

Abstract
Two important problems affect the ability of asexual populations to accumulate beneficial mutations and hence to adapt. First, clonal interference causes some beneficial mutations to be outcompeted by more-fit mutations that occur in the same genetic background. Second, multiple mutations occur in some individuals, so even mutations of large effect can be outcompeted unless they occur in a good genetic background that contains other beneficial mutations. In this article, we use a Monte Carlo simulation to study how these two factors influence the adaptation of asexual populations. We find that the results depend qualitatively on the shape of the distribution of the fitness effects of possible beneficial mutations. When this distribution falls off slower than exponentially, clonal interference alone reasonably describes which mutations dominate the adaptation, although it gives a misleading picture of the evolutionary dynamics. When the distribution falls off faster than exponentially, an analysis based on multiple mutations is more appropriate. Using our simulations, we are able to explore the limits of validity of both of these approaches, and we explore the complex dynamics in the regimes where neither one is fully applicable.
THE accumulation of beneficial mutations drives adaptation and evolutionary innovation. Yet despite its central importance, the evolutionary dynamics by which a population accumulates such mutations are poorly understood. To better understand adaptation in any particular system, we must ask two questions. First, what is the range of beneficial mutations that are possible given the particular environmental challenge and genetic state of the population? Second, given this set of possibilities, what will actually happen probabilistically?
The first of these questions is fundamentally empirical, although Orr and Gillespie have argued on general theoretical grounds that the distribution of fitness effects of beneficial mutations should be exponential (Gillespie 1983, 1984, 1991; Orr 2002, 2003). A variety of recent experimental studies are roughly consistent with this exponential expectation (Imhof and Schlotterer 2001; Rozen et al. 2002; Sanjuan et al. 2004; Depristo et al. 2005; Lunzer et al. 2005; Rokyta et al. 2005; Kassen and Bataillon 2006). However, beneficial mutations are rare and their fitness effects are difficult to measure precisely, so these experimental studies are generally based on relatively few total mutations and have correspondingly limited resolution. The tail of the distribution, which refers to the rare mutations that confer a very large fitness benefit, is particularly hard to measure. Further, the spectrum of beneficial mutations available to a population is likely to vary with genetic background, history, and the environment, so it is unclear how far we can generalize from individual experimental studies. Thus it is still unknown whether in general the distribution of mutant effects, particularly of large-effect mutations, is exponential.
Even if we knew the precise distribution of mutational possibilities, it is not clear how a population would evolve. Because mutations are random events, there will inevitably be some randomness in how a given population adapts. What we want to understand is the statistics of which beneficial mutations are more or less likely to contribute to adaptation and the dynamics by which they do so. That is, given a set of things that are possible, what is the probability that any given one of them will actually occur and contribute to the adaptation of the population? What is the evolutionary dynamics by which they do so? In this article, we focus on how the distribution of mutations that actually occur and spread through the entire population (i.e., fix), ρf(s), depends on the distribution of mutations that are possible, ρ(s), where s is the fitness benefit from a single mutation. We explore these features as a function of the population size N and the overall mutation rate U. Besides its importance in understanding adaptation, this question is relevant in practical attempts to measure the distribution of possible mutations, since ρf(s) is much easier to measure experimentally than ρ(s). We also examine some aspects of the dynamics by which the mutations that fix do so.
There are a number of effects that make the distribution of mutations that fix different from the distribution of all possible mutations. First, most beneficial mutations that occur are lost rapidly by random genetic drift. If a beneficial mutation is particularly lucky, it will avoid this stochastic loss and reach a high enough frequency that thereafter its dynamics become dominated by selection rather than by drift. We refer to this process as the establishment of the beneficial mutation. Mutations of larger effect are more likely to survive random drift—they have a higher establishment probability—so this will tend to bias the distribution of mutations that actually fix toward larger-effect mutations, relative to the distribution of mutations that are possible (Haldane 1927; Rozen et al. 2002).
Once a mutation has become established, it will fix provided that nothing else interferes. However, this fixation takes time, and other beneficial mutations can become established in individuals without the original mutation before the original mutation can fix. In an asexual population, if one or more of these other mutations has a larger fitness benefit than the original mutation, the original mutation will eventually be outcompeted and driven to extinction. This process is known as clonal interference (Gerrish and Lenski 1998; Gerrish 2001; Wilke 2004). The same process also operates in a sexual population, where it is referred to as the Hill–Robertson effect, but is mitigated because the two competing mutations can potentially recombine onto the same genome and fix together (Hill and Robertson 1966). In this article, we focus exclusively on asexual populations, where this effect is strongest.
In a small population with a small to modest mutation rate, the establishment of a beneficial mutation is an extremely rare event. Thus clonal interference is unlikely to occur, and the distribution of mutations that fix is simply the distribution of mutations that establish. In a larger population, or one with a higher mutation rate, however, clonal interference can be extremely common. This is true whenever the population size times the beneficial mutation rate is large compared to one (the precise condition is actually slightly weaker than this), so clonal interference is likely common in a wide range of microbial and viral populations, especially in light of recent studies showing that beneficial mutation rates in bacteria and yeast are relatively high (Joseph and Hall 2004; Perfeito et al. 2007). Because small-effect mutations are more likely to be interfered with than large-effect mutations, clonal interference biases the distribution of mutations that fix toward those of large effect. This bias has been analyzed in detail both theoretically (Gerrish and Lenski 1998; Wilke 2004) and experimentally (De Visser et al. 1999; De Visser and Rozen 2005).
These analyses of clonal interference consider only mutations that occur in the wild-type population; they assume that the largest such mutation is the one that fixes. The possibility of double mutations in a single organism is neglected. But, in fact, even if a more-fit mutation B occurs before an earlier but less-fit mutation A fixes, A may still survive, because an individual with mutation A can get another mutation C such that the A-C double mutant is more fit than B. Recently, Desai and Fisher (2007) showed that whenever clonal interference is important, these multiple mutations are also at least of comparable importance—and, in fact, many large asexual populations will often routinely have triple or quadruple mutations (Desai et al. 2007). Because small-effect mutations are more common than mutations of larger effect, they are more likely to form double mutants. Thus the possibility of multiple mutations biases the distribution of mutations that fix back toward those of smaller effect. In short, it will often be the case that getting two small-effect mutations is more common than getting a single (rarer) large-effect mutation. This effect depends on the shape of the distribution of mutational effects: the rarer large-effect mutations are compared to small-effect ones, the stronger the multiple-mutation effect should be. The importance of this effect also depends on population size and mutation rate, though in a somewhat different way than clonal interference does.
In addition to affecting the distribution of mutations that fix, multiple mutations also have an important impact on the evolutionary dynamics. Different individuals have different numbers and strengths of beneficial mutations, so a large population can maintain substantial variation in fitness. It is only those mutations that occur in the most-fit individuals that have the best chance of surviving and contributing to the long-term adaptation of the population. Thus the dynamics of adaptation are slowed down, limited by the rate at which good mutations occur in good backgrounds.
Because the distribution of beneficial mutations that fix depends in a subtle way on both clonal interference and multiple-mutation effects, it cannot be fully understood without a complete model that includes both. No analytical results from such a model yet exist, though Kim and Orr (2005) have analyzed a model that includes some aspects of both effects and Pepin and Wichman (2008) have studied an experimental system in which both appear to occur. In this article, we address this question using Monte Carlo simulations of the full evolutionary dynamics of large asexual populations, including both clonal interference and multiple-mutation effects. Our analysis is in the spirit of Park and Krug (2007), though we focus more directly on the interplay between clonal interference and multiple mutations. We consider several distributions of possible mutational effects ρ(s) and determine the distribution of mutations that fix, ρf(s), across a range of population sizes and mutation rates. We find that clonal interference analysis provides a good approximation for some aspects of ρf(s) when large-effect mutations are sufficiently common relative to small-effect ones. When large-effect mutations are more rare, we find that an approximation focusing on multiple-mutation effects, proposed by Desai and Fisher (2007), is more appropriate.
We next turn to the evolutionary dynamics by which beneficial mutations fix. Using our Monte Carlo approach, we simulate the evolutionary dynamics. We show that multiple-mutation dynamics involving mutations within a narrow range of fitness effects describe the evolution. We describe how this range of fitness effects depends on ρ(s) and the other parameters and how the mutations within this range accumulate.
MODEL AND SIMULATION METHODS
We consider an asexual population of N haploid individuals with an overall mutation rate Ub toward beneficial mutations. Our model also applies to asexual diploids, where the fitness effects of mutations refer to their effects in the individual in which they occur (i.e., if the mutation creates a heterozygote the relevant fitness effect is given by the difference in fitness between the original state and the newly mutated heterozygote, and analogously if the mutation creates a homozygote). Given that a beneficial mutation occurs, we assume that its fitness effect is between s and s + ds (i.e., the fitness of the organism increases by a factor between es and es+ds) with probability
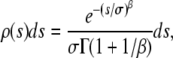 |
(1) |
where the two parameters σ and β characterize the shape of the distribution and Γ is the Gamma function. This form for ρ(s) allows us to explore the importance of the shape of the tail of the distribution of mutant effects—that is, the relative rareness of large-effect mutations compared to small-effect ones. When β = 1, the distribution of mutant effects is exponential with mean σ. When β > 1, the distribution of mutant effects falls off faster than exponentially (i.e., large-effect mutations are more rare), and when β < 1 the distribution of mutation effects falls off more slowly than exponentially (large-effect mutations are more common). Note that this distribution is normalized to 1, so beneficial mutations with effect between s and s + ds occur at a rate Ubρ(s)ds. The average fitness effect of a beneficial mutation, 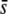 , is
, is
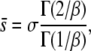 |
(2) |
so σ is exactly equal to the average effect only for β = 1. We assume that there is no epistasis, so that an individual with two mutations of effect s1 and s2 has fitness 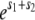 (or more generally for n mutations, the fitness is
(or more generally for n mutations, the fitness is 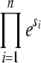 ).
).
We neglect deleterious mutations, as Rouzine et al. (2003) and Desai and Fisher (2007) have shown theoretically that they are not expected to qualitatively affect the rate or dynamics by which a large asexual population adapts when beneficial mutations are relatively common, which is the situation we study here (Rouzine et al. 2003 verified this expectation with simulations). When deleterious mutations become extremely common, beneficial mutations are sufficiently rare, or the population sizes are small enough that Muller's ratchet becomes important, this approximation breaks down. We do not study any of these situations here, but several other authors have analyzed them using both theoretical and simulation approaches (Orr 2000; Johnson and Barton 2002; Rouzine et al. 2003; Campos and De Oliveira 2004).
We assume dynamics with discrete generations. In each generation, we first randomly select which individuals will survive to the next generation, weighted by each individual's fitness. The overall survival probabilities are normalized so that on average half of the population will survive to the next generation. Each surviving individual then duplicates to create two identical individuals in the next generation. Finally, each of these individuals in the next generation has a probability Ub of acquiring a new beneficial mutation. We then repeat this algorithm for the subsequent generation. We record all the information about the genetic state of the population at each step. All simulations were checked to ensure that the results were extracted after a steady state had been achieved. Note that this algorithm does not enforce an exact population size N at each step, but rather keeps the average population size equal to N.
These evolutionary dynamics have the advantage of being fast to simulate, allowing us to explore a greater range of parameters in a reasonable amount of computation time. However, they are slightly different from standard Wright–Fisher dynamics. It is not clear which of these models is the best representation of any particular population, but our model might be expected to correspond to a bacterial or yeast population that divides by binary fission in a chemostat (or that fluctuates in size, with an appropriately defined effective population size). The differences between models do lead to small differences in the stochastic dynamics while lineages are rare and thus are expected to cause minor modifications to the evolutionary dynamics. However, we have also simulated the standard Wright–Fisher dynamics for many of the parameters we describe in this article, and the results are all qualitatively similar.
To test the assumption that deleterious mutations can be neglected, we have also carried out a more limited set of simulations of a model that includes deleterious mutations. For these simulations, we assume that deleterious mutations occur at an overall rate Ud. Given that a deleterious mutation occurs, we assume that it has a fitness cost drawn at random from an exponential distribution. The results from these simulations are nearly identical to those from simulations that neglect them, justifying our assumption, with a few minor differences that are noted in the results section.
PREVIOUS THEORY
In large asexual populations, the effects of clonal interference and multiple mutations interact in a subtle way. As noted in the Introduction, analytic results are difficult to obtain in this complex situation, and no complete theory yet exists. Instead, two different theoretical approaches have been proposed. The first focuses exclusively on the clonal interference effect, while the second focuses on multiple mutation dynamics. In this section, we briefly summarize the relevant theoretical predictions given by each of these two approaches, as well as our expectations for small populations where neither effect matters. We then compare these predictions to our simulations in subsequent sections.
Definitions and small populations:
We begin by considering populations that are sufficiently small that neither clonal interference nor multiple mutations will occur [this requires 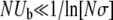 (Desai and Fisher 2007)]. In these populations, the distribution of mutations that fix, ρf(s), equals the distribution of mutations that establish. We expect
(Desai and Fisher 2007)]. In these populations, the distribution of mutations that fix, ρf(s), equals the distribution of mutations that establish. We expect
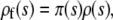 |
(3) |
valid for small N and Ub, where π(s) is the establishment probability. Note that by convention we assume ρf(s) is not normalized; this makes various calculations much more transparent. In our model, for 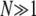 this establishment probability is given by
this establishment probability is given by
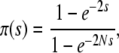 |
(4) |
where s is the fitness advantage relative to the background population. Note that despite slightly different stochastic dynamics, this establishment probability is identical to that of the Wright–Fisher model. When 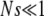 , we have
, we have 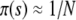 , while for
, while for 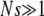 but
but 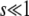 we have π(s) ≈ 2s. This small-population limit of ρf(s), which is the distribution of mutations that establish, is sometimes called the distribution of contending mutations (Rozen et al. 2002). We denote it by ρc(s).
we have π(s) ≈ 2s. This small-population limit of ρf(s), which is the distribution of mutations that establish, is sometimes called the distribution of contending mutations (Rozen et al. 2002). We denote it by ρc(s).
To determine how beneficial mutations of different effects contribute to the overall adaptation of the population, we need to weight mutations by their fitness effects. That is, a single fixed mutation with effect 2s contributes the same amount to the adaptation of the population as two mutations with effect s. Thus we define R(s) ≡ sρf(s) as the distribution of the relative contributions to the overall adaptation as a function of s. The integral of R(s) from s1 to s2 is the total contribution of mutations of size between s1 and s2 to the adaptation.
Using R(s), we can study which mutations are most important. We expect that mutations of very small effect will not contribute substantially to adaptation, because they confer such a small advantage (and in larger populations they are also strongly suppressed by clonal interference). On the other hand, mutations of very large effect will be too rare to contribute substantially (and in larger populations may be impeded by multiple smaller mutations). Thus we expect that typically R(s) will have a peak at some intermediate value of s, with some range of mutations around this that contribute substantially to adaptation. This is indeed what we find. We characterize R(s) by its mean, which we call 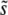 [to be precise, we define
[to be precise, we define 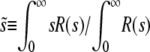 ]. We estimate the width of the range of mutations around the mean that contribute substantially to the evolution by the standard deviation of R(s) around
]. We estimate the width of the range of mutations around the mean that contribute substantially to the evolution by the standard deviation of R(s) around 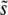 , which we call SD(s). From ρf(s), we can easily calculate the R(s), and hence also the
, which we call SD(s). From ρf(s), we can easily calculate the R(s), and hence also the 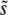 and SD(s), relevant for populations so small that neither clonal interference nor multiple mutations occur.
and SD(s), relevant for populations so small that neither clonal interference nor multiple mutations occur.
As beneficial mutations accumulate, the population adapts. We define the rate of adaptation, v, to be the rate at which the average fitness of the population increases. In the small-population limit, v is simply the rate at which mutations destined to establish occur times the fitness effect of these mutations.
Clonal interference analysis:
We now turn to a description of the first theoretical approach to the adaptation of large populations, which focuses on clonal interference and neglects the possibility of multiple mutations (Gerrish and Lenski 1998; Wilke 2004). In this analysis the probability that a beneficial mutation fixes is the probability that it establishes and then fixes before another more-fit mutation establishes. Gerrish and Lenski (1998) found that given that a mutation of effect s has established, the expected number of more-fit mutations establishing before the original mutation fixes is roughly
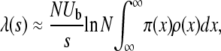 |
(5) |
assuming that all mutations arise in the wild-type population. Thus the distribution of mutations that fix is
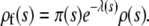 |
(6) |
From Equation 6 it is also straightforward to calculate the expected 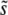 and SD(s). Because clonal interference becomes more likely as either population size or mutation rate increases, increasing either of these parameters is expected to increase
and SD(s). Because clonal interference becomes more likely as either population size or mutation rate increases, increasing either of these parameters is expected to increase 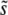 .
.
The average rate at which mutations fix equals the rate at which those destined to fix occur. Thus clonal interference analysis predicts that the average fixation rate 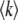 is
is
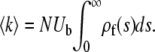 |
(7) |
This means that the rate of adaptation v is
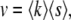 |
(8) |
where 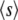 is the average fitness of mutations that fix.
is the average fitness of mutations that fix.
Multiple-mutations analysis:
We now turn to a description of the second theoretical framework, proposed by Desai and Fisher (2007), which focuses primarily on multiple-mutation effects. These and other authors studied a model where all beneficial mutations have the same fitness advantage s (Ridgway et al. 1998; Rouzine et al. 2003, 2008; Desai and Fisher 2007). They calculated the rate at which these mutations accumulate, v(s), as a function of population size and mutation rate. We do not go through this calculation here, but note that in this idealized single-s model, we can describe the state of the population as a distribution of the number of mutations each individual has. Some lucky most-fit individuals have more than the average number of mutations, and it is only additional mutations within this small subpopulation that will contribute to the long-term evolution of the population; others will eventually go extinct because they are handicapped by their relatively poor genetic background. The multiple-mutations analysis focuses on the rate at which new mutations occur in this small highly fit subpopulation and how this subpopulation evolves relative to the rest of the population. We define the lead, q, to be the number of beneficial mutations the most-fit individual possesses in excess of that of the average individual (more precisely, q − 1 is defined to be the difference in number of mutations between the most-fit class of established individuals and the mean individual). By definition, multiple mutations are important when q > 1.
In a more general situation where beneficial mutations have a range of fitness effects, Desai and Fisher (2007) argued that under many conditions SD(s) should be small compared to 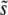 , so that mutations within a narrow range of fitness effects dominate the evolution. If this is true, it is natural to expect that multiple mutations of effect of order
, so that mutations within a narrow range of fitness effects dominate the evolution. If this is true, it is natural to expect that multiple mutations of effect of order 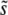 may routinely appear and that their accumulation can be described roughly by the single-s model, provided one chooses that single s to be
may routinely appear and that their accumulation can be described roughly by the single-s model, provided one chooses that single s to be 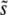 and chooses the beneficial mutation rate to these mutations,
and chooses the beneficial mutation rate to these mutations, 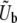 , to be the total mutation rate to all mutations within ∼ ±SD(s) of
, to be the total mutation rate to all mutations within ∼ ±SD(s) of 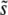 . This approximation should be valid as long as SD(s) is relatively narrow—at most of order
. This approximation should be valid as long as SD(s) is relatively narrow—at most of order 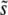 . As we will see for large populations and mutation rates, our simulations show that this is indeed the case, as do recent experimental studies in Saccharomyces cerevisiae and Escherichia coli (Hegreness et al. 2006; Desai et al. 2007). Note that we can define the lead q in this more complex situation as the fitness of the most-fit individual minus the fitness of the mean individual, divided by
. As we will see for large populations and mutation rates, our simulations show that this is indeed the case, as do recent experimental studies in Saccharomyces cerevisiae and Escherichia coli (Hegreness et al. 2006; Desai et al. 2007). Note that we can define the lead q in this more complex situation as the fitness of the most-fit individual minus the fitness of the mean individual, divided by 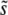 (more precisely, q − 1 is defined to be the difference in the fitness of the most-fit class of established individuals and the mean individual, divided by
(more precisely, q − 1 is defined to be the difference in the fitness of the most-fit class of established individuals and the mean individual, divided by 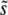 ). That is, q is the number of extra
). That is, q is the number of extra 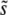 -sized mutations that the most-fit individual has compared to the average individual.
-sized mutations that the most-fit individual has compared to the average individual.
Of course, the appropriate values of 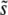 and
and 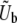 depend on ρ(s), N, and the overall mutation rate. A full understanding of this depends in a subtle way on both clonal interference and multiple-mutation effects, but Desai and Fisher (2007) proposed a simple approximation. They first calculate v(s) for each possible value of s, assuming that only mutations of this size are possible. To do this, one must specify an appropriate mutation rate to mutations of this size,
depend on ρ(s), N, and the overall mutation rate. A full understanding of this depends in a subtle way on both clonal interference and multiple-mutation effects, but Desai and Fisher (2007) proposed a simple approximation. They first calculate v(s) for each possible value of s, assuming that only mutations of this size are possible. To do this, one must specify an appropriate mutation rate to mutations of this size, 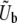 . Desai and Fisher (2007) made the ad hoc assumption that
. Desai and Fisher (2007) made the ad hoc assumption that 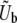 should be the total mutation rate to mutations of order s (i.e., within roughly a factor of 2 of s). They then calculated v(s). This v(s) expresses the contribution of mutations of effect s to the overall evolution and thus should equal R(s), up to normalization. From this R(s), they calculate
should be the total mutation rate to mutations of order s (i.e., within roughly a factor of 2 of s). They then calculated v(s). This v(s) expresses the contribution of mutations of effect s to the overall evolution and thus should equal R(s), up to normalization. From this R(s), they calculate 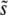 . They find
. They find
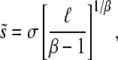 |
(9) |
where 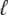 is related to the overall mutation rate by
is related to the overall mutation rate by
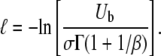 |
(10) |
This expression for 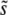 is valid only for β > 1; for distributions of mutational effects that fall off exponentially or slower the behavior is more complicated and the analysis breaks down. These results were derived using a continuous-time version of our model; see Desai and Fisher (2007) for details. Note that this analysis makes no precise prediction for ρf(s), but does imply that R(s) will be a narrow distribution centered around
is valid only for β > 1; for distributions of mutational effects that fall off exponentially or slower the behavior is more complicated and the analysis breaks down. These results were derived using a continuous-time version of our model; see Desai and Fisher (2007) for details. Note that this analysis makes no precise prediction for ρf(s), but does imply that R(s) will be a narrow distribution centered around 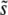 .
.
Using Equation 9 and their analysis of the single-s model, Desai and Fisher (2007) calculated how the rate of adaptation v and the lead q should depend on N and Ub and the shape of the distribution of mutational effects by plugging in the appropriate values of 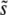 and
and 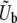 . They found
. They found
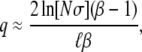 |
(11) |
and
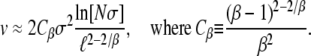 |
(12) |
As with Equation 9, these expressions are valid only for β > 1.
RESULTS
We now describe the results of our Monte Carlo simulations for the evolutionary dynamics in large populations when both clonal interference and multiple mutations are present and compare these results to the theoretical predictions described above.
The distribution of mutations that fix:
We begin in Figure 1 by showing several examples of the distribution of mutations that fix, ρf(s), as compared to the distribution of possible and contending mutations, ρ(s) and ρc(s), respectively. The predictions of clonal interference analysis, Equation 6, are also shown.
Figure 1.—

Examples of the distribution of possible mutations ρ(s) (black line), the distribution of contending mutations ρc(s) (red line), and the distribution of fixed mutations ρf(s) from our simulations (yellow histogram). Also shown is the distribution of mutations that occurred in the simulations (white histogram). Additionally the distribution of predicted fixed mutations ρf(s) from a clonal interference calculation is shown (blue line). Note the logarithmic scale. In all examples, σ = 0.02. (a) A small population, where ρf(s) = ρc(s) except for the smallest-effect mutations. Here N = 3 × 104, Ub = 10−5, and β = 1.0. (b) A large population with β = 10. Here N = 107 and Ub = 10−5. Note that small-effect mutations are suppressed by clonal interference effects, while large-effect mutations are suppressed by multiple-mutation effects. (c) A large population with β = 1. Here N = 107 and Ub = 10−5. Note that small-effect mutations are suppressed by clonal interference effects, but less strongly than clonal interference analysis alone predicts. (d) A large population with β = 0.5. Here N = 1 × 107 and Ub = 10−5.
In small populations, we have seen that neither clonal interference nor multiple mutations will occur. Thus we expect the distribution of mutations that fix to equal the distribution of mutations that establish, ρf(s) = ρc(s). This is indeed the case (Figure 1a), except for extremely small s (these s are so small that clonal interference prevents them from fixing even in these small populations). We find similar results for small populations regardless of β (data not shown).
In larger populations, the behavior depends on β. In all cases, small-effect mutations are suppressed quite dramatically by clonal interference effects (Figure 1, b–d). Note, however, that clonal interference analysis predicts that this suppression of small-effect mutations should be even stronger than we observe. This is because some of these small-effect mutations are able to fix in multiple-mutation combinations with those of larger effect. As we would expect, this effect is more dramatic for larger β, where multiple mutations play a larger role.
For large-effect mutations, clonal interference analysis predicts that ρf(s) should equal ρc(s), regardless of β, since clonal interference cannot suppress the fixation of the largest-effect mutations. This is indeed the case for small β (Figure 1, c and d). However, for large β the largest-effect mutations are also suppressed (Figure 1b). This is consistent with the multiple-mutations approach of Desai and Fisher (2007), who suggest that whenever β > 1 multiple small-s mutations can suppress the fixation of large-s mutations, exactly as we observe.
We have also studied the distribution of mutations that fix using a model that includes deleterious mutations (see model and simulation methods section). We find that the distributions of beneficial mutations that fix, ρf(s), are nearly identical to those shown in Figure 1, even in the presence of deleterious mutations (data not shown). In addition to these beneficial mutations, a few deleterious mutations also fix. This occurs when a small-effect deleterious mutation occurs in an individual with a large beneficial mutation. However, even for deleterious mutation rates 1000-fold higher than the beneficial mutation rate, the number of deleterious mutations that fix is a tiny fraction of the number of beneficial mutations that fix, and these deleterious mutations have a fitness cost that is small compared to the average fitness advantage of the beneficial mutations that fix.
Which mutations contribute to adaptation:
To analyze our results across a broad range of parameters, we shift to considering the distribution of mutations that fix weighted by their contributions to adaptation, R(s). Above we have claimed that R(s) will have a peak at some intermediate value 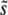 , with some range of mutations around this that contribute substantially to adaptation. This is indeed what we find. In Figure 2, we show how
, with some range of mutations around this that contribute substantially to adaptation. This is indeed what we find. In Figure 2, we show how 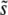 in our simulated populations depends on N and Ub for several different values of β. We compare these simulated results to the predictions of clonal interference analysis alone and, for β > 1, to multiple-mutations analysis. Note that clonal interference provides accurate estimates of
in our simulated populations depends on N and Ub for several different values of β. We compare these simulated results to the predictions of clonal interference analysis alone and, for β > 1, to multiple-mutations analysis. Note that clonal interference provides accurate estimates of 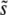 for β ≤ 1, but breaks down for larger N and Ub, especially when β > 1. The multiple-mutations analysis, on the other hand, more accurately predicts
for β ≤ 1, but breaks down for larger N and Ub, especially when β > 1. The multiple-mutations analysis, on the other hand, more accurately predicts 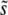 when N and Ub are large and β > 1, but does not provide a prediction for the case β ≤ 1.
when N and Ub are large and β > 1, but does not provide a prediction for the case β ≤ 1.
Figure 2.—

The average scaled fitness effect of mutations that fix, 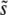 . In all cases σ = 0.02. (a) Simulation results for
. In all cases σ = 0.02. (a) Simulation results for 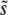 for N = 107 as a function of Ub for β = 0.5 (solid black circles), β = 1 (open red circles), and β = 10 (open blue squares). Predictions of clonal interference analysis are shown as dotted lines, and the predictions of the multiple-mutation analysis (for β = 10) are shown as a solid line. Note that for β = 10,
for N = 107 as a function of Ub for β = 0.5 (solid black circles), β = 1 (open red circles), and β = 10 (open blue squares). Predictions of clonal interference analysis are shown as dotted lines, and the predictions of the multiple-mutation analysis (for β = 10) are shown as a solid line. Note that for β = 10, 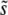 decreases with Ub for large Ub. (b) Simulation results for
decreases with Ub for large Ub. (b) Simulation results for 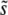 for Ub = 10−5 as a function of N for β = 0.5 (solid black circles), β = 1 (open red circles), and β = 10 (open blue squares). Predictions of clonal interference analysis are shown as dotted lines, and the predictions of the multiple-mutation analysis (for β = 10) are shown as a solid line.
for Ub = 10−5 as a function of N for β = 0.5 (solid black circles), β = 1 (open red circles), and β = 10 (open blue squares). Predictions of clonal interference analysis are shown as dotted lines, and the predictions of the multiple-mutation analysis (for β = 10) are shown as a solid line.
In Figure 3, we show 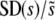 as a function of N and Ub, again for several values of β, compared to the predictions of clonal interference analysis. Note that
as a function of N and Ub, again for several values of β, compared to the predictions of clonal interference analysis. Note that 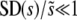 corresponds to the case where a narrow range of mutations around
corresponds to the case where a narrow range of mutations around 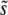 dominate the evolution. We see that this is typically the case, regardless of whether the distribution of mutational effects falls off faster or more slowly than exponentially, except for small N or Ub. However, SD(s) is not nearly as small as clonal interference analysis predicts for β > 1. This is due to multiple-mutation effects and has consequences for the predictions of the clonal interference approach, as we describe in the discussion.
dominate the evolution. We see that this is typically the case, regardless of whether the distribution of mutational effects falls off faster or more slowly than exponentially, except for small N or Ub. However, SD(s) is not nearly as small as clonal interference analysis predicts for β > 1. This is due to multiple-mutation effects and has consequences for the predictions of the clonal interference approach, as we describe in the discussion.
Figure 3.—

The scaled width of the weighted distribution of mutations that fix, 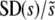 . In all cases σ = 0.02. (a) Simulation results for
. In all cases σ = 0.02. (a) Simulation results for 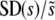 for N = 107 as a function of Ub for β = 0.5, β = 1, and β = 10. Predictions of clonal interference analysis are shown as dotted lines. (b) Simulation results for
for N = 107 as a function of Ub for β = 0.5, β = 1, and β = 10. Predictions of clonal interference analysis are shown as dotted lines. (b) Simulation results for 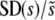 for Ub = 10−5 as a function of N for β = 0.5, β = 1, and β = 10. Predictions of clonal interference analysis are shown as dotted lines.
for Ub = 10−5 as a function of N for β = 0.5, β = 1, and β = 10. Predictions of clonal interference analysis are shown as dotted lines.
Note that although for clarity we only show one case where β > 1 (namely, β = 10) in all the figures, we have also simulated the case β = 2, and the results are qualitatively the same as those shown for β = 10.
The dynamics of adaptation:
We now turn to the dynamics by which these important mutations accumulate. We have seen that multiple mutations are important in determining 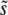 for β > 1, but not so important for β ≤ 1. Despite this, mutations of effect around
for β > 1, but not so important for β ≤ 1. Despite this, mutations of effect around 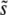 , which dominate the overall adaptation of the population, may accumulate via multiple-mutations dynamics even when β ≤ 1, although mutations of much larger effect fix whenever they establish. As we have seen, this will be true whenever the lead q > 1.
, which dominate the overall adaptation of the population, may accumulate via multiple-mutations dynamics even when β ≤ 1, although mutations of much larger effect fix whenever they establish. As we have seen, this will be true whenever the lead q > 1.
In Figure 4, we show how q depends on N and Ub for several different values of β. We see that even for β ≤ 1, these multiple mutations are important to the dynamics (where values of q = 3–5 are reached). For β = 10, we also show the predictions of the multiple-mutations analysis. Note that clonal interference analysis implicitly assumes no multiple mutations. Thus it implies that q is between 1 and 2, depending on whether an established mutant group is sweeping to fixation.
Figure 4.—

The effective lead q. (a) Simulation results for q as a function of Ub for β = 0.5, β = 1, and β = 10. Other parameters are as in Figure 3a. Predictions of the multiple-mutations analysis (for β = 10) are shown as a solid blue line. Note that clonal interference predicts q ≈ 1, independent of the parameters. (b) Simulation results for q as a function of N for β = 0.5, β = 1, and β = 10. Other parameters are as in Figure 3b. Predictions of the multiple-mutations analysis (for β = 10) are shown as a solid blue line. As before, clonal interference predicts q ≈ 1, independent of the parameters.
As beneficial mutations accumulate, the population adapts. In Figure 5, we show how the rate at which the average fitness of the population increases, v, depends on N and Ub, again for several values of β. We compare this to the predictions of clonal interference theory alone and to the multiple-mutations analysis of Desai and Fisher (2007).
Figure 5.—

The average rate of adaptation, v. (a) Simulation results for v as a function of Ub for β = 0.5, β = 1, and β = 10. Other parameters are as in Figure 3a. Predictions of clonal interference analysis are shown as dotted lines, and the predictions of the multiple-mutations analysis (for β = 10) are shown as a solid blue line. (b) Simulation results for v as a function of N for β = 0.5, β = 1, and β = 10. Other parameters are as in Figure 3b. Predictions of clonal interference analysis are shown as dotted lines, and the predictions of the multiple-mutations analysis (for β = 10) are shown as a solid blue line.
As for Figures 2 and 3, we have also carried out simulations for the case β = 2, and the results for q and v are qualitatively similar to those for β = 10. In addition, we have performed a limited set of simulations that include deleterious mutations. Our results for v and q are the same as those shown in Figures 4 and 5 for the model that excludes deleterious mutations, even when the deleterious mutation rate is orders of magnitude larger than the beneficial mutation rate.
DISCUSSION
Our Monte Carlo simulation approach allows us to study the evolutionary dynamics of adaptation in large asexual populations, where both clonal interference and multiple mutations are important. We have described two theoretical approaches that each focus on one of these two effects. Using our simulations, we can now assess the usefulness and generality of these methods.
In its original form, which neglects the possibility of multiple mutations, clonal interference analysis predicts the distribution of mutations that contribute to adaptation, ρf(s), according to Equation 6. From this we can also calculate clonal interference analysis predictions for 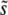 and SD(s). Because clonal interference becomes more likely as either population size or mutation rate increases, increasing either of these parameters is expected to increase
and SD(s). Because clonal interference becomes more likely as either population size or mutation rate increases, increasing either of these parameters is expected to increase 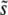 . These predictions are shown in Figures 2 and 3. Finally, we have seen that the rate of adaptation, v, predicted by clonal interference analysis is given in Equation 8. This prediction is shown in Figure 5.
. These predictions are shown in Figures 2 and 3. Finally, we have seen that the rate of adaptation, v, predicted by clonal interference analysis is given in Equation 8. This prediction is shown in Figure 5.
For β = 0.5 and β = 1, we see from Figure 2 that clonal interference yields reasonable estimates of 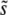 . This makes sense, as in this regime multiple mutations do not suppress the fixation of larger-effect mutations. For β = 10, clonal interference systematically overestimates
. This makes sense, as in this regime multiple mutations do not suppress the fixation of larger-effect mutations. For β = 10, clonal interference systematically overestimates 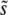 , by an amount that increases with the population size and mutation rate. This also makes sense, as in large populations for β = 10 multiple mutations in fact suppress the fixation of large-effect mutations.
, by an amount that increases with the population size and mutation rate. This also makes sense, as in large populations for β = 10 multiple mutations in fact suppress the fixation of large-effect mutations.
Although clonal interference accurately predicts 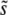 for small β, we can see from Figure 4 that for all values of β, q > 2 when N and Ub are large, pointing to the importance of multiple mutations in the dynamics. Clonal interference analysis, by contrast, assumes that all mutations occur and fix in the wild-type population, which implies q between 1 and 2. This underlies the calculation of the fixation rate in Equation 7, which assumes that mutations in any individual can contribute to adaptation, when in fact for q > 1 it is only the mutations in the relatively rare individuals that already have other beneficial mutations that contribute. Thus we expect that neglecting the importance of multiple mutations should lead clonal interference analysis to overestimate the rate of adaptation v, for all β. This is indeed what we find for β = 0.5, but we see from Figure 5 that clonal interference analysis accurately predicts v for β = 1 and actually underestimates the rate of adaptation for β = 10.
for small β, we can see from Figure 4 that for all values of β, q > 2 when N and Ub are large, pointing to the importance of multiple mutations in the dynamics. Clonal interference analysis, by contrast, assumes that all mutations occur and fix in the wild-type population, which implies q between 1 and 2. This underlies the calculation of the fixation rate in Equation 7, which assumes that mutations in any individual can contribute to adaptation, when in fact for q > 1 it is only the mutations in the relatively rare individuals that already have other beneficial mutations that contribute. Thus we expect that neglecting the importance of multiple mutations should lead clonal interference analysis to overestimate the rate of adaptation v, for all β. This is indeed what we find for β = 0.5, but we see from Figure 5 that clonal interference analysis accurately predicts v for β = 1 and actually underestimates the rate of adaptation for β = 10.
The reason for this discrepancy is apparent from Figure 3, which shows that for β = 1 and β = 10, clonal interference tends to underestimate SD(s). This problem gets worse as N and Ub increase. The reason for this underestimate is that clonal interference analysis assumes that the largest mutation that occurs before any other fixes goes to fixation by itself. This strongly suppresses the fixation of mutations that have a substantially smaller fitness effect and leads to a prediction of a very small SD(s). But in fact, mutations with a variety of smaller effects will sometimes happen to occur in individuals that have this larger-effect mutation, and these will sweep to fixation together. This broadens the distributions of mutations that fix and hence increases the actual SD(s), as is apparent in Figure 3 and in the distributions shown in Figure 1, b and c. This means that clonal interference assumes that only mutations within a much narrower range of fitness effects contribute to adaptation than is actually the case, which should lead to underestimates of the rate of adaptation. In other words, although only mutations that happen in very fit individuals can contribute (which slows adaptation), many mutations of various effects occur in these fit individuals and can all fix together (speeding adaptation). The underestimate of v from this latter effect is more severe for larger β, because larger β corresponds to a larger underestimate of SD(s). We see from Figure 5 that for β = 1, this underestimate of v roughly cancels the overestimate of v caused by the assumption that mutations in any individual can contribute to adaptation. For β = 0.5, the underestimate of SD(s) is less severe, so it only partially cancels the overestimate, and in sum clonal interference overestimates the rate of adaptation. For β = 10, the reverse is true.
Together, these results allow us to study the parameter regimes in which different aspects of clonal interference analysis are accurate. The key parameter is β. For β > 1, clonal interference analysis does not accurately predict either 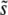 or the shape of the distribution of mutations that fix, or the dynamics of adaptation in large populations. For β = 1 it correctly estimates
or the shape of the distribution of mutations that fix, or the dynamics of adaptation in large populations. For β = 1 it correctly estimates 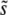 , but still misunderstands the distribution of mutations that fix. For β < 1, clonal interference fares best, accurately estimating both
, but still misunderstands the distribution of mutations that fix. For β < 1, clonal interference fares best, accurately estimating both 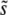 and the distribution of mutations that fix. However, even here it still does not give an accurate analysis of all quantities of interest—it wrongly asserts q < 2 and overestimates the rate of adaptation.
and the distribution of mutations that fix. However, even here it still does not give an accurate analysis of all quantities of interest—it wrongly asserts q < 2 and overestimates the rate of adaptation.
It should be noted that for small populations, where 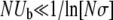 , clonal interference gives an accurate picture of all aspects of the evolution of the populations, regardless of the value of β. This is largely because this is the regime where the small-population analysis is correct for the mutations of interest—that is, clonal interference analysis is accurate because it correctly predicts that clonal interference will not occur, except possibly among extremely small-effect mutations.
, clonal interference gives an accurate picture of all aspects of the evolution of the populations, regardless of the value of β. This is largely because this is the regime where the small-population analysis is correct for the mutations of interest—that is, clonal interference analysis is accurate because it correctly predicts that clonal interference will not occur, except possibly among extremely small-effect mutations.
We have described above an alternative theoretical approach, focusing primarily on multiple-mutation effects, which we expect to be valid when SD(s) is small compared to 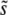 , so that mutations within a narrow range of fitness effects dominate the evolution. We can see from our simulations that this is indeed the case (Figure 3). Desai and Fisher (2007) calculated
, so that mutations within a narrow range of fitness effects dominate the evolution. We can see from our simulations that this is indeed the case (Figure 3). Desai and Fisher (2007) calculated 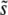 using this approach, finding Equation 9 when β > 1; the approximation broke down for β ≤ 1. Using this
using this approach, finding Equation 9 when β > 1; the approximation broke down for β ≤ 1. Using this 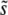 , and assuming that the mutation rate relevant to v(s) is the total mutation rate to mutations of order s, Desai and Fisher (2007) also calculated the lead q and the rate of adaptation v, Equation 11 and Equation 12, respectively, for β > 1.
, and assuming that the mutation rate relevant to v(s) is the total mutation rate to mutations of order s, Desai and Fisher (2007) also calculated the lead q and the rate of adaptation v, Equation 11 and Equation 12, respectively, for β > 1.
We compare these theoretical predictions to our simulation results in Figures 2, 4, and 5. We see that for this β > 1 case, and large N and Ub, they give qualitatively the correct behavior for 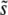 , the lead q, and the rate of adaptation v, though they do systematically overestimate v and underestimate q (see below). In this regime, these results are more accurate than those given by clonal interference analysis. We see from Figure 4 that indeed q can become >2, indicating that multiple mutations are often important to the dynamics. For small N and Ub, corresponding to q ≤ 2, the multiple-mutations results are less accurate, as expected because the above results are valid only when q ≳ 2 (Desai and Fisher 2007).
, the lead q, and the rate of adaptation v, though they do systematically overestimate v and underestimate q (see below). In this regime, these results are more accurate than those given by clonal interference analysis. We see from Figure 4 that indeed q can become >2, indicating that multiple mutations are often important to the dynamics. For small N and Ub, corresponding to q ≤ 2, the multiple-mutations results are less accurate, as expected because the above results are valid only when q ≳ 2 (Desai and Fisher 2007).
The theoretical predictions for q and v, Equations 11 and 12, rely on the ad hoc assumption that the appropriate mutation rate 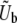 to the mutations around
to the mutations around 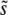 that dominate the dynamics is the total mutation rate to mutations of order
that dominate the dynamics is the total mutation rate to mutations of order 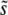 (i.e., within roughly a factor of 2 of
(i.e., within roughly a factor of 2 of 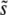 ). As we have found here, SD(s) is often much smaller than
). As we have found here, SD(s) is often much smaller than 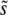 (Figure 3), so the range of mutational effects that contributes substantially to the evolution is much smaller than assumed. This means that the intuitive picture behind the multiple-mutations model of the dynamics is correct: there is indeed a narrow range of mutations that contribute to the evolution, and their accumulation is well described by a corresponding single-s model of the dynamics. However, the estimate of the appropriate mutation rate
(Figure 3), so the range of mutational effects that contributes substantially to the evolution is much smaller than assumed. This means that the intuitive picture behind the multiple-mutations model of the dynamics is correct: there is indeed a narrow range of mutations that contribute to the evolution, and their accumulation is well described by a corresponding single-s model of the dynamics. However, the estimate of the appropriate mutation rate 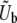 is too large, since a narrower range contributes than was assumed. In addition to this, the estimate of
is too large, since a narrower range contributes than was assumed. In addition to this, the estimate of 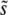 in Equation 9 is also slightly too large (Figure 2). Both of these effects mean that the prediction for v in Equation 12 should be an overestimate, as we observe in Figure 5. On the other hand, while its overestimate of
in Equation 9 is also slightly too large (Figure 2). Both of these effects mean that the prediction for v in Equation 12 should be an overestimate, as we observe in Figure 5. On the other hand, while its overestimate of 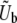 should mean that theory overestimates q, its overestimate of
should mean that theory overestimates q, its overestimate of 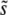 leads to a compensating underestimate of q (because q is defined in terms of
leads to a compensating underestimate of q (because q is defined in terms of 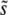 ). The net result of these two effects is that the theory underestimates q (Figure 4). To correct these problems, we need to understand what determines SD(s). Unfortunately neither clonal interference analysis nor the multiple-mutations approach does this well; it remains an important topic for future analytical work.
). The net result of these two effects is that the theory underestimates q (Figure 4). To correct these problems, we need to understand what determines SD(s). Unfortunately neither clonal interference analysis nor the multiple-mutations approach does this well; it remains an important topic for future analytical work.
Another feature of our simulation results is particularly striking. Both the multiple-mutations and the clonal interference analyses predict that 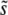 should increase with N, because increasing the population size increases the probability of clonal interference but does not dramatically change the relative importance of large-effect mutations compared to multiple smaller-effect ones. However, for large β, there is a qualitative difference between the predictions of the two approaches for the relationship between
should increase with N, because increasing the population size increases the probability of clonal interference but does not dramatically change the relative importance of large-effect mutations compared to multiple smaller-effect ones. However, for large β, there is a qualitative difference between the predictions of the two approaches for the relationship between 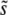 and Ub. Clonal interference analysis alone predicts that
and Ub. Clonal interference analysis alone predicts that 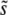 increases with Ub, because higher mutation rates make clonal interference more common. However, higher mutation rates also increase the importance of multiple small-effect mutations relative to large-effect ones. From Equation 9 we see that for β > 1 this should mean that
increases with Ub, because higher mutation rates make clonal interference more common. However, higher mutation rates also increase the importance of multiple small-effect mutations relative to large-effect ones. From Equation 9 we see that for β > 1 this should mean that 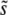 actually decreases with Ub for large Ub. Although the effect is small, we do in fact observe this decrease in our simulations (Figure 2a). For small β, on the other hand, we expect that multiple-mutation effects typically do not impede the fixation of large-effect mutations. Thus clonal interference analysis gives a qualitatively accurate picture of how
actually decreases with Ub for large Ub. Although the effect is small, we do in fact observe this decrease in our simulations (Figure 2a). For small β, on the other hand, we expect that multiple-mutation effects typically do not impede the fixation of large-effect mutations. Thus clonal interference analysis gives a qualitatively accurate picture of how 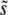 depends on N and Ub, as observed.
depends on N and Ub, as observed.
While the clonal interference and the multiple-mutations approximations together help us to form a more complete understanding of the dynamics, both leave much to be desired. Clonal interference processes appear to be the main determinant of 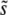 when β ≤ 1, as demonstrated by Figure 2. But even for these small β, we see that q is often >2, and hence clonal interference analysis gives the wrong picture for the dynamics. Further, it misses the possibility of smaller-effect mutations fixing together with those of effect ∼
when β ≤ 1, as demonstrated by Figure 2. But even for these small β, we see that q is often >2, and hence clonal interference analysis gives the wrong picture for the dynamics. Further, it misses the possibility of smaller-effect mutations fixing together with those of effect ∼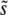 and hence drastically underestimates SD(s) even for β = 1. On the other hand, while the multiple-mutations analysis provides the right picture of the dynamics for β > 1, and accurately predicts
and hence drastically underestimates SD(s) even for β = 1. On the other hand, while the multiple-mutations analysis provides the right picture of the dynamics for β > 1, and accurately predicts 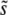 , q, and v, it provides no way to estimate SD(s) nor any specific predictions when β ≤ 1. The simulation approach we have taken in this article sheds some light on where and why these two different approaches work and has highlighted the regimes where neither one provides a satisfactory picture. A more detailed understanding will require analysis of a general model that explicitly incorporates both clonal interference and multiple mutations, to produce a theory that has the correct picture of the dynamics in these difficult regimes. Given that both of these effects appear likely to be widespread in microbial and viral populations (Bollback and Huelsenbeck 2007; Desai et al. 2007; Pepin and Wichman 2008), this is an important topic for future work.
, q, and v, it provides no way to estimate SD(s) nor any specific predictions when β ≤ 1. The simulation approach we have taken in this article sheds some light on where and why these two different approaches work and has highlighted the regimes where neither one provides a satisfactory picture. A more detailed understanding will require analysis of a general model that explicitly incorporates both clonal interference and multiple mutations, to produce a theory that has the correct picture of the dynamics in these difficult regimes. Given that both of these effects appear likely to be widespread in microbial and viral populations (Bollback and Huelsenbeck 2007; Desai et al. 2007; Pepin and Wichman 2008), this is an important topic for future work.
An interesting result of our simulations is that the general shape of the distribution of the mutations that contribute to adaptation, R(s), is rather universal. R(s) is always a relatively narrow distribution with a clear peak at some 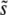 . The distribution of mutations that fix, ρf(s), has a similar shape with a sharp peak near
. The distribution of mutations that fix, ρf(s), has a similar shape with a sharp peak near 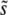 . This means that if we do a single experiment at a given population size and mutation rate, the observed ρf(s) will not provide much information about the underlying ρ(s). This lack of sensitivity of experimental adaptation to the distribution of mutational effects has been noted in a related context by Hegreness et al. (2006). However, our simulations also show that the scaling of various aspects of ρf(s) (such as
. This means that if we do a single experiment at a given population size and mutation rate, the observed ρf(s) will not provide much information about the underlying ρ(s). This lack of sensitivity of experimental adaptation to the distribution of mutational effects has been noted in a related context by Hegreness et al. (2006). However, our simulations also show that the scaling of various aspects of ρf(s) (such as 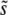 ) with population size and mutation rate does depend strongly on ρ(s). Most important is the shape of the tail of ρ(s); as we have seen, the way in which
) with population size and mutation rate does depend strongly on ρ(s). Most important is the shape of the tail of ρ(s); as we have seen, the way in which 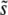 depends on N and Ub is strongly dependent on β. Thus careful experiments that are carried out at a range of population sizes or mutation rates may make it possible to infer important aspects of ρ(s) from measurements of ρf(s) or the rate of adaptation v.
depends on N and Ub is strongly dependent on β. Thus careful experiments that are carried out at a range of population sizes or mutation rates may make it possible to infer important aspects of ρ(s) from measurements of ρf(s) or the rate of adaptation v.
As our simulations make clear, the actual values of β applicable to natural asexual populations are of central importance to the dynamics by which these populations adapt. The values of β found in natural populations may also tell us something about the evolutionary history of these populations. Orr and Gillespie have argued that if an individual is at a random high-fitness genotype, the distribution of more-fit genotypes is exponential, so we should expect β = 1 (Gillespie 1983, 1984, 1991; Orr 2002, 2003). However, as a population adapts it is natural to expect ρ(s) to change. For example, a population may face a static challenge and gradually deplete the available beneficial mutations as it adapts. If the population were small enough that ρf(s) = 2sρ(s), the distribution ρ(s) should converge to an exponential as this adaptation progresses. But if clonal interference and multiple-mutation effects cause large-effect mutations to be depleted much faster than small-effect ones, we expect that β should increase as the available mutations become depleted. Thus large observed values of β may be indicative of this type of adaptation. Of course, this increase in β could be avoided if the accumulation of beneficial mutations tends to open up new possibilities for further adaptation. In other words, the structure of fitness landscapes has an important role in determining typical values of β and how these change as populations adapt. Given these values of β, our simulations provide a way to understand, in a statistical sense, which mutations will tend to contribute to adaptation and the dynamics by which they will do so.
Acknowledgments
J.L.N. and C.F. acknowledge support from the University of Colorado and the Undergraduate Research Opportunity Program at the University of Colorado. M.M.D. acknowledges support from Center grant P50GM071508 from the National Institute of General Medical Science to the Lewis-Sigler Institute.
References
- Bollback, J. P., and J. P. Huelsenbeck, 2007. Clonal interference is alleviated by high mutation rates in large populations. Mol. Biol. Evol. 24 1397–1406. [DOI] [PubMed] [Google Scholar]
- Campos, P. R. A., and V. M. de Oliveira, 2004. Mutational effects on the clonal interference phenomenon. Evolution 58 932–937. [DOI] [PubMed] [Google Scholar]
- DePristo, M. A., D. M. Weinreich and D. Hartl, 2005. Missense meanderings in sequence space: a biophysical view of protein evolution. Nat. Rev. Genet. 6 678–687. [DOI] [PubMed] [Google Scholar]
- de Visser, J. A. G. M., and D. E. Rozen, 2005. Limits to adaptation in asexual populations. J. Evol. Biol. 18 779–788. [DOI] [PubMed] [Google Scholar]
- de Visser, J., C. W. Zeyl, P. J. Gerrish, J. L. Blanchard and R. E. Lenski, 1999. Diminishing returns from mutation supply rate in asexual populations. Science 283 404–406. [DOI] [PubMed] [Google Scholar]
- Desai, M. M., and D. S. Fisher, 2007. Beneficial mutation-selection balance and the effect of linkage on positive selection. Genetics 176 1759–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai, M. M., D. S. Fisher and A. W. Murray, 2007. The speed of evolution and maintenance of variation in asexual populations. Curr. Biol. 17 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish, P., 2001. The rhythm of microbial adaptation. Nature 413 299–302. [DOI] [PubMed] [Google Scholar]
- Gerrish, P., and R. Lenski, 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102/103 127–144. [PubMed] [Google Scholar]
- Gillespie, J. H., 1983. A simple stochastic gene substitution model. Theor. Popul. Biol. 23 202–215. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H., 1984. Molecular evolution over the mutational landscape. Evolution 38 1116–1129. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H., 1991. The Causes of Molecular Evolution. Oxford University Press, Oxford.
- Haldane, J. B. S., 1927. The mathematical theory of natural and artificial selection, part v: selection and mutation. Proc. Camb. Philos. Soc. 23 838–844. [Google Scholar]
- Hegreness, M., N. Shoresh, D. Hartl and R. Kishony, 2006. An equivalence principle for the incorporation of favorable mutations in asexual populations. Science 311 1615–1617. [DOI] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1966. Effect of linkage on limits to artificial selection. Genet. Res. 8 269–294. [PubMed] [Google Scholar]
- Imhof, M., and C. Schlotterer, 2001. Fitness effects of advantageous mutations in evolving Escherichia coli populations. Proc. Natl. Acad. Sci. USA 98 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, T., and N. H. Barton, 2002. The effect of deleterious alleles on adaptation in asexual organisms. Genetics 162 395–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph, S. B., and D. W. Hall, 2004. Spontaneous mutations in diploid Saccharomyces cerevisiae: more beneficial than expected. Genetics 168 1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen, R., and T. Bataillon, 2006. Distribution of fitness effects among beneficial mutations before selection in experimental populations of bacteria. Nat. Genet. 38 484–488. [DOI] [PubMed] [Google Scholar]
- Kim, Y., and H. A. Orr, 2005. Adaptation in sexuals vs. asexuals: clonal interference and the Fisher–Muller model. Genetics 171 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunzer, M., S. P. Miller, R. Felsheim and A. M. Dean, 2005. The biochemical architecture of an ancient adaptive landscape. Science 310 499–501. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2000. The rate of adaptation in asexuals. Genetics 155 961–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 2002. The population genetics of adaptation: the adaptation of DNA sequences. Evolution 56 1317–1330. [DOI] [PubMed] [Google Scholar]
- Orr, H. A., 2003. The distribution of fitness effects among beneficial mutations. Genetics 163 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, S., and J. Krug, 2007. Clonal interference in large populations. Proc. Natl. Acad. Sci. USA 104 18135–18140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin, K. M., and H. A. Wichman, 2008. Experimental evolution and genome sequencing reveal variation in levels of clonal interference in large populations of bacteriophage φx174. BMC Evol. Biol. 8 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfeito, L., L. Fernandes, C. Mota and I. Gordo, 2007. Adaptive mutations in bacteria: high rate and small effects. Science 317 813–815. [DOI] [PubMed] [Google Scholar]
- Ridgway, D., H. Levine and D. Kessler, 1998. Evolution on a smooth landscape: the role of bias. J. Stat. Phys. 90 191. [Google Scholar]
- Rokyta, D. R., P. Joyce, S. B. Caudle and H. A. Wichman, 2005. An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat. Genet. 37 441–444. [DOI] [PubMed] [Google Scholar]
- Rouzine, I., J. Wakeley and J. M. Coffin, 2003. The solitary wave of asexual evolution. Proc. Natl. Acad. Sci. USA 100 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine, I., E. Brunet and C. O. Wilke, 2008. The traveling-wave approach to asexual evolution: Muller's ratchet and speed of adaptation. Theor. Popul. Biol. 73 24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen, D. E., J. de Visser and P. J. Gerrish, 2002. Fitness effects of fixed beneficial mutations in microbial populations. Curr. Biol. 12 1040–1045. [DOI] [PubMed] [Google Scholar]
- Sanjuan, R., A. Moya and S. F. Elena, 2004. The distribution of fitness effects caused by single-nucleotide substitutions in an RNA virus. Proc. Natl. Acad. Sci. USA 101 8396–8401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke, C. O., 2004. The speed of adaptation in large asexual populations. Genetics 167 2045–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]