly lower than the neutral expectation Fst/(1 − Fst) (estimated and predicted C.I.'s do not overlap, left) and the P-value distribution of the Bartlett test is significantly different from uniform [0, 1] (right). However, in a large proportion of simulations, proportionality is not rejected, although false (high type II error risk, indicated on the graph). (b) Heterogeneous selection for distinct optima in each population: 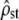 is significantly higher than Fst/(1 − Fst) (left) and the P-value distribution is not uniform [0, 1] but again there is a high type II error risk (right). (c) Mixture of both selection regimes with two traits under heterogeneous selection and three traits under homogeneous selection: this time,
is significantly higher than Fst/(1 − Fst) (left) and the P-value distribution is not uniform [0, 1] but again there is a high type II error risk (right). (c) Mixture of both selection regimes with two traits under heterogeneous selection and three traits under homogeneous selection: this time, 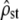 is not significantly different from Fst/(1 − Fst) (left), but proportionality is systematically rejected (low type II error risk, right ). In all cases, the theoretical C.I. of
is not significantly different from Fst/(1 − Fst) (left), but proportionality is systematically rejected (low type II error risk, right ). In all cases, the theoretical C.I. of  is relatively accurate (compare shaded bars and shaded dashed lines), although MS matrices are not proportional.
is relatively accurate (compare shaded bars and shaded dashed lines), although MS matrices are not proportional.
Accuracy of the test on sample covariance matrices:
We first checked that the proportionality test was accurate on simulated phenotype distributions of a metapopulation that corresponded to the assumptions of the test: proportionality of the within- and between-population covariance matrices (G and D, respectively), with a known value of ρ. To this end, we drew samples of individual phenotypes, from several subpopulations, into multivariate normal distributions that G and D matrices were set to be proportional such that D = ρG. First, a p × p covariance matrix G between p = 5 phenotypic traits was created as a Wishart deviate (the choice of the Wishart, here, is simply a null model of covariance matrix and bears no importance to the test). Then a second p × p covariance matrix D proportional to G was created as D = ρG. For each population i, the mean phenotype of the population was drawn as a p × 1 vector  from a multivariate normal distribution with mean 0 and covariance matrix D. Then N = 100 individual phenotype vectors were drawn into multivariate normal distributions with mean
from a multivariate normal distribution with mean 0 and covariance matrix D. Then N = 100 individual phenotype vectors were drawn into multivariate normal distributions with mean 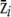 and covariance matrix G. Therefore, on average, the resulting phenotype distribution across populations had a within-population covariance equal to G and a between-population covariance equal to
and covariance matrix G. Therefore, on average, the resulting phenotype distribution across populations had a within-population covariance equal to G and a between-population covariance equal to 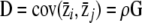 . Finally to simulate empirical sampling, samples of nf = 20 individuals from each subpopulation were drawn randomly. From this sample across the metapopulation, estimates of the between- and within-population mean square matrices (MSb and MSw) and covariance matrices
. Finally to simulate empirical sampling, samples of nf = 20 individuals from each subpopulation were drawn randomly. From this sample across the metapopulation, estimates of the between- and within-population mean square matrices (MSb and MSw) and covariance matrices 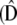 and
and 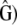 were computed by MANOVA, and the corresponding degrees of freedom were estimated. Using the proportionality test presented above, the proportionality coefficient
were computed by MANOVA, and the corresponding degrees of freedom were estimated. Using the proportionality test presented above, the proportionality coefficient 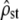 was estimated (test i, Equation 4) and the proportionality between MSb and MSw was tested (test ii, Equations 6 and 7). To check for the importance of the Bartlett adjustment (Eriksen 1987), we estimated the P-value of the proportionality test ii for both the likelihood ratio (X2, Equation 6) and the Bartlett-adjusted ratio (B1X2).
was estimated (test i, Equation 4) and the proportionality between MSb and MSw was tested (test ii, Equations 6 and 7). To check for the importance of the Bartlett adjustment (Eriksen 1987), we estimated the P-value of the proportionality test ii for both the likelihood ratio (X2, Equation 6) and the Bartlett-adjusted ratio (B1X2).
This process was replicated 3000 times (with the same value of ρ = 1.5, G and D) to measure the distribution of 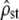 estimates and that of the P-values of the proportionality test (either the χ2-test or the Bartlett-adjusted test). First, we checked whether the mean of estimated
estimates and that of the P-values of the proportionality test (either the χ2-test or the Bartlett-adjusted test). First, we checked whether the mean of estimated 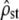 was equal to its simulated value ρ = 1.5 and lay within its predicted range, obtained by inverting Equation 5,
was equal to its simulated value ρ = 1.5 and lay within its predicted range, obtained by inverting Equation 5, 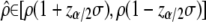 at the α = 5% level. Second, we checked that the distribution of the P-values obtained from the proportionality test (asymptotic χ2 of Equation 7 or Bartlett-adjusted version) did not differ from a uniform over [0, 1], as expected when the null hypothesis is true.
at the α = 5% level. Second, we checked that the distribution of the P-values obtained from the proportionality test (asymptotic χ2 of Equation 7 or Bartlett-adjusted version) did not differ from a uniform over [0, 1], as expected when the null hypothesis is true.
Accuracy of the test on simulated evolution:
The accuracy of the neutrality tests was checked in a similar way as above, but with individual-based simulations of population divergence under mutation, drift, recombination, and optionally selection. The simulation scheme was meant to correspond to realistic (although large) empirical schemes (e.g., Chapuis et al. 2008). We simulated the phenotypic evolution (on five traits) of haploid individuals in 10 isolated subpopulations of equal size N = 100, during 100 generations of divergence. An initial population was created by simulating 300 generations of mutation starting from an isomorphic population of size N. This ancestral population served as the initial state of each subpopulation before divergence. Then selection (optionally), drift, pleiotropic mutation, and reproduction (with free recombination between loci) were simulated in this order, in each isolated subpopulation (see details in the next section). Every 20 generations, sets of nf = 20 individuals were sampled in each subpopulation, and, as in the previous section, their phenotype distribution across populations was used to estimate the MS matrices (MANOVA), 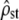 (Equations 4 and 11), and test for proportionality between MSb and MSw (using only the Bartlett-adjusted test, which is more accurate). This process was replicated 100 times starting from the same initial population, to compute the distribution of
(Equations 4 and 11), and test for proportionality between MSb and MSw (using only the Bartlett-adjusted test, which is more accurate). This process was replicated 100 times starting from the same initial population, to compute the distribution of 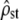 every 20 generations and of the P-value of the proportionality test. As the P-value distribution did not change over time, we studied the pooled values over the whole course of divergence (i.e., on a total of 100 × 5 = 500 replicate values).
every 20 generations and of the P-value of the proportionality test. As the P-value distribution did not change over time, we studied the pooled values over the whole course of divergence (i.e., on a total of 100 × 5 = 500 replicate values).
In parallel, the expected value of Fst was computed using the theoretical recursion from one generation to the next: Fst(t + 1) = (1 − U/L)2(1/N + (1 − 1/N)Fst(t)), starting at Fst(0) = 0, where U and L are the per-generation genomic mutation rate and the number of loci determining the traits, respectively. This recursion corresponds to haploid individuals in isolated populations undergoing mutation (infinite-allele model) and drift. Because we use U and L at the quantitative trait loci, it gives Fst at these loci under neutrality, but should not differ from Fst estimated from molecular markers in the limit of a low per-locus mutation rate of both QTL and markers (Whitlock 1999). In our examples, we simulated L = 25 loci and U = 0.1 so that, per locus, μ = U/L = 0.1/25 = 0.004.
As in the above section, the accuracy of test i was studied by comparing the mean and the range of 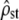 -estimates from replicate simulations at each 20-generations time interval to their predicted value under neutrality: a mean
-estimates from replicate simulations at each 20-generations time interval to their predicted value under neutrality: a mean 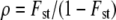 (for haploids) and range
(for haploids) and range  . To study the accuracy of test ii, the distribution of the P-values of the (Bartlett-adjusted) proportionality test in replicate simulations and over the 100 generations of divergence was compared to the uniform [0, 1], expected under neutrality (Equation 10, MSb and MSw are proportional under neutrality, H0 should be true).
. To study the accuracy of test ii, the distribution of the P-values of the (Bartlett-adjusted) proportionality test in replicate simulations and over the 100 generations of divergence was compared to the uniform [0, 1], expected under neutrality (Equation 10, MSb and MSw are proportional under neutrality, H0 should be true).
These simulation checks were performed under pure neutrality (only mutation, recombination, and drift) and under three selection regimes: homogeneous selection for a new optimum (the same in each population), heterogeneous selection for distinct new optima in each population, and a mixed regime where three traits were under homogeneous selection and the two remaining were under heterogeneous selection.
Individual-based simulations:
Mutation and selection on individual phenotypes were modeled according to the classic multivariate pleiotropic model of Lande (1979), except that allele states after mutation were independent of the ancestral allele state (house-of-cards model, Kingman 1978). Each individual consisted of L haploid loci. Mutation followed the K-allele model with a large number (K = 2000) of possible alleles per locus, which is effectively equivalent to the classic “infinite-allele model” (Kimura and Crow 1964). For each individual, at each generation, the number of new mutations in the genome (occurring at a random subset of the loci) was drawn into a Poisson distribution with parameter U, the genomic mutation rate. The index of each mutant allele was drawn into an integer uniform distribution in the range [1:2000]. Each allele had completely pleiotropic effects on the five quantitative traits, and allelic effects were additive across loci. A new mutant allele replaced the current allele effect at the locus considered (no memory of the previous allele state, house-of-cards model). For simplicity, the same set of 2000 possible allele effects was used for all loci, by drawing 2000 vectors dz (of pleiotropic effects on all five traits) into a multivariate Gaussian distribution with mean 0 and mutational covariance matrix M.
To model selection, the phenotype z of a multilocus genotype was computed as the sum of the effects of its alleles at each locus l (z = Σldzl, additivity), and the fitness W of this genotype was computed as a multivariate Gaussian function of z around an optimum zo:  , where S is the matrix of selective covariances.
, where S is the matrix of selective covariances.
In the simulations with selection, the strength of selection was controlled by the parameter 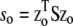 : the log fitness of the optimal phenotype (z = zo) relative to the mean fitness of the initial population (Martin and Lenormand 2006). It was set to so = 0.7 in all cases, such that mean fitness increased by <1% per generation, which corresponds to an intermediate level of selection. Random mating of haplotypes and free recombination occurred at each generation within each subpopulation. Drift and (optionally) selection were jointly simulated each generation by sampling (with replacement) into each subpopulation of parents to produce the next generation with sampling probability equal to the fitness of each genotype in the parent population. This corresponds to the Wright–Fisher model of genetic drift. Under neutrality, all fitnesses were set to W = 1.
: the log fitness of the optimal phenotype (z = zo) relative to the mean fitness of the initial population (Martin and Lenormand 2006). It was set to so = 0.7 in all cases, such that mean fitness increased by <1% per generation, which corresponds to an intermediate level of selection. Random mating of haplotypes and free recombination occurred at each generation within each subpopulation. Drift and (optionally) selection were jointly simulated each generation by sampling (with replacement) into each subpopulation of parents to produce the next generation with sampling probability equal to the fitness of each genotype in the parent population. This corresponds to the Wright–Fisher model of genetic drift. Under neutrality, all fitnesses were set to W = 1.
The selective and mutational covariance matrices S and M were created randomly by drawing into Wishart distributions and evenly scaled so as to get a given distribution of the fitness effects of single deleterious mutations [i.e., known value of the average E(s) and variance V(s)], as in Martin and Lenormand (2006). This allows parameterizing the simulations according to known mutational parameters in model species. Here, parameters roughly corresponded to Drosophila melanogaster: U = 0.1, E(s) = 0.1, and V(s) = 0.012. With these settings, there are fairly strong mutational and selective correlations and heterogeneity among traits for both mutational and selective (co)variances.
RESULTS
Detection of the expected pattern under neutrality:
Accuracy of the proportionality test as a function of sampling effort:
Figure 1 shows the impact of the number of populations sampled (nb + 1) on the accuracy of the proportionality test. From simulations of phenotype distributions across sets of populations with proportional between- and within-covariance matrices, we estimated the 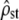 -estimates between G and D and compared them with their expected mean (ρ = 1.5) and range (see methods). Figure 1a shows that, even for the smallest values of nb, the sample distribution of the proportionality coefficient
-estimates between G and D and compared them with their expected mean (ρ = 1.5) and range (see methods). Figure 1a shows that, even for the smallest values of nb, the sample distribution of the proportionality coefficient  (3000 replicate simulations) is accurately predicted by the application (to MANOVA estimates, Equation 11) of the Flury approach (Equations 4 and 5). This suggests that test i (
(3000 replicate simulations) is accurately predicted by the application (to MANOVA estimates, Equation 11) of the Flury approach (Equations 4 and 5). This suggests that test i (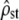 vs. Fst/(1 − Fst)) should be valid even when very few populations are sampled empirically, although the test would have reduced power (larger C.I. for
vs. Fst/(1 − Fst)) should be valid even when very few populations are sampled empirically, although the test would have reduced power (larger C.I. for 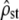 with small nb).
with small nb).
Figure 1b shows the distribution of the P-values of the test of proportionality between MSb and MSw (test ii, Equation 10) over the same 3000 replicate simulations. We show two options for testing proportionality: the classic asymptotic χ2-test proposed by Flury (1988) and used in the CPC software (Phillips and Arnold 1999) and the version proposed by Eriksen (1987) with a Bartlett adjustment for small samples. Figure 1b, left, shows that the asymptotic χ2-test is not very accurate for realistic numbers of sampled populations (i.e., up to 20 populations, an already large sampling effort). Indeed, the distribution of P-values is not the uniform U[0, 1] expected under the null hypothesis (which was simulated), and rejection of the true H0 hypothesis occurs more often than the expected α = 5% level (type I error). This occurs because the null (χ2-) distribution of X2 predicted in Equation 7 is valid only when both degrees of freedom n1 and n2 are large. In our case, nb < 20 appears to be too small for the asymptotic distribution to be accurate. However, Figure 1b, right, shows that the Bartlett-adjusted test (B1X2, see methods and Eriksen 1987) is accurate for realistic numbers of sampled populations (≥10 populations), as the distribution of P-values is uniform [0, 1] as expected. On the basis of our simulations (not shown), the test seems to be more accurate when p ≪ nb, where p is the number of traits (e.g., p ≤ 2nb). Detailed study of the power of the test for a given data set can be easily performed by simple simulations such as those presented here, so we did not delve into these aspects any further.
Overall, it appears that the Bartlett-adjusted test is necessary and sufficient to test proportionality between MSb and MSw among realistic samples based on MANOVA estimates. Furthermore, it appears that a moderate to large number of sampled populations are required to obtain a reliable proportionality test (test ii), the more so when many traits are measured, while the sampling distribution of  (test i) is correctly predicted even with samples from only very few populations (although its C.I. is then large).
(test i) is correctly predicted even with samples from only very few populations (although its C.I. is then large).
Accuracy of the neutrality test under neutral evolution:
We checked whether the relationship predicted under neutral divergence (Equations 10 and 11) was correct using individual-based simulations and with the same approach as in the previous section (Figure 1). For each of 100 replicate simulations, every 20 generations, MSb and MSw were estimated by MANOVA, their proportionality was tested (Bartlett adjustment), and  was estimated following Equation 11:
was estimated following Equation 11:
Test i: Figure 2, left, shows that, for all time intervals (corresponding to distinct values of Fst in the x-axis),
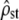 -estimates lie within their predicted range (dashed lines) with mean Fst/(1 − Fst) as predicted under neutrality (test i). Note also that the range of
-estimates lie within their predicted range (dashed lines) with mean Fst/(1 − Fst) as predicted under neutrality (test i). Note also that the range of 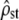 -values is reasonably small, smaller than most estimates of Qst (means over traits) found in the literature (e.g., McKay and Latta 2002).
-values is reasonably small, smaller than most estimates of Qst (means over traits) found in the literature (e.g., McKay and Latta 2002).Test ii: Figure 2, right, shows the distribution of P-values for the proportionality test (test ii), pooled over all time intervals (100 replicate simulations × 5 time intervals = 500 P-values). The distribution does not differ from a uniform [0, 1] [Kolmogorov–Smirnov (KS) test, P = 0.156] as appears in Figure 2, and the type I error is equal to that predicted at the α = 5% level (type I error probability = 5.6%). Overall, this shows that the hypothesis of proportionality between MSb and MSw (Equation 11) under neutral divergence (only drift, recombination, and mutation are occurring) is correct and that the (Bartlett-corrected) proportionality test accurately detects it.
Detecting selection by departures from the neutral pattern (joint use of the two tests):
Figure 3 corresponds to Figure 2 for the case of nonneutral divergence between populations. Figure 3a shows the behavior of the two tests under homogeneous selection (for the same optimum in each population). It appears that test i (Figure 3a, left) accurately rejects neutrality as the estimated 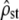 from simulations all lay below their expected neutral range. On the contrary (Figure 3a, right, test ii), although the distribution of P-values for the proportionality test differed from a uniform (KS test, P < 10−4), as expected because MSb and MSw are not proportional, a large number of the tests failed to (correctly) reject proportionality (i.e., there was a large type II error probability = 84.4%).
from simulations all lay below their expected neutral range. On the contrary (Figure 3a, right, test ii), although the distribution of P-values for the proportionality test differed from a uniform (KS test, P < 10−4), as expected because MSb and MSw are not proportional, a large number of the tests failed to (correctly) reject proportionality (i.e., there was a large type II error probability = 84.4%).
Similarly, Figure 3b shows that in the case of heterogeneous selection, test i (left) accurately rejects neutrality as  from simulations all lay above their expected neutral range, but that test ii (right) was not very powerful to detect selection. Again, although a uniform distribution of P-values is clearly rejected (KS test, P < 10−4, H0 is false), many of the P-values were above the rejection level (type II error probability = 91.5%).
from simulations all lay above their expected neutral range, but that test ii (right) was not very powerful to detect selection. Again, although a uniform distribution of P-values is clearly rejected (KS test, P < 10−4, H0 is false), many of the P-values were above the rejection level (type II error probability = 91.5%).
Finally, in some situations, the set of traits under study may be under a “mixed” selection regime, where some of the traits are under directional selection while the others are under stabilizing selection. In this case, the average divergence of quantitative traits under opposite forces may equal that of neutral markers, so that a comparison of a mean Qst among traits with Fst leaves selection undetected. However, using the multivariate approach, this selection regime can be detected, as a mixed selection regime does not keep an overall proportionality between G and D. Figure 3c illustrates this: the mixed regime can hardly be distinguished from pure neutrality only on the basis of the values of 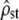 , which remain close to or within the range expected under neutrality (test i, left). However, proportionality between MSb and MSw is clearly rejected in almost all simulations (test ii, right: type II error probability = 1%), which allows identifying the presence of selection.
, which remain close to or within the range expected under neutrality (test i, left). However, proportionality between MSb and MSw is clearly rejected in almost all simulations (test ii, right: type II error probability = 1%), which allows identifying the presence of selection.
Overall, the joint use of both tests allows properly detecting all selection regimes, while still correctly retaining H0 under neutral conditions. Homogeneous and heterogeneous selection on all traits is efficiently detected by test i in a way similar to classic Qst–Fst comparisons, with 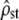 lying respectively below or above its expected value under neutrality (Fst/(1 − Fst)). On the other hand, the case where some traits are under homogeneous and others under heterogeneous selection is detected through test ii as the proportionality between MSb and MSw is strongly rejected. Furthermore, it is noteworthy that the predicted C.I. for
lying respectively below or above its expected value under neutrality (Fst/(1 − Fst)). On the other hand, the case where some traits are under homogeneous and others under heterogeneous selection is detected through test ii as the proportionality between MSb and MSw is strongly rejected. Furthermore, it is noteworthy that the predicted C.I. for  based on its estimate (Equation 5, shaded dashed lines in Figure 3) is still fairly accurate under selective divergence. It need not be the case, as the C.I. is predicted under the assumption of proportionality between MS matrices, which fails when selection is occurring. This point is useful as test i is based on comparing an empirical C.I. with a predicted C.I. under neutrality and would fail to be accurate under selection if the empirical C.I. was not well predicted by Equation 5 in this case. Note, however, that in the case of homogeneous selection (Figure 3c) the predicted C.I. tended to underestimate the observed C.I., so that care should be taken when only a small gap between the neutral and the observed C.I. is observed, before concluding to the influence of homogeneous selection.
based on its estimate (Equation 5, shaded dashed lines in Figure 3) is still fairly accurate under selective divergence. It need not be the case, as the C.I. is predicted under the assumption of proportionality between MS matrices, which fails when selection is occurring. This point is useful as test i is based on comparing an empirical C.I. with a predicted C.I. under neutrality and would fail to be accurate under selection if the empirical C.I. was not well predicted by Equation 5 in this case. Note, however, that in the case of homogeneous selection (Figure 3c) the predicted C.I. tended to underestimate the observed C.I., so that care should be taken when only a small gap between the neutral and the observed C.I. is observed, before concluding to the influence of homogeneous selection.
The results of the test in each type of selection regime are summarized in Table 1, which shows that the joint use of both tests allows detecting every selection regime.
TABLE 1.
Results of the neutrality tests according to the selection regime
| Selection regime | Test i: 
|
Test ii: MSb and MSw proportional |
|---|---|---|
| Neutral | Minimal rejection | Minimal rejection |
| Homogeneous selection | High rejection | Low rejectiona |
| Heterogeneous selection | High rejection | Low rejectiona |
| Mixed regime | Low rejectiona | High rejection |
For all divergence scenarios considered in Figures 2 and 3 the outcome of the two neutrality tests in our simulations is summarized. Both tests are accurate under neutrality but in all cases of selection, one of them fails. However, at least one test is accurate so that the joint use of tests i and ii allows detecting all selection regimes.
An outcome that does not correspond to the correct situation (in all cases it corresponds to a high type II error risk).
DISCUSSION
In this article, we devised and tested a new neutrality test for quantitative traits, which is a multivariate extension of the classic Qst–Fst comparison. The idea is to compare the among-population (D) and within-population (G) covariance matrices and to test the neutral pattern of D = Fst/(1 − Fst)G (for haploids, as in our simulations, and with a factor 2 for diploids). The test is twofold: (i) testing for equality between an estimate of the proportionality coefficient ( , Equation 11) and its expectation (Fst/(1 − Fst)) from neutral markers and (ii) testing for proportionality itself between D and G. The first test (test i) is very close to the classic Qst–Fst comparisons but in a rigorous multivariate framework, while test ii is more akin to the approaches proposed in the studies of G matrix evolution (e.g., Schluter 1996). Our tests make use of Flury's (1988) framework of sample covariance matrix comparisons (CPC analysis), with a Bartlett correction for small sample sizes proposed by Eriksen (1987), which proves essential for realistic sampling designs (<20 populations sampled, Figure 1). Note that the software CPC (Phillips and Arnold 1999), although a pioneer in divulging the CPC framework among evolutionary biologists, does not use the Bartlett adjustment, which could be problematic for at least some data sets in evolutionary studies, with typically relatively small sample sizes. When using the proportionality test in our particular context, the correction will always be necessary, as one of the degrees of freedom is given by the number of populations (rarely exceeding 10).
, Equation 11) and its expectation (Fst/(1 − Fst)) from neutral markers and (ii) testing for proportionality itself between D and G. The first test (test i) is very close to the classic Qst–Fst comparisons but in a rigorous multivariate framework, while test ii is more akin to the approaches proposed in the studies of G matrix evolution (e.g., Schluter 1996). Our tests make use of Flury's (1988) framework of sample covariance matrix comparisons (CPC analysis), with a Bartlett correction for small sample sizes proposed by Eriksen (1987), which proves essential for realistic sampling designs (<20 populations sampled, Figure 1). Note that the software CPC (Phillips and Arnold 1999), although a pioneer in divulging the CPC framework among evolutionary biologists, does not use the Bartlett adjustment, which could be problematic for at least some data sets in evolutionary studies, with typically relatively small sample sizes. When using the proportionality test in our particular context, the correction will always be necessary, as one of the degrees of freedom is given by the number of populations (rarely exceeding 10).
Our simulations suggest that, with a realistic sampling design (10 populations, 20 families per population, five traits measured), the predicted neutral pattern is exact when only mutation and drift (and potentially migration) affect the trait (Figure 2) and that different forms of selection can efficiently be detected (Figure 3). The efficiency of this neutrality test stems from the fact that two independent tests are used that are complementary in detecting different types of departures from the neutral pattern (Figure 3 and Table 1). We believe the combined tests have more power than the classic mean Qst–Fst comparisons for three reasons. First, the confidence intervals of 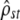 -estimates appear to be fairly reduced even with the limited data sets of our simulations, and the effect of sampling on these C.I.'s appears to be accurately predicted by the CPC framework. This is often not the case with classic mean Qst estimates over several traits, which have notoriously large C.I.'s and for which the correct statistical inference of the C.I. is problematic, both for univariate Qst (as in O'Hara and Merila 2005) and for their mean over traits (often with the implicit and wrong assumption of independence between traits). Second, the C.I. for
-estimates appear to be fairly reduced even with the limited data sets of our simulations, and the effect of sampling on these C.I.'s appears to be accurately predicted by the CPC framework. This is often not the case with classic mean Qst estimates over several traits, which have notoriously large C.I.'s and for which the correct statistical inference of the C.I. is problematic, both for univariate Qst (as in O'Hara and Merila 2005) and for their mean over traits (often with the implicit and wrong assumption of independence between traits). Second, the C.I. for  (Equation 5) is a simple product of
(Equation 5) is a simple product of 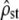 by a factor that is determined only by the sampling design (nw, nb, p), which allows making straightforward power analysis before any empirical study (detailed below). Finally and maybe more importantly, averaging over traits that are under opposite selective forces (mixture regime, Figure 3c) can lead to the erroneous acceptation of neutrality in the classic Qst approach. Logically, it is also the case based on test i, which is akin to the Qst–Fst comparison; however, test ii efficiently rejects neutrality in this case as the existence of opposite selection forces breaks down the proportionality between G and D. Overall the combined tests i and ii give a fairly powerful and statistically rigorous framework to detect various selection regimes, including some that are undetected by the classic Qst approach. Below, we discuss the robustness of our theoretical and statistical results, practical implications for empirical designs, and future developments of this approach to study multivariate evolution in natural populations.
by a factor that is determined only by the sampling design (nw, nb, p), which allows making straightforward power analysis before any empirical study (detailed below). Finally and maybe more importantly, averaging over traits that are under opposite selective forces (mixture regime, Figure 3c) can lead to the erroneous acceptation of neutrality in the classic Qst approach. Logically, it is also the case based on test i, which is akin to the Qst–Fst comparison; however, test ii efficiently rejects neutrality in this case as the existence of opposite selection forces breaks down the proportionality between G and D. Overall the combined tests i and ii give a fairly powerful and statistically rigorous framework to detect various selection regimes, including some that are undetected by the classic Qst approach. Below, we discuss the robustness of our theoretical and statistical results, practical implications for empirical designs, and future developments of this approach to study multivariate evolution in natural populations.
Robustness of the theoretical expectation and the statistical method:
Our simulations made precise assumptions for the genetic basis of the quantitative traits under study. Some are required for the theoretical prediction (Equation 2) to be valid (additivity and neutrality of the loci, no linkage disequilibrium), and some are not. For instance, as for its univariate version in Whitlock (1999), Equation 2, being derived from coalescent theory, is independent of the ecological mechanism of neutral population divergence (migration between demes of arbitrary sizes, isolation by distance, extinction/colonization dynamics, etc.), although we simulated only the simpler case of identical and isolated demes. As an example, this is confirmed in simulations of a finite-island model with migration, provided in supplemental Figure 1. Similarly, heterogeneity among loci for mutational effects, or the number of loci itself, does not influence our results based only on the total mutational variance, summed over loci (Equation 1); this was confirmed by simulations (not shown), for the number of loci. The choice of an alternative mutation model to the house of cards with Gaussian distribution of effects in our simulations should also not influence the results, as long as the mutational covariance between traits remains independent of the current state of the individual (Whitlock 1999) and as long as the breeding value distribution for each trait remains approximately Gaussian. For example, Equation 2 should still be valid in the classic infinite-allele model (Kimura and Crow 1964), where new mutation effects add up to the current allele effect at each locus, but are still drawn into a distribution that is independent of the current state. Our results should in fact still be valid when mutational covariances change through time, but the net input of mutational covariance averaged over generations is the same along every branch of the coalescent (i.e., within each subpopulation). Finally, because Equation 2 is based on coalescent theory, wrong identification of the subpopulation units in the field (e.g., if one of the samples includes in fact several biological subunits) should not invalidate the prediction under neutrality. Under selective divergence, however, such erroneous sampling would lead to including part of the among-population divergence in the within-population level. This may tend to artificially align G with D and could favor the neutrality hypothesis, when selection is in fact occurring. In this case, 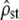 should still be different from its neutral expectation (test i), but this difference may not be significant anymore. Therefore, correct identification of biological subpopulation units is recommended, as is the case for many methods in metapopulation studies. Note that molecular markers could be used to this end, with identification of units via Bayesian clustering algorithms (e.g., Pritchard et al. 2000).
should still be different from its neutral expectation (test i), but this difference may not be significant anymore. Therefore, correct identification of biological subpopulation units is recommended, as is the case for many methods in metapopulation studies. Note that molecular markers could be used to this end, with identification of units via Bayesian clustering algorithms (e.g., Pritchard et al. 2000).
Moreover, discrepancies with basic assumptions of the model may not always invalidate its results. Linkage disequilibrium between loci determining the traits is assumed to be negligible in the coalescent approach we used (Whitlock 1999). However, as detailed in (Le Corre and Kremer 2003), significant linkage disequilibrium need not affect the Qst = Fst relationship (nor its multivariate equivalent in Equation 2) provided that it affects the between-population and the within-population covariances similarly. There are two possible sources of genetic covariance between traits: linkage disequilibrium and pleiotropic mutation. In this article, the mechanism studied is pleiotropy, but interestingly, when linkage disequilibrium, not pleiotropic mutation, is the source of the genetic covariance between traits, Equation 2 was also obtained by Rogers and Harpending (1983), although only for diallelic and nonpleiotropic loci and in an island model. However, we can expect that when neutral divergence occurs with an initial negative linkage disequilibrium buildup by past selection (Bulmer effect), the value of ρ may tend to fall below its neutral expectation (which assumes linkage equilibrium), as predicted by Le Corre and Kremer (2003) for univariate Qst measures. This effect should be even stronger with asexual populations. Finally, as shown in methods, in the case of species with some level of inbreeding, the test can still be performed but a correction must be applied by changing the expected ratio in Equation 2 to 2Fst/((1 − Fst)(1 + Fis)), where Fis is the inbreeding coefficient, also estimated from the neutral marker data.
Regarding Fst estimation, the relevant Fst value in Equation 2 should be that of the loci encoding the quantitative traits (QTL) under study and may differ from that empirically measured on neutral markers. However, as discussed in Whitlock (1999), when the influence of mutation on Fst is weak relative to that of demography and the genetic system (strong drift and/or migration relative to the mutation rate for both markers and QTL), Fst is mainly determined by demography, common to both markers and QTL, so that the neutral pattern can be tested with Fst from neutral markers.
We can see two key assumptions to which the results should be sensitive: normality of the breeding values and additivity across loci encoding the traits. The method should be fairly strongly dependent on the normality assumption, as is the case for both the CPC analysis framework (Flury 1988) and the parametric Qst estimation methods (O'Hara and Merila 2005). Consequently, proper transformation of the phenotypic data to conform to the Gaussian assumption is recommended before analyzing the empirical data. We did not study the effect of nonnormality on the tests presented here, but one prediction can be made. When traits are non-Gaussian, the deviance of all models in the CPC analysis should be lower (as the Wishart distribution does not give a good fit to the sample covariance estimates), whereas the number of parameters for each model will remain unchanged. Therefore, (i) the proportionality test may reject the neutral pattern even when it is in fact correct (type I error), and (ii) the estimate of ρ and its C.I. may be incorrect. The first point should artificially favor selective interpretations against neutral ones. The second one may induce any unwanted effect (type I or II error) and we did not study this impact here. Our simulations (Figure 3) revealed that the C.I. for ρ was fairly correctly predicted by Equation 5, even under nonneutral conditions for which the proportionality assumption was violated. Consequently, we may suspect that test i based on the comparison of ρ with 2Fst/(1 − Fst) should also be more robust to nonnormality than test ii. However, again, proper standardization of the data to approach normality is a general recommendation in many multivariate analyses, and this method is no exception.
Nonadditivity of the quantitative traits, even neutral, is an acknowledged source of bias in Qst–Fst comparisons, leading, in most cases, to a downward bias of Qst relative to Fst. Both additive-by-additive variance (Whitlock 1999) and dominance variance (Goudet and Buchi 2006) affecting a neutral trait are expected to lower Qst relative to Fst, which in our case would lead to a value of ρ < 2Fst/(1 − Fst). Note that this effect is expected in many but not all cases when there is only dominance variance (discussed in Goudet and Martin 2007; Lopez-Fanjul et al. 2007). Together with the probable negative bias induced by negative linkage disequilibria due to past selection, these results suggest that a pattern of ρ < 2Fst/(1 − Fst) should be interpreted with some caution, as previously outlined (Whitlock 1999; Goudet and Buchi 2006) for the univariate Qst–Fst comparison. Such a pattern (if weak, at least) may reflect nonadditivity or linkage disequilibria affecting otherwise approximately neutral traits. On the contrary, the opposite pattern ρ > 2Fst/(1 − Fst) can be taken with greater reliability as evidence of heterogeneous selection.
Finally, in some studies, the phenotypic covariance matrix P is used as a proxy for the genetic covariance matrix G, because it is easier to estimate. If our tests are applied to phenotypic distributions instead of breeding value distributions, environmental variance might influence the result. Let E be the environmental covariance matrix within subpopulations. Then the within-population phenotypic covariance is Pw = G + E while the between-population phenotypic covariance is Pb = D + E/nb (nb is the number of populations), so that the environmental variance increases Pw more than Pb and creates a bias. In the neutral case where D = Fst/(1 − Fst)G, the relationship on phenotypic covariances will be Pb < Fst/(1 − Fst)Pw, with nonproportionality between Pw and Pb, thus mimicking the effect of stabilizing selection. The impact of this bias is larger with smaller Fst and larger environmental variance E. Overall it is therefore advisable to apply the test on breeding values, and when applied on phenotypic values and rejecting proportionality with a pattern ρ < 2Fst/(1 − Fst), the results will again have to be interpreted with caution, as with nonadditivity (above).
We did not simulate all departures from our model assumptions. However, on the basis of the above theoretical arguments and our simulations, we believe that the test presented in this article is fairly robust to several genetic and ecological details underlying quantitative variation in a metapopulation. Yet, we note that some departures from the basic assumptions of the model may lead to erroneous conclusions. In this regard, the test on  -values (test i) is probably more robust than the proportionality test (test ii).
-values (test i) is probably more robust than the proportionality test (test ii).
Best experimental design and method to detect selection:
The analytic expression of the C.I. for ρ (Equation 5) gives a straightforward basis for optimization of any empirical sampling design. Two conclusions can be drawn from this equation and from our study in general. First, the number of traits (p) and the effect of sampling [harmonic mean of sampling sizes between and within populations (1/nw + 1/nb)] act multiplicatively to reduce the sample variance of 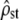 , so that it is not necessary to pool numerous traits together if the sampling design is large enough. This is important in the perspective of being able to distinguish between different types of traits: indeed, by choosing a biologically coherent set of traits (e.g., morphological, life history, etc.), which may be rather small, the results can be interpreted with more relevance. Second, the sampling effort should be balanced as much as possible between the among-population and the within-population levels, as the sampling variance decreases with the harmonic mean of relative sample sizes (1/nw + 1/nb). Furthermore, the χ2-distribution of the log-likelihood ratio (Equation 7) under proportionality, even with the Bartlett correction of Eriksen (1987), is an asymptotic result that is more accurate when both nb and nw are large (in our simulations, at least 10, Figure 1). Overall, this means that sampling of many populations is advisable to get a more accurate measure of
, so that it is not necessary to pool numerous traits together if the sampling design is large enough. This is important in the perspective of being able to distinguish between different types of traits: indeed, by choosing a biologically coherent set of traits (e.g., morphological, life history, etc.), which may be rather small, the results can be interpreted with more relevance. Second, the sampling effort should be balanced as much as possible between the among-population and the within-population levels, as the sampling variance decreases with the harmonic mean of relative sample sizes (1/nw + 1/nb). Furthermore, the χ2-distribution of the log-likelihood ratio (Equation 7) under proportionality, even with the Bartlett correction of Eriksen (1987), is an asymptotic result that is more accurate when both nb and nw are large (in our simulations, at least 10, Figure 1). Overall, this means that sampling of many populations is advisable to get a more accurate measure of 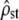 (test i) and a more reliable proportionality test (test ii). On the contrary, the number of families per population may be rather small if many populations are sampled: the within-population degree of freedom in the MANOVA (nw) will still remain large. Finally, the required number of populations to be sampled increases with the number of traits studied for the proportionality test (at least nb > p), which again argues in favor of avoiding the study of too many traits together. Overall, to detect selection, we recommend that sampling efforts in studies of metapopulations be oriented to considering more populations (e.g., >10–20), even at the cost of a reduced number of families per population or of a reduced number of traits measured. Again, power analysis can be carried out very easily as the sample variance of ρ has a simple close form (Equation 5), a main advantages of this test compared to several classic methods of Qst estimation.
(test i) and a more reliable proportionality test (test ii). On the contrary, the number of families per population may be rather small if many populations are sampled: the within-population degree of freedom in the MANOVA (nw) will still remain large. Finally, the required number of populations to be sampled increases with the number of traits studied for the proportionality test (at least nb > p), which again argues in favor of avoiding the study of too many traits together. Overall, to detect selection, we recommend that sampling efforts in studies of metapopulations be oriented to considering more populations (e.g., >10–20), even at the cost of a reduced number of families per population or of a reduced number of traits measured. Again, power analysis can be carried out very easily as the sample variance of ρ has a simple close form (Equation 5), a main advantages of this test compared to several classic methods of Qst estimation.
Because it is directly based on the estimation of variance components from the data set by MANOVA, the method can be extended to hierarchical designs (e.g., populations within habitats, etc.), using the corresponding mean square matrices (MS) and degrees of freedom. However, the power and the accuracy of the tests will be reduced at higher levels of the hierarchy for which the degrees of freedom are very small (often two habitats so that d.f. = 1). This makes the proportionality test (test ii) irrelevant as it requires d.f. > p for all MS matrices. However, test i could still be applied as the estimate and C.I. of 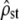 seems valid even for very low degrees of freedom (Figure 1a). Nevertheless,
seems valid even for very low degrees of freedom (Figure 1a). Nevertheless,  at the habitat level will likely have a very large C.I. in this case, which would also greatly reduce the power of the test. Improvements of the test to gain power in the study of a small number of groups will be necessary in the future to provide powerful tests based on quantitative genetic data from, e.g., two distinct habitats. Meanwhile, it may be best to study larger sets of habitat types (e.g., using several thresholds along a gradient).
at the habitat level will likely have a very large C.I. in this case, which would also greatly reduce the power of the test. Improvements of the test to gain power in the study of a small number of groups will be necessary in the future to provide powerful tests based on quantitative genetic data from, e.g., two distinct habitats. Meanwhile, it may be best to study larger sets of habitat types (e.g., using several thresholds along a gradient).
Further issues:
The main problem common to all multivariate Qst analyses (means over traits or our method) is that different traits are pooled and the net effect of evolutionary forces on the whole trait set is the only information available. This problem is partly overcome by our approach, which can detect when distinct subsets of traits are under qualitatively different selection regimes. However, it remains a “pooled-traits” approach for which individual traits information is lost. The alternative of using single-trait Qst is obviously worse, as such, because in most cases the C.I. for each Qst is of the order of [0, 1] and is sometimes not easy to compute (O'Hara and Merila 2005), so that a test against the value of Fst has no power and may be inaccurate. To allow the proper biological interpretation of the type of multitrait analysis presented here, it is therefore best to consider sets of biologically coherent traits for which similar evolutionary forces are expected to act. Such an a priori interpretation is always difficult to state, but because the method can detect mixed selection regimes, it may be possible to conduct exploratory studies and find the set of biologically coherent traits to be analyzed.
In molecular evolution, the same problem applies: neutrality tests have been designed that use comparisons of diversity between populations (divergence between species) and within populations (polymorphism within species) such as the McDonald–Kreitman test (McDonald and Kreitman 1991), in a way akin to the Qst–Fst method. An inherent limit to these methods is that information from different nucleotide sites/genes is pooled. Therefore, improvements have been proposed by including an explicit model of selection on distinct sites (Yang et al. 2000).
In a similar way, an alternative to the approach presented here (and to all neutrality tests in quantitative genetics) would be to model selection on the set of traits under study, together with drift and mutation. Fst estimates could then be used to correct for the effect of drift, then test for a significant component due to selection, and finally estimate this selection effect if it is significant. This would provide insights into the strength of selection, on each trait of the set. Such multivariate models exist: under the assumption of Gaussian breeding values, the evolution of multivariate genetic covariances among isolated populations (i.e., of D) under the joint action of drift, selection, and mutation has been modeled, first for Gaussian stabilizing selection (Lande 1980) and later for various other types of selection regimes [both directional and stabilizing and homogeneous and heterogeneous (Hansen and Martins 1996)]. These models, which relate D to G, provide a powerful framework for the detection and the estimation of the impacts of drift and selection on sets of quantitative traits. However, as such, they assume constancy of G over time, are not expressed in terms of Fst, and consider independent lineages as they have been developed in the context of speciation theory. However, the general framework proposed in these articles could be applied to not only test for, but also estimate selection effects. Such an approach seems possible and we believe is essential if we are to understand the interplay of drift and adaptation in wild populations, for the largest class of traits affecting fitness: continuous polygenic traits.
Acknowledgments
We thank Patrice David, Thomas Lenormand, Mike Whitlock, and particularly Frédéric Guillaume for helpful discussions and comments on our approaches in this article. Laurent Excoffier and two anonymous reviewers made useful suggestions to improve the manuscript. This work was carried out in part when J.G. was hosted by François Balloux in the Department of Genetics at Cambridge University. J.G. was under the benefit of a visiting professor Underwood fellowship from the United Kingdom Biotechnology and Biological Sciences Research Council. G.M. was supported by grant 31-108194/1 from the Swiss National Science Foundation to J.G and by a postdoctoral grant from the Centre National de la Recherche Scientifique to Sylvain Gandon.
References
- Blows, M. W., 2007. A tale of two matrices: multivariate approaches in evolutionary biology. J. Evol. Biol. 20 1–8. [DOI] [PubMed] [Google Scholar]
- Chapuis, E., G. Martin and J. Goudet, 2008. Effects of selection and drift on G matrix evolution in a heterogeneous environment: a multivariate Qst–Fst test with the freshwater snail Galba truncatula. Genetics 180 2151–2161. [DOI] [PMC free article] [PubMed]
- Chapuis, E., S. Trouve, B. Facon, L. Degen and J. Goudet, 2007. High quantitative and no molecular differentiation of a freshwater snail (Galba truncatula) between temporary and permanent water habitats. Mol. Ecol. 16 3484–3496. [DOI] [PubMed] [Google Scholar]
- Eriksen, P. S., 1987. Proportionality of covariance matrices. Ann. Stat. 15 732–748. [Google Scholar]
- Flury, B., 1988. Common Principal Components and Related Multivariate Models. Wiley, New York.
- Goudet, J., and L. Buchi, 2006. The effects of dominance, regular inbreeding and sampling design on Q(ST), an estimator of population differentiation for quantitative traits. Genetics 172 1337–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudet, J., and G. Martin, 2007. Under neutrality, QST ≤ FST when there is dominance in an island model. Genetics 176 1371–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman, I., D. Y. Kim and I. Olkin, 1985. Statistical inference for constants of proportionality, pp. 257–280 in Multivariate Analysis–VI., edited by P. R. Krishnaiah. North-Holland, New York.
- Hansen, T. F., and E. P. Martins, 1996. Translating between microevolutionary process and macroevolutionary patterns: the correlation structure of interspecific data. Evolution 50 1404–1417. [DOI] [PubMed] [Google Scholar]
- Hill, W. G., and R. Thompson, 1978. Probabilities of non-positive definite between-group or genetic covariance matrices. Biometrics 34 429–439. [Google Scholar]
- Ihaka, R., and R. Gentleman, 1996. R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5 299–314. [Google Scholar]
- Kimura, M., and J. F. Crow, 1964. The number of alleles that can be maintained in a finite population. Genetics 49 725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1978. Simple-model for balance between selection and mutation. J. Appl. Probab. 15 1–12. [Google Scholar]
- Kremer, A., A. Zanetto and A. Ducousso, 1997. Multilocus and multitrait measures of differentiation for gene markers and phenotypic traits. Genetics 145 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande, R., 1979. Quantitative genetic analysis of multivariate evolution, applied to brain:body size allometry. Evolution 33 402–416. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1980. Genetic-variation and phenotypic evolution during allopatric speciation. Am. Nat. 116 463–479. [Google Scholar]
- Latta, R. G., 2004. Gene flow, adaptive population divergence and comparative population structure across loci. New Phytol. 161 51–58. [Google Scholar]
- Le Corre, V., and A. Kremer, 2003. Genetic variability at neutral markers, quantitative trait loci and trait in a subdivided population under selection. Genetics 164 1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Fanjul, C., A. Fernandez and M. Toro, 2007. The effect of dominance on the use of the Qst–Fst contrast to detect natural selection on quantitative traits. Genetics 176 725–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, G., and T. Lenormand, 2006. A general multivariate extension of Fisher's geometrical model and the distribution of mutation fitness effects across species. Evolution 60 893–907. [PubMed] [Google Scholar]
- McDonald, J. H., and M. Kreitman, 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351 652–654. [DOI] [PubMed] [Google Scholar]
- McGuigan, K., 2006. Studying phenotypic evolution using multivariate quantitative genetics. Mol. Ecol. 15 883–896. [DOI] [PubMed] [Google Scholar]
- McGuigan, K., S. F. Chenoweth and M. W. Blows, 2005. Phenotypic divergence along lines of genetic variance. Am. Nat. 165 32–43. [DOI] [PubMed] [Google Scholar]
- McKay, J. K., and R. G. Latta, 2002. Adaptive population divergence: markers, QTL and traits. Trends Ecol. Evol. 17 285–291. [Google Scholar]
- Merila, J., 1997. Quantitative trait and allozyme divergence in the greenfinch (Carduelis chloris, Aves: Fringillidae). Biol. J. Linn. Soc. 61 243–266. [Google Scholar]
- Merila, J., and P. Crnokrak, 2001. Comparison of genetic differentiation at marker loci and quantitative traits. J. Evol. Biol. 14 892–903. [Google Scholar]
- O'Hara, R. B., and J. Merila, 2005. Bias and precision in Qst estimates: problems and some solutions. Genetics 171 1331–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, P. C., and S. J. Arnold, 1999. Hierarchical comparison of genetic variance-covariance matrices. I. Using the Flury hierarchy. Evolution 53 1506–1515. [DOI] [PubMed] [Google Scholar]
- Phillips, P. C., M. C. Whitlock and K. Fowler, 2001. Inbreeding changes the shape of the genetic covariance matrix in Drosophila melanogaster. Genetics 158 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pigliucci, M., 2006. Genetic variance-covariance matrices: a critique of the evolutionary quantitative genetics research program. Biol. Philos. 21 1–23. [Google Scholar]
- Porcher, E., T. Giraud and C. Lavigne, 2006. Genetic differentiation of neutral markers and quantitative traits in predominantly selfing metapopulations: confronting theory and experiments with Arabidopsis thaliana. Genet. Res. 87 1–12. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roff, D., 2000. The evolution of the G matrix: selection or drift? Heredity 84 135–142. [DOI] [PubMed] [Google Scholar]
- Rogers, A. R., and H. C. Harpending, 1983. Population structure and quantitative characters. Genetics 105 985–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter, D., 1996. Adaptive radiation along genetic lines of least resistance. Evolution 50 1766–1774. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1995. A measure of population subdivision based on microsatellite allele frequencies. Genetics 139 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokal, R. R., and F. J. Rolfh, 1981. Biometry. W. H. Freeman, New York.
- Spitze, K., 1993. Population-structure in Daphnia-obtusa - quantitative genetic and allozymic variation. Genetics 135 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steppan, S. J., P. C. Phillips and D. Houle, 2002. Comparative quantitative genetics: evolution of the G matrix. Trends Ecol. Evol. 17 320–327. [Google Scholar]
- Waldmann, P., and S. Andersson, 1999. Multilocus and multitrait differentiation of populations of the locally rare plant Scabiosa canescens and the more common S-columbaria. Hereditas 130 341–343. [Google Scholar]
- Whitlock, M. C., 1999. Neutral additive genetic variance in a metapopulation. Genet. Res. 74 215–221. [DOI] [PubMed] [Google Scholar]
- Yang, Z. H., R. Nielsen, N. Goldman and A. M. K. Pedersen, 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]