

Abstract
Antipsychotic drugs are increasingly being prescribed for children and adolescents, and are used in pregnant women without a clear demonstration of safety in these populations. Global effects of these drugs on neurodevelopment (e.g., decreased brain size) have been reported in rats, but detailed knowledge about neuronal effects and mechanisms of action are lacking. Here we report on the evaluation of a comprehensive panel of antipsychotic drugs in a model organism (Caenorhabditis elegans) that is widely used to study neuronal development. Specifically, we examined the effects of the drugs on neuronal migration and axonal outgrowth in mechanosensory neurons visualized with green fluorescent protein expressed from the mec-3 promoter. Clozapine, fluphenazine, and haloperidol produced deficits in the development and migration of ALM neurons and axonal outgrowth in PLM neurons. The defects included failure of neuroblasts to migrate to the proper location, and excessive growth of axons past their normal termination point, together with abnormal morphological features of the processes. Although the antipsychotic drugs are potent antagonists of dopamine and serotonin receptors, the neurodevelopmental deficits were not rescued by co-incubation with serotonin or the dopaminergic agonist, quinpirole. Other antipsychotic drugs, risperidone, aripiprazole, quetiapine, trifluoperazine and olanzapine, also produced modest, but detectable, effects on neuronal development. This is the first report that antipsychotic drugs interfere with neuronal migration and axonal outgrowth in a developing nervous system.
Keywords: antipsychotic drugs, axon outgrowth, neuronal migration
1. Introduction
Antipsychotic drugs are used to treat symptoms associated with several psychiatric illnesses including schizophrenia, bipolar disorder, and psychotic depression. Increasingly, these drugs are prescribed off label to adolescents and children as young as 2 years old (Findling et al., 1998; Zito et al., 2000), and to pregnant women (Lanczik et al., 1998; Yaeger et al., 2006). It is unclear whether early developmental exposure to antipsychotic drugs results in permanent changes in the brain that affect cognitive function or behavior later in life. Antipsychotic drugs have been reported to produce developmental deficits and neurological symptoms in humans. For example, in utero exposure to clozapine has been associated with floppy infant and jittery baby syndrome (Di Michele et al., 1996; Lanczik et al., 1998), and lower developmental scores (Yoshida et al., 1998). Moreover, it was recently reported that clozapine, but not haloperidol, produced long-lasting changes in the behavior of mice when given during gestation (Wang et al., 2006). The FDA classifies antipsychotic drugs as class C because the risk of developmental defects cannot be excluded. More studies are needed to determine whether these drugs produce lasting adverse effects on the developing nervous system.
The neurotransmitters serotonin and dopamine play important roles during development of the nervous system. Serotonin acts as a trophic factor early in neurodevelopment, where it regulates neurogenesis, differentiation, neuronal migration, and synaptic plasticity (Azmitia, 2001; Luo et al., 2003). Dopamine mediates synaptogenesis (Spencer et al., 1998), and the expression of axon guidance molecules in neuronal cells (Jassen et al., 2006). Antipsychotic drugs bind to and block dopamine and serotonin receptors. Thus, the antipsychotic drugs may produce neurodevelopmental defects related to antagonism of these neurotransmitter receptors. The second-generation antipsychotic drugs have higher affinities for serotonin receptors than first-generation antipsychotics (Meltzer et al., 1989). Therefore, antipsychotic drugs may differentially disrupt normal neurodevelopment in relation to their pharmacological properties. A major goal of these studies was to test the hypothesis that antipsychotic drugs affect neurodevelopment through their actions on known signaling pathways, including dopamine and serotonin receptors, and calmodulin.
To evaluate this hypothesis, we characterized the effects of the antipsychotic drugs on neuronal development in the model organism, Caenorhabditis elegans. This model system was chosen because previous work showed that antipsychotic drugs interfered with normal development in this simple invertebrate (Donohoe et al, 2006). Moreover, C. elegans has been widely used to study neuronal migration and axonal pathfinding (Branda and Stern, 1999; Merz and Culotti, 2000). The gentle touch response in C. elegans is mediated by 5 mechanosensory neurons, ALML/R, AVM, and PLML/R (Chalfie et al., 1985). These neurons and their processes can be visualized with green fluorescent protein (GFP) markers such as MEC-3::GFP (e.g., see Toms et al., 2001). The ALM neuroblasts migrate from near the terminal pharyngeal bulb to the midline of the animal and then send a long axon to the tip of the head with a short branch near the nerve ring. The PLM neurons reside in the tail region and extend their main neurite in the anterior direction with termination near the midline, and a second short neurite, which projects to the tip of the tail. The distance traversed by the ALM neuroblast and the length of the ALM and PLM axons offer a unique opportunity to study in detail the regulation of neuroblast migration and axonal pathfinding in vivo.
Antipsychotic drugs have been reported to produce developmental delay and brain abnormalities in rats (Holson et al., 1994), although the mechanism of action was not explored. The drugs also delayed reproductive maturation in C. elegans via dopamine and serotonin receptor-independent mechanisms that appear to include calmodulin (Donohoe et al., 2006). Antipsychotic drugs, at a concentration of 1–50 µM, effectively antagonize calmodulin (Weiss et al., 1980). This corresponds to the physiological concentration of these drugs in brain tissue due to accumulation (Baldessarini et al., 1993; Dahl, 1986; Jann et al., 1993; Kornhuber et al., 1999). Upon binding Ca2+, calmodulin activates Ca2+-calmodulin-dependent protein kinase II (CaMKII) and the protein phosphatase calcineurin (Klee, 1991). CaMKII and calcineurin modulate axonal pathfinding through a switch-like mechanism that controls directional changes in growth cone guidance (Wen et al., 2004). Therefore, antipsychotic drugs that antagonize calmodulin might be expected to affect neurodevelopmental processes regulated by this protein, including axonal outgrowth.
In this study, we used a C. elegans strain that expresses MEC-3::GFP in the touch receptor (mechanosensory) neurons and monitored changes in the migration of ALM neurons, and PLM axonal outgrowth in animals exposed to antipsychotic drugs from fertilization to the third larval stage. The antipsychotic drugs, to varying degrees, disrupted both neuroblast migration and axonal outgrowth of the ALM and PLM neurons. To our knowledge, this is the first study showing that antipsychotic drugs disrupt neuroblast migration and axonal outgrowth, in vivo.
2. Experimental procedures
2.1. C. elegans culture
C. elegans strains were grown and maintained at 20 °C on NGM plates seeded with Escherichia coli strain (OP50) as previously described (Brenner, 1974). The mec-3::gfp strain, TU2562, was generously provided by Dr. Martin Chalfie. The tph-1(mg280) strain was obtained from the Caenorhabditis Genetics Center (Minneapolis, MN).
2.2. Preparation of drug plates
Serotonin, quinpirole, clozapine, risperidone, haloperidol, fluphenazine, chlorpromazine, trifluoperazine, and calmidazolium were purchased from Sigma-Aldrich (St. Louis, MO). Quetiapine was obtained as a gift from Astra-Zeneca. Aripiprazole and olanzapine were provided by Bristol-Myers Squibb and Eli Lilly and Company, respectively. All drugs were prepared as described by Donohoe et al. (2006). For stock solutions (made at 30 mM), drugs were dissolved in DMSO, except risperidone, haloperidol, and aripiprazole, which were dissolved in ethanol. Serotonin was prepared in water slightly acidified with acetic acid (1:10,000) to obtain a stock concentration of 50 mM.
2.3. Drug treatment
Animals in the fourth larval stage (prior to reproductive maturation) were placed on control (solvent alone) or drug plates and allowed to mature and lay eggs to ensure that developing embryos were exposed to drug. Adult animals were removed and their progeny were allowed to develop to the third larval stage, at which time they were mounted on microscope slides for fluorescence microscopy.
2.4. Fluorescence microscopy
For each experimental condition, animals were collected from the plates and mounted on microscope slides containing 50 mM sodium azide (to paralyze the worms). Microscopy was performed with a Nikon Eclipse TE300 inverted microscope with epi-fluorescence attachment, Nomarski optics, and a CoolSNAP monochrome CCD camera. Neuronal migration was scored by two methods: 1) the position of the neuronal cell body was scored by eye according to a scale where the back of the terminal bulb was position 0 and the start of the vulva was position 10 (Toms et al., 2001), and 2) by measuring both the distance from the pharyngeal grinder to the ALM cell body and the total body length with the program Image J (Bethesda, MD) in merged differential interference contrast (DIC) and fluorescence micrographs. Migration was then calculated as the fraction of the body length traversed by the ALM neuroblasts. The migration data show a skewed distribution because neuroblasts are never observed beyond position 10, and, in control animals, most reach positions 7–9. The PLM axon length was analyzed with Image J (Bethesda, MD) by measuring the distance in µm from the PLM cell body to the termination point of the axon. Twenty synchronized animals (2nd larval stage) were evaluated for each condition.
2.5. Statistical analysis
A one-way ANOVA followed by a post hoc Tukey test was used to evaluate statistical significance between control and drug groups with respect to parametric measures of ALM position and PLM axon length. The data regarding classification of neuronal migration according to the rating scale and the deviation from control values (Fig. 3) were analyzed for statistical significance according to the goodness-of-fit test of Kolmogorov-Smirnov. This test can be used to fit data with a non-normal distribution. Each experiment was performed with duplicate plates, and was repeated at least twice. Differences between drug-treated and control groups were considered significant with a p-value < 0.05.
Fig. 3.

Antipsychotic drugs affect the distance migrated by ALM neuroblasts. Transgenic animals (n = 25) expressing mec-3::gfp, were incubated on plates containing DMSO (control), clozapine (160 µM), fluphenazine (160 µM), chlorpromazine (160 µM), trifluoperazine (80 µM), haloperidol (160 µM), risperidone (160 µM), olanzapine (160 µM), quetiapine (160 µM), or aripiprazole (160 µM), from fertilization until L3, and the distance from the grinder (see Fig. 1A) to the ALM neuron was measured as a function of body length. The data are plotted as the percentage of neurons versus the deviation from their normal position relative to controls.
3. Results
We previously found that various antipsychotic drugs (the first generation drugs and clozapine) produced developmental delay in C. elegans that was not explained by their actions on dopamine and serotonin receptors (Donohoe et al., 2006). To extend these findings, we tested whether the antipsychotic drugs that slowed maturation also disrupted neurodevelopment. Specifically, in the present study, we set out to characterize the effects of the antipsychotic drugs on neuronal migration of the ALM neuroblasts in C. elegans.
For these experiments, transgenic animals (expressing mec-3::gfp) were exposed to antipsychotic drugs from fertilization until the third larval stage, at which time neuronal migration and axonal pathfinding were analyzed. The concentrations tested in the present study (final concentration was 80–160 µM on the agar plate) reflect the amount of drug required to block dopamine receptors, in C. elegans (Donohoe et al., 2006).
3.1. Antipsychotic drugs disrupt ALM neuroblast migration
The ALM neuroblasts are born just posterior to the pharynx and migrate to the middle of the animal during embryogenesis (Fig. 1A). We observed that animals exposed to clozapine, fluphenazine, and haloperidol had fewer detectable ALM neurons in comparison to control animals as judged by a failure to routinely observe two bright cell bodies in their correct positions (Fig. 1C). In addition, the ALM neurons that were detected frequently failed to migrate to their proper position (Fig. 1B). To further characterize these effects, we scored the position of the ALM neurons in animals exposed to just the solvent, DMSO (control) or antipsychotic drugs. We established that the antipsychotic drugs, to varying degrees, altered neuroblast migration as compared to control animals (Fig. 2). In DMSO-treated animals, ALM neuroblasts migrate to approximately position 8 with little variability, whereas animals exposed to clozapine, fluphenazine, and haloperidol had a significantly higher percentage of ALM neurons that were scored as 4–7, and there was greater variability in the positioning. Animals exposed to olanzapine and quetiapine had a higher percentage of ALM neurons located at 8 and 9 as compared to the controls, which suggests that these drugs induce a different defect.
Fig. 1.

Antipsychotic drugs disrupt normal development of ALM neurons. (A) Diagram of ALM neuroblast migration and positional scoring system, from 0–10, used to assess ALM neuroblast migration deficits in animals exposed to DMSO (control) and drug. (B) Fluorescence micrographs of animals expressing the mec-3::gfp transgene and exposed to DMSO, clozapine (160 µM) or fluphenazine (160 µM). (C) Analysis of the number of ALM neurons expressing mec-3::gfp at the third larval stage (L3) in 100 animals chronically exposed to DMSO, clozapine (160 µM), clozapine-N-oxide (160 µM), fluphenazine (160 µM), chlorpromazine (160 µM), haloperidol (160 µM) olanzapine (160 µM), and quetiapine (160 µM). Error bars represent the standard deviations from the mean. Significant differences from the control group are indicated by asterisks: *p < 0.05, **p < 0.01.
Fig. 2.

Effect of antipsychotic drugs on the migration of ALM neurons. ALM neuron position was scored from 0–10 (see Fig. 1A) in 100 animals per experimental condition. Animals were incubated from fertilization to the L3 stage with DMSO (control), or 160 µM clozapine, clozapine-N-oxide, fluphenazine, chlorpromazine, haloperidol, olanzapine, or quetiapine. Data from the groups exposed to clozapine, fluphenazine, haloperidol, olanzapine, and quetiapine were significantly different from the control values (p < 0.05).
To independently confirm the effects of the drugs on neuroblast migration, we repeated the experiments on another set of animals and measured the distance from the ALM cell body to the grinder of each worm (Fig. 1A) rather than assigning a migration score. Again, ALM neurons showed greater disruption of migration in animals exposed to antipsychotic drugs than in controls, and were typically observed anterior to their normal location (Fig. 3). This resulted in a 15–75 % increase in the percentage of ALM neurons that were located at a distance greater than 2 standard deviations from the mean of control animals. Clozapine and fluphenazine produced significant disruption of ALM neuroblast migration (p < 0.05). Other antipsychotic drugs, including haloperidol, risperidone, quetiapine, aripiprazole, and chlorpromazine (at 80–160 µM), clearly affected neuroblast migration, however, the distance measurements in Fig. 3 were not significantly different from the controls. These studies demonstrate that the antipsychotic drugs differentially interfere with normal neurodevelopmental processes controlling ALM migration.
3.2. Abnormal PLM axonal outgrowth induced by antipsychotic drugs
In addition to disrupting ALM migration, we observed that the antipsychotic drugs also affected PLM axonal outgrowth. The PLM neurons are located in the tail of the animal and send their axons along the side of the animal toward the head. The axons terminate at the midline of the animal, just posterior to the ALM cell bodies (Fig. 1A). Some of the animals exposed to clozapine and fluphenazine had PLM axons that extended all the way to the head (Fig. 4A). Several animals treated with clozapine had PLM axons that reversed direction or displayed abnormal zig-zag morphology (Fig. 4B).
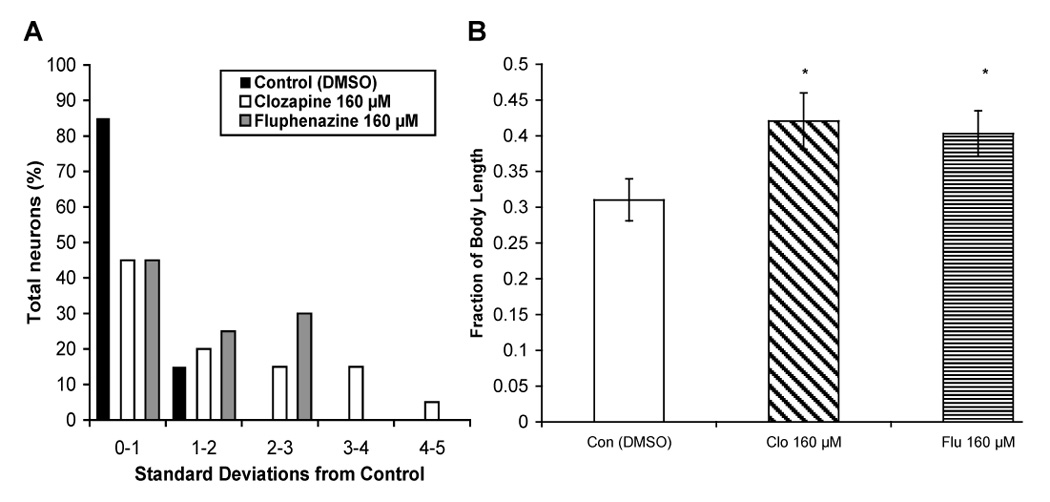
Fig. 4.

Antipsychotic drugs affect PLM axonal outgrowth. (A) Fluorescence photomicrographs showing extended PLM axons in animals exposed to clozapine (160 µM) and fluphenazine (160 µM). (B) Morphological defects (misdirected axons) produced by clozapine (160 µM). (C) Quantitative measurement of axon length (cell body to axon termination point) in relation to total body length in animals (n = 25) exposed to DMSO (Con), clozapine (Clo; 160 µM), fluphenazine (Flu; 160 µM), trifluoperazine (Tri; 80 µM), haloperidol (Hal; 160 µM), olanzapine (Ola; 160 µM), quetiapine (Que; 160 µM), risperidone (Ris; 160 µM), and aripiprazole (Ari; 160 µM). Error bars represent standard deviation from the mean. Significant differences are indicated with asterisks: *p > 0.05, **p >0.01.
To quantify the effects of various antipsychotic drugs on PLM axonal outgrowth, we measured the distance from the PLM cell body to the axon termination point (expressed as a fraction of the entire body length of the animal) in both DMSO (control) and drug-treated animals. Data in Figure 4C show that animals exposed to clozapine and fluphenazine had abnormally long PLM axons in comparison to control animals. Frequently, animals with abnormal PLM axons also had misplaced ALM neurons; however, deficits in ALM neuroblast migration were not always accompanied by PLM abnormalities. These data demonstrate that antipsychotic drugs, clozapine and fluphenazine in particular, produce defects in neuroblast migration and axonal outgrowth. We did not observe the same frequency of aberrant axonal outgrowth in animals exposed to similar concentrations of trifluoperazine, haloperidol, olanzapine, quetiapine, risperidone, and aripiprazole. Because clozapine and fluphenazine inhibit calmodulin, it was possible that this calcium-signaling protein was involved in the effect of the drugs on neurodevelopment. Therefore, we tested calmidazolium, a specific calmodulin antagonist, for its effects on ALM migration and PLM axonal outgrowth. Although calmidazolium (80 µM) slowed the growth of C. elegans as expected, it did not affect neuroblast migration or axonal outgrowth (data not shown).
3.3. Effects of the drugs on neurodevelopment are serotonin independent
Previously, Kindt et al. (2002) reported that serotonin-deficient mutants (tph-1) manifested deficits in neuronal migration that could be rescued through application of serotonin to the culture plates. The antipsychotic drugs block the actions of serotonin at a variety of 5-HT receptors (Meltzer et al., 2003). Therefore, we sought to evaluate whether the effects of the antipsychotic drugs on neurodevelopment are due to inhibition of serotonin receptors. To characterize the potential role of serotonin in these effects, we attempted to rescue neuronal migration and axonal pathfinding deficits in animals grown on drug-plates that also contained serotonin. Serotonin (5 mM) failed to rescue the effects of clozapine (Fig. 5A) and fluphenazine (Fig. 5B) on neuronal migration and PLM axonal outgrowth (Fig. 5C). Animals exposed to clozapine or fluphenazine in the presence of serotonin still had a higher percentage of ALM neurons located greater than 1 standard deviation from the mean distance measured in control animals, and had longer PLM axons. Thus, serotonin appears to play a limited role in the effects of antipsychotic drugs on ALM and PLM neurons. Moreover, whereas serotonin deficiency is associated with abnormal migration of the ALM, SDQR, and AVM neurons (Kindt et al., 2002), no defects in PLM axons have been reported.
Fig. 5.

Serotonin fails to rescue the deficits in ALM neuroblast migration and PLM axonal pathfinding produced by clozapine and fluphenazine. ALM neuroblast migration was assessed as before in Fig. 3 in transgenic animals expressing mec-3::gfp that were incubated with: (A) clozapine (160 µM) or (B) fluphenazine (160 µM) alone or in combination with 5 mM serotonin. (C) PLM axonal outgrowth was analyzed in animals exposed to clozapine (160 µM) and fluphenazine (160 µM) alone or in combination with 5 mM serotonin. Error bars represent the standard deviation from the mean. Significant differences are indicated as before.
Additional evidence against a major role for serotonin in the deficits induced by antipsychotic drugs was obtained from studies of the tph-1 strain, which lacks serotonin (Sze et al., 2000). Because the tph-1 mutants are deficient in serotonin, the antipsychotic drugs cannot further block serotonin signaling in this strain. Therefore, we tested whether the antipsychotic drugs still produced the same spectrum of neurodevelopmental effects in tph-1 mutants. Both clozapine and fluphenazine produced significant disruption of ALM migration (Fig. 6A) and PLM axonal outgrowth (Fig. 6B) in drug-treated tph-1 animals as compared to control animals exposed to DMSO alone. Taken together with the serotonin rescue data, these experiments suggest that the antipsychotic drugs mainly affect neuronal development through serotonin-independent mechanism(s).
Fig. 6.
Effect of clozapine and fluphenazine on ALM neuroblast migration and PLM axonal outgrowth in tph-1 mutants. (A) The distance traveled by ALM neuroblasts during migration was measured in tph-1(mg280) mutants (n = 25) expressing the mec-3::gfp transgene in the presence of DMSO (control), clozapine (160 µM) (A), and fluphenazine (160 µM). (B) Measurement of defects in PLM axonal outgrowth in tph-1(mg280) mutants exposed to DMSO (control), clozapine (160 µM), and fluphenazine (160 µM). Error bars represent the standard deviation from the mean.
3.4. Quinpirole fails to rescue effects of the drugs on neurodevelopment
The ALM and PLM touch receptor neurons express the D1-like dopamine receptor, DOP-1. Because clozapine and fluphenazine block dopamine receptors, it is possible that the neurodevelopmental effects of antipsychotic drugs result from antagonism of DOP-1 or other C. elegans dopamine receptors. We sought to test this hypothesis by preventing antagonism of dopamine receptors with the agonist quinpirole at concentrations established to be effective in behavioral assays (Donohoe et al., 2006). Data shown in Figure 7A & B demonstrate that addition of the dopamine receptor agonist, quinpirole (200 µM), to drug-plates does not rescue the defects in ALM neuroblast migration produced by clozapine and fluphenazine. Furthermore, quinpirole (200 µM) also failed to rescue the effects of clozapine and fluphenazine on PLM axonal pathfinding (Fig. 7C). Therefore, the effects of antipsychotic drugs on neuronal development are not primarily mediated through antagonism of dopamine receptors.
Fig. 7.
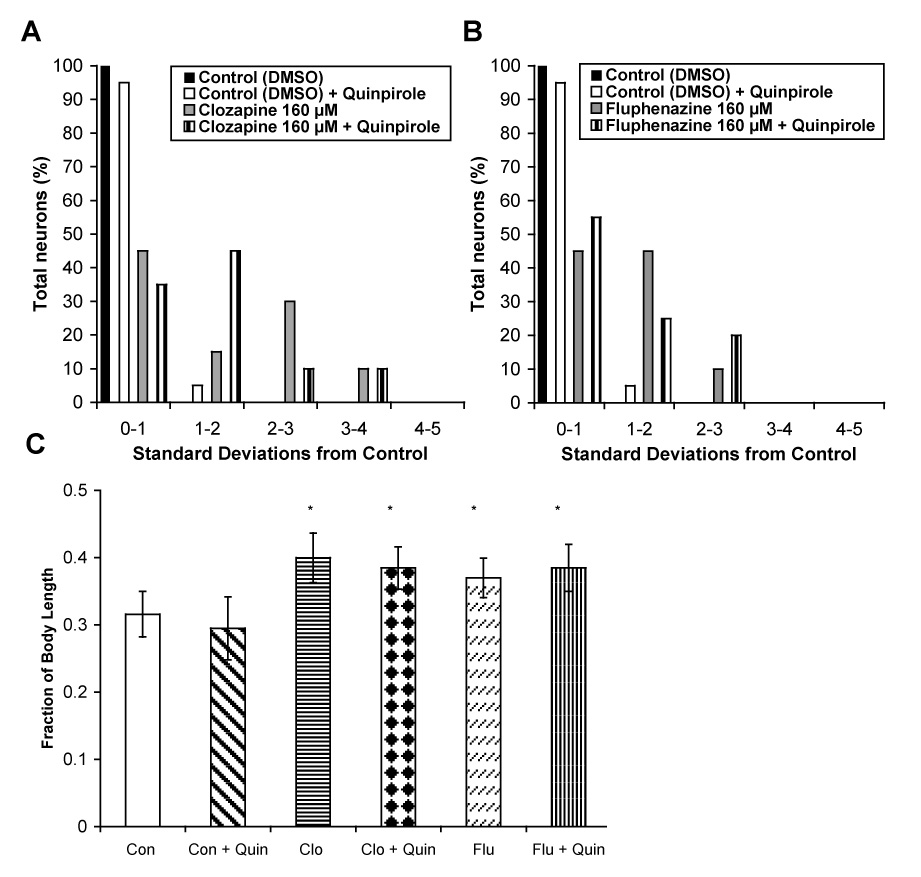
Quinpirole failed to rescue deficits in ALM neuroblast migration and PLM axonal outgrowth produced by clozapine and fluphenazine. ALM neuroblast migration was assessed as before in animals incubated with: (A) clozapine (160 µM) and (B) fluphenazine (160 µM) either alone or in combination with quinpirole (750 µM). (C) PLM axonal outgrowth in animals exposed to clozapine (160 µM) and fluphenazine (160 µM), either alone or in combination with quinpirole (750 µM). Error bars represent the standard deviation from the mean. Significant differences are indicated as before.
4. Discussion
Relatively little is known about changes in neurodevelopment that result from exposure to antipsychotic drugs early in life. Even less is known about whether the antipsychotic drugs alter specific processes such as neuronal migration and axonal pathfinding. In the present study, animals exposed to clozapine or fluphenazine during embryonic development showed striking deficits in the migration of ALM neuroblasts and axonal growth in PLM neurons. The ALM neurons were often anterior to their proper position, which reflects a failure to migrate far enough on their trajectory. Clozapine and fluphenazine produced the greatest deficits in neuronal development in C. elegans. Other antipsychotic drugs, e.g., haloperidol, quetiapine, olanzapine, risperidone and aripiprazole, mainly affected ALM migration with little perturbation of PLM axonal outgrowth. Clozapine and fluphenazine may simply be more potent than the other drugs or perhaps they target additional pathways that are not affected by the other antipsychotics at the concentrations used in these studies.
Antipsychotic drugs are potent antagonists of dopamine and serotonin receptors. Serotonin has been reported to regulate ALM migration in C. elegans (Kindt et al., 2002), which makes serotonin receptors an attractive target for the neurodevelopmental effects of the drugs on ALM neurons. However, serotonin has not been implicated in the regulation of PLM axonal outgrowth. We tested whether the effects of clozapine and fluphenazine on neuronal migration and axonal outgrowth were mediated via serotonin signaling and found that the neurodevelopmental deficits were not rescued or reduced in the presence of biologically-active concentrations of serotonin. Furthermore, clozapine and fluphenazine induced abnormal ALM migration and PLM axonal outgrowth in tph-1 mutants, which lack serotonin. Taken together, the evidence suggests that the neurodevelopmental effects of clozapine and fluphenazine are largely mediated via serotonin-independent mechanism(s).
A similar analysis revealed that the contribution of dopamine receptors to the effects of clozapine and fluphenazine was limited. Specifically, quinpirole (a dopamine receptor agonist) failed to reverse the deficits in ALM migration and PLM axonal pathfinding produced by clozapine and fluphenazine. In addition, haloperidol has been shown to block the dopamine receptor, DOP-1, which is expressed on the ALM and PLM neurons (Tsalik et al., 2003; Sanyal et al., 2004). However, haloperidol produced modest effects on ALM migration and PLM axonal outgrowth. Thus, it is unlikely that the drugs disrupt neurodevelopment through antagonism of dopamine receptors. These findings force us to consider other possible molecular targets of the antipsychotic drugs.
Fluphenazine and clozapine both antagonize calmodulin (Weiss et al., 1980), although fluphenazine is the more potent inhibitor. Calmodulin has been shown to regulate growth cone direction through activation of CaMKII and calcineurin (Wen et al., 2004), which would be consistent with the nature of the deficits in axonal outgrowth induced by the drugs. However, we determined that calmidazolium, a potent calmodulin antagonist, did not produce significant effects on neuroblast migration and axonal outgrowth. Therefore, we conclude that the antipsychotic drugs must target other proteins besides calmodulin to disrupt normal development of ALM and PLM neurons.
Another possible mechanism that might explain the more extensive effects of clozapine and fluphenazine is altered signaling via the aryl hydrocarbon receptor. The aryl hydrocarbon receptor, known as AHR-1 in C. elegans, is a ligand-activated transcription factor that regulates migration of the ALM neurons (Qin and Powell-Coffman, 2004). Furthermore, mutant animals that lack AHR-1 have PLM axons that extend past the normal termination point (Qin and Powell-Coffman, 2004). Clozapine binds to the human aryl hydrocarbon receptor at physiologically-relevant concentrations (Heiser et al., 2003; Meyer et al., 1996; Talaska et al., 2006). Thus, AHR-1 represents an intriguing target because mutant strains lacking this receptor phenocopy the effects of clozapine on ALM migration and PLM axonal outgrowth. It is not known whether clozapine also binds to AHR-1 in C. elegans. Furthermore, fluphenazine has not been established as a ligand for the mammalian aryl hydrocarbon receptor, although the structure of this drug - a halogenated aromatic ring system - closely matches the motif involved in binding to the aryl hydrocarbon receptor. Therefore, fluphenazine may also bind to AHR-1 of C. elegans. Future studies will explore in greater detail the role of serotonin and dopamine receptor-independent mechanisms such as calmodulin and AHR-1 in the neurodevelopmental deficits induced by antipsychotic drugs.
Irrespective of the exact mechanism of action, our studies show that certain antipsychotic drugs significantly disrupt normal development of the nervous system. Neuronal migration and axonal pathfinding are complex processes that involve multiple signaling and recognition pathways. In C. elegans, the anterior-posterior neuronal migration and axonal trajectories are controlled by a diverse set of genes that includes unc-53 (Hekimi and Kershaw, 1993), vab-8, unc-73, and sax-3 (Hao et al., 2001; Watari-Goshima, 2007; Wolf et al., 1998). Animals with mutations in unc-5 (netrin receptor), sax-3 (robo receptor), and unc-73 (trio) phenocopy the effects of clozapine, fluphenazine, and haloperidol on ALM migration. Interestingly, stable integration (and overexpression) of vab-8l (a kinesin-related protein) produces excessive posterior migration of ALM neurons (Watari-Goshima et al., 2007), which is similar to the effects of olanzapine and quetiapine. We speculate that the antipsychotic drugs affect signaling pathways that intersect with systems that control neuronal migration and guidance. Many genes involved in neuronal migration in C. elegans also regulate cell migration in higher vertebrates (Hatten, 2002). Consequently, C. elegans may be a valuable model system for studying clinically-relevant effects of antipsychotic drugs on neuronal development.
Previously, post-natal exposure of mice to fluoxetine, a serotonin reuptake inhibitor, was shown to produce long-lasting behavioral alterations that persisted to adulthood (Ansorge et al., 2004). Similarly, prenatal exposure to clozapine has been reported to alter behaviors, such as memory acquisition and scopolamine-induced impairment of memory in mice later in life (Wang et al., 2006). Thus, early exposure to antipsychotic drugs may permanently alter neuronal development with a lasting impact on behavior. The significance of early neurodevelopmental insults is illustrated by heterozygous reeler mice, which have aberrant neuronal migration and impairments in behavior that include learning and sensorimotor deficits in adult animals (Brigman et al., 2006; Carboni et al. 2004). In view of the increased use of antipsychotic medications in children, our data suggest that further studies are warranted to examine whether antipsychotic drugs produce similar changes in neuronal migration in vertebrate nervous systems.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ansorge MS, Zhou M, Lira A, Hen R, Gingrich JA. Early-life blockade of the 5-HT transporter alters emotional behavior in adult mice. Science. 2004;306:879–881. doi: 10.1126/science.1101678. [DOI] [PubMed] [Google Scholar]
- Azmitia EC. Modern views on an ancient chemical: serotonin effects on cell proliferation, maturation, and apoptosis. Brain Res. Bull. 2001;56:413–424. doi: 10.1016/s0361-9230(01)00614-1. [DOI] [PubMed] [Google Scholar]
- Baldessarini RJ, Centorrino F, Flood JG, Volpicelli SA, Huston-Lyons D, Cohen BM. Tissue concentrations of clozapine and its metabolites in the rat. Neuropsychopharmacology. 1993;9:117–124. doi: 10.1038/npp.1993.50. [DOI] [PubMed] [Google Scholar]
- Branda CS, Stern MJ. Cell migration and axon growth cone guidance in Caenorhabditis elegans. Curr. Opin. Genet. Dev. 1999;9:479–484. doi: 10.1016/S0959-437X(99)80073-2. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigman JL, Padukiewicz KE, Sutherland ML, Rothblat LA. Executive functions in the heterozygous reeler mouse model of schizophrenia. Behav. Neurosci. 2006;120:984–988. doi: 10.1037/0735-7044.120.4.984. [DOI] [PubMed] [Google Scholar]
- Carboni G, Tueting P, Tremolizzo L, Sugaya I, Davis J, Guidotti A. Enhanced dizocilpine efficacy in heterozygous reeler mice relates to GABA turnover downregulation. Neuropharmacology. 2004;46:1070–1081. doi: 10.1016/j.neuropharm.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Chalfie M, Sulston JE, Whire JG, Southgate E, Thomson JN, Brenner S. The neural circuit for touch sensitivity in Caenorhabditis elegans. J. Neurosci. 1985;5:956–964. doi: 10.1523/JNEUROSCI.05-04-00956.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl SG. Plasma level monitoring of antipsychotic drugs. Clinical utility. Clin. Pharmacokinet. 1986;11:336–361. doi: 10.2165/00003088-198611010-00003. [DOI] [PubMed] [Google Scholar]
- Di Michele V, Ramenghi LA, Sabatino G. Clozapine and lorazepam administration in pregnancy. Eur. Psychiatry. 1996;11:214. doi: 10.1016/0924-9338(96)88396-9. [DOI] [PubMed] [Google Scholar]
- Donohoe DR, Aamodt EJ, Osborn E, Dwyer DS. Antipsychotic drugs disrupt normal development in Caenorhabditis elegans via additional mechanisms besides dopamine and serotonin receptors. Pharmacol. Res. 2006;54:361–372. doi: 10.1016/j.phrs.2006.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findling RL, Schulz SC, Reed MD, Blumer JL. The antipsychotics. A pediatric perspective. Pediatr. Clin. North Am. 1998;45:1205–1232. doi: 10.1016/s0031-3955(05)70070-5. [DOI] [PubMed] [Google Scholar]
- Hao JC, Yu TW, Fujisawa K, Culotti JG, Gengyo-Ando K, Mitani S, Moulder G, Barstead R, Tessier-Lavigne M, Bargmann CI. C. elegans slit acts in midline, dorsal-ventral, and anterior-posterior guidance via the SAX-3/Robo receptor. Neuron. 2001;32:25–38. doi: 10.1016/s0896-6273(01)00448-2. [DOI] [PubMed] [Google Scholar]
- Hatten ME. New directions in neuronal migration. Science. 2002;297:1660–1663. doi: 10.1126/science.1074572. [DOI] [PubMed] [Google Scholar]
- Heiser P, Schuler-Springorum M, Schulte E, Hausmann C, Remschmidt H, Krieg JC, Vedder H. Pharmacokinetics of clozapine and its metabolites in hippocampal HT22 cells. Eur. J. Pharmacol. 2003;476:167–172. doi: 10.1016/s0014-2999(03)02176-9. [DOI] [PubMed] [Google Scholar]
- Hekimi S, Kershaw D. Axonal guidance defects in a Caenorhabditis elegans mutant reveal cell-extrinsic determinants of neuronal morphology. J. Neurosci. 1993;13:4254–4271. doi: 10.1523/JNEUROSCI.13-10-04254.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holson RR, Webb PJ, Grafton TF, Hansen DK. Prenatal neuroleptic exposure and growth stunting in the rat: an in vivo and in vitro examination of sensitive periods and possible mechanisms. Teratology. 1994;50:125–136. doi: 10.1002/tera.1420500207. [DOI] [PubMed] [Google Scholar]
- Jann MW, Grimsley SR, Gray EC, Chang WH. Pharmacokinetics and pharmacodynamics of clozapine. Clin. Pharmacokinet. 1993;24:161–176. doi: 10.2165/00003088-199324020-00005. [DOI] [PubMed] [Google Scholar]
- Jassen AK, Yang H, Miller GM, Calder E, Madras BK. Receptor regulation of gene expression of axon guidance molecules: implications for adaptation. Mol. Pharmacol. 2006;70:71–77. doi: 10.1124/mol.105.021998. [DOI] [PubMed] [Google Scholar]
- Kindt KS, Tam T, Whiteman S, Schafer WR. Serotonin promotes G(o)-dependent neuronal migration in Caenorhabditis elegans. Curr. Biol. 2002;12:1738–1747. doi: 10.1016/s0960-9822(02)01199-5. [DOI] [PubMed] [Google Scholar]
- Klee CB. Concerted regulation of protein phosphorylation and dephosphorylation by calmodulin. Neurochem. Res. 1991;16:1059–1065. doi: 10.1007/BF00965851. [DOI] [PubMed] [Google Scholar]
- Kornhuber J, Schultz A, Wiltfang J, Meineke I, Gleiter CH, Zoechling R, Boissl K, Leblhuber F, Riederer P. Persistence of haloperidol in human brain tissue. Am. J. Psychiatry. 1999;156:885–890. doi: 10.1176/ajp.156.6.885. [DOI] [PubMed] [Google Scholar]
- Lanczik M, Knoche M, Fritze J. Psychopharmacotherapy during pregnancy and lactation. I. Pregnancy. Nervenarzt. 1998;69:1–9. doi: 10.1007/s001150050232. [DOI] [PubMed] [Google Scholar]
- Luo X, Persico AM, Lauder JM. Serotonergic regulation of somatosensory cortical development: lessons from genetic mouse models. Dev. Neurosci. 2003;25:173–183. doi: 10.1159/000072266. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Li Z, Kaneda Y, Ichikawa J. Serotonin receptors: their key role in drugs to treat schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2003;27:1159–1172. doi: 10.1016/j.pnpbp.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Meltzer HY, Matsubara S, Lee JC. The ratios of serotonin 2 and dopamine 2 affinities differentiate atypical and typical antipsychotic drugs. Psychopharmacol. Bull. 1989;25:390–392. [PubMed] [Google Scholar]
- Merz DC, Culotti JG. Genetic analysis of growth cone migration in Caenorhabditis elegans. J. Neurobiol. 2000;44:281–288. [PubMed] [Google Scholar]
- Meyer MC, Baldessarini RJ, Goff DC, Centorrino F. Clinically significant interactions of psychotropic agents with antipsychotic drugs. Drug Saf. 1996;15:333–346. doi: 10.2165/00002018-199615050-00004. [DOI] [PubMed] [Google Scholar]
- Qin H, Powell-Coffman JA. The Caenorhabditis elegans aryl hydrocarbon receptor, AHR-1, regulates neuronal development. Dev. Biol. 2004;270:44–75. doi: 10.1016/j.ydbio.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Wintle RF, Kindt KS, Nuttley WM, Arvan R, Fitzmaurice P, Bigras E, Merz DC, Herbert TE, van der Kooy D, Schafer WR, Culotti JG, Van Tol HH. Dopamine modulates the plasticity of mechanosensory responses in Caenorhabditis elegans. EMBO J. 2004;23:473–482. doi: 10.1038/sj.emboj.7600057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer GE, Klumperman J, Syed NI. Neurotransmitters and neurodevelopment. Role of dopamine in neurite outgrowth, target selection, and specific synapse formation. Perspect. Dev. Neurobiol. 1998;5:451–467. [PubMed] [Google Scholar]
- Sze JY, Victor M, Loer C, Shi Y, Ruvkun G. Food and metabolic signaling defects in a Caenorhabditis elegans serotonin-synthesis mutant. Nature. 2000;403:560–564. doi: 10.1038/35000609. [DOI] [PubMed] [Google Scholar]
- Talaska G, Ginsburg D, LaDow K, Puga A, Dalton T, Warshawsky D. Impact of Cyp1a2 or Ahr gene knockout in mice: implications for biomonitoring studies. Toxicol. Lett. 2006;162:246–249. doi: 10.1016/j.toxlet.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Toms N, Cooper J, Patchen B, Aamodt E. High coy arrays containing a sequence upstream of mec-3 alter cell migration and axonal morphology in C. elegans. BMC Dev. Biol. 2001;1:2. doi: 10.1186/1471-213X-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalik EL, Niacaris T, Wenick AS, Pau K, Avery L, Hobert O. LIM homeobox gene-dependent expression of biogenic amine receptors in restricted regions of the C. elegans nervous system. Dev. Biol. 2003;263:81–102. doi: 10.1016/s0012-1606(03)00447-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Yang JZ, Wilson FA, Ma YY. Differently lasting effects of prenatal and postnatal chronic clozapine/haloperidol on activity and memory in mouse offspring. Pharmacol. Biochem. Behav. 2006;84:468–478. doi: 10.1016/j.pbb.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Watari-Goshima N, Ogura K, Wolf FW, Goshima Y, Garriga G. C. elegans VAB-8 and UNC-73 regulate the SAX-3 receptor to direct cell and growth-cone migrations. Nat. Neurosci. 2007;10:169–176. doi: 10.1038/nn1834. [DOI] [PubMed] [Google Scholar]
- Weiss B, Prozialeck W, Cimino M, Barnette MS, Wallace TL. Pharmacological regulation of calmodulin. Ann. N. Y. Acad. Sci. 1980;356:319–345. doi: 10.1111/j.1749-6632.1980.tb29621.x. [DOI] [PubMed] [Google Scholar]
- Wen Z, Guirland C, Ming GL, Zheng JQ. A CaMKII/calcineurin switch controls the direction of Ca(2+)-dependent growth cone guidance. Neuron. 2004;43:835–846. doi: 10.1016/j.neuron.2004.08.037. [DOI] [PubMed] [Google Scholar]
- Wolf FW, Hung MS, Wightman B, Way J, Garriga G. vab-8 is a key regulator of posteriorly directed migrations in C. elegans and encodes a novel protein with kinesin motor similarity. Neuron. 1998;20:655–666. doi: 10.1016/s0896-6273(00)81006-5. [DOI] [PubMed] [Google Scholar]
- Yaeger D, Smith HG, Altshuler LL. Atypical antipsychotics in the treatment of schizophrenia during pregnancy and postpartum. Am. J. Psychiatry. 2006;163:2064–2070. doi: 10.1176/ajp.2006.163.12.2064. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Smith B, Craggs M, Kumar R. Neuroleptic drugs in breast-milk: a study of pharmacokinetics and of possible adverse effects in breast-fed infants. Psychol. Med. 1998;28:81–91. doi: 10.1017/s0033291797005965. [DOI] [PubMed] [Google Scholar]
- Zito JM, Safer DJ, dos Reis S, Gardner JF, Boles M, Lynch F. Trends in the prescribing of psychotropic medications to preschoolers. JAMA. 2000;283:1025–1030. doi: 10.1001/jama.283.8.1025. [DOI] [PubMed] [Google Scholar]