

Abstract
Peroxisome proliferators (PPs) are a diverse class of chemicals, which cause a dramatic increase in the size and number of hepatic peroxisomes in rodents and eventually lead to the development of hepatic tumors. Nuclear factor-κB (NF-κB) is a transcription factor activated by reactive oxygen and is involved in cell proliferation and apoptosis. Previously we found that the peroxisome proliferator ciprofibrate (CIP) activates NF-κB and that dietary vitamin E decreases CIP-induced NF-κB DNA binding. We therefore hypothesized that inhibition of NF-κB by vitamin E is necessary for effects of vitamin E on CIP-induced cell proliferation and the inhibition of apoptosis by CIP. Sixteen B6129 female mice (p50+/+) and twenty mice deficient in the p50 subunit of NF-κB (p50−/−) were fed a purified diet containing 10 or 250 mg/kg vitamin E (α-tocopherol acetate) for 28 days. At that time, half of the mice were placed on the same diet with 0.01% CIP for 10 days. CIP treatment increased the DNA binding activity of NF-κB and cell proliferation, but had no significant effect on apoptosis. Compared to wild-type mice, the p50−/− mice had lower NF-κB activation, higher basal levels of cell proliferation and apoptosis, and a lower ratio of reduced glutathione to oxidized glutathione (GSH/GSSG). There was approximately a 60% reduction in cell proliferation in the CIP-treated p50−/− mice fed higher vitamin E in comparison to the p50−/− mice fed lower vitamin E. Dietary vitamin E also inhibited the DNA binding activity of NF-κB, increased apoptosis, and increased the GSH/GSSG ratio. This study shows the effects of vitamin E on cell growth parameters do not appear to be solely through decreased NF-κB activation, suggesting that vitamin E is acting by other molecular mechanisms.
Keywords: vitamin E, ciprofibrate, NF-κB, cell proliferation
INTRODUCTION
Peroxisome proliferators (PPs), also known as peroxisome proliferator activated receptor-α (PPARα) agonists, are a diverse class of chemicals, which cause a dramatic increase in the size and number of hepatic peroxisomes in rodents and cause hepatic tumors upon long-term administration (Reddy and Lalwani, 1983). Hepatomegaly occurs within days of administration of PPs as a result of hypertrophy and hyperplasia. The hyperplasia peaks at one week, yet the hypertrophy continues with PP administration (Rao and Reddy, 1991). PPs are classified as nongenotoxic carcinogens and exert their tumor promoting capabilities by activating PPARα (Rao and Reddy, 1991; Desvergne and Wahli, 1999). PPARs are ligand-activated transcription factors, which modulate transcription by binding to PP response elements (PPREs) in target genes (Desvergne and Wahli, 1999). Target genes include peroxisomal and microsomal fatty-acid oxidizing enzymes (Desvergne and Wahli, 1999), and the persistent alterations of these enzymes are hypothesized to contribute to the development of hepatic tumors (Rao and Reddy, 1991). Specifically, the activity of the H2O2-producing fatty acyl-CoA oxidase (FAO) enzyme is increased 6- to 30-fold following PP-treatment with PPs (Reddy and Lalwani, 1983; Lock et al., 1989). PPs induce other enzymes, which can also produce H2O2 or superoxide anion as by-products, including urate oxidase and the cytochrome P-450 4A family (Lock et al., 1989; Corton et al., 2000). PP-treatment results in a 2-fold increase in the H2O2-detoxifying enzyme catalase and a decrease in the activity of glutathione peroxidase (Reddy et al., 1974; O'Brien et al., 2005). This imbalance is hypothesized to result in oxidative stress (Reddy and Lalwani, 1983); some studies have shown that PP treatment leads to oxidative DNA damage or lipid peroxidation but others have not (O'Brien et al., 2005). Oxidative stress can also alter signal transduction pathways and several transcription factors (Klaunig et al., 1995; Cimino et al., 1997; Lander, 1997; Palmer and Paulson, 1997; Dalton et al., 1999).
The exact mechanism by which PPs lead to hepatocarcinogenesis has not been elucidated, but could be a result of increased cell replication and/or suppressed apoptosis (Roberts et al., 2000). PPs do not directly interact with DNA, but they may amplify the consequences of mutagenic events that would otherwise be repaired (Rao and Reddy, 1991). A possible mechanism by which PPs alter hepatocyte cell growth is by the activation of the oxidative stress-sensitive transcription factor, nuclear factor κB (NF-κB), which has been shown to be antiapoptotic and to increase cell proliferation (Baichwal and Baeuerle, 1997; Chen and Li, 1998; Barkett and Gilmore, 1999; Guttridge et al., 1999; Hinz et al., 1999; Lee et al., 1999). Several studies have shown NF-κB DNA binding activity to be increased with PP-treatment in mice and rats (Li et al., 1996; Nilakantan et al., 1998; Tharappel et al., 2001). PPs increase the DNA binding activity of NF-κB starting several days after administration, and this increase has been shown to be maintained for 90 days (Li et al., 1996; Nilakantan et al., 1998; Tharappel et al., 2001).
NF-κB is sequestered in the cytoplasm in quiescent cells as a dimer bound to specific inhibitors, the IκBs. Currently, five members of the NF-κB/Rel family exist, which include NF-κB1 (p50 and its precursor p105), NF-κB2 (p52 and its precursor p100), c-Rel, Rel A (p65), and RelB. Heterodimers of p65:p50 were the first forms identified and are also the most abundant in cells (Karin and Ben-Neriah, 2000), yet NF-κB members can form other homo- or heterodimers. Translocation of NF-κB to the nucleus requires phosphorylation and proteolytic degradation of the inhibitory subunit (IκB). NF-κB has been shown to be activated in numerous cell types by various stimuli including proinflammatory cytokines, T- and B-cell mitogens, radiation, bacterial lipopolysaccharide (LPS), viruses, physical and chemical stresses, and oxidative stress (Meyer et al., 1993; Karin and Ben-Neriah, 2000). Several studies have shown antioxidants to reduce NF-κB DNA binding (Glauert, 2007). We previously showed that increased dietary α-tocopherol acetate in the diet decreased ciprofibrate- and phenobarbital-induced NF-κB activation (Calfee-Mason et al., 2002; Calfee-Mason et al., 2004). NF-κB activation and cell proliferation were reduced in mice deficient in the p50 subunit of NF-κB (p50 −/−) compared to wild-type mice (p50 +/+) after CIP-treatment (Tharappel et al., 2003). Despite the link between vitamin E and NF-κB, whether dietary vitamin E-induced changes in cell proliferation, apoptosis, and oxidative stress are mediated by NF-κB has not been investigated.
In this study, we hypothesized that increased dietary vitamin E reduces CIP-induced oxidative stress and cell proliferation and the inhibition of apoptosis by reducing the activation of NF-κB in mice. Therefore, we tested this hypothesis by feeding p50−/− and p50+/+ mice 10 or 250 mg/kg dietary α-tocopherol acetate for 28 days; then half of the mice were fed 0.01% CIP for 10 days. We found that the p50−/− mice, compared to p50+/+ mice, had decreased NF-κB activation, higher basal levels of cell proliferation and apoptosis, and increased oxidative stress parameters. CIP-treatment increased cell proliferation and the NF-κB DNA binding of the p50:p65 heterodimer, but not the DNA binding activity of the p65:p65 homodimer. Higher dietary vitamin E decreased oxidative stress parameters, decreased cell proliferation, and increased apoptosis, and did so independently from its action on NF-κB.
MATERIALS AND METHODS
Chemicals
Ciprofibrate was a gift from Sterling Drug Inc., Rensselaer, NY. The tocopherol-stripped corn oil was from Acros Organics (Morris Plains, NJ). All dry constituents of the purified diet were from Teklad Test Diets (Madison, WI). The dl-α-tocopherol acetate was from Sigma Chemical Co. (St. Louis, MO) as were all other reagents, unless specifically stated otherwise.
Experimental design
Sixteen B6129 female mice and twenty homozygous NF-κB1 (p50) deficient mice (8–10 weeks old) were obtained from our breeding colony (originally from the Jackson Laboratory, Bar Harbor, ME). The mice were housed with no more than four mice per microisolator cage in a temperature- and light-controlled room. The mice were fed a purified diet (Table 1) containing either 10 or 250 mg α-tocopherol acetate/kg diet administered ad libitum in feeding cups; the 10 and 250 ppm α-tocopherol acetate represented diets lower and higher than the recommended dietary level of vitamin E, respectively. Calfee-Mason et al. ( 2004) previously found that rats fed the 250 ppm level had significantly lower NF-κB activation than those fed the 10 ppm level; the 50 ppm level (the recommended dietary level) produced an intermediate response. The mice were fed the diet for 28 days, at which time half of the p50+/+ and half the p50−/− mice were placed on the same diet with 100 mg/kg ciprofibrate for 10 days. Three days before sacrifice, each mouse had an Alzet model # 1003D osmotic pump (Alza Corporation, Palo Alto, CA) surgically implanted s.c. containing a total volume of 100 µl of 20 mg/ml 5-bromo-2’-deoxyuridine (BrdU) with a flow rate of 1 µl/hour. Ten days after starting the CIP-treated diets, all mice were euthanized by CO2 inhalation, and the livers were immediately removed and weighed. A piece of liver was randomly sliced from three different lobes of the liver and placed in a tissue cassette (Sakura Finetek, Torrance, CA), and placed in 10% buffered formalin. Another random piece of liver was removed, weighed, and homogenized in a 1:10 dilution (w/v) of 5% 5-sulfosalicyclic acid. The liver homogenates were immediately used for glutathione analysis. The remainder of liver was quick frozen in liquid nitrogen and stored at −80°C until time of assay. All mice were weighed once a week and at the end of the study. This study was approved by the University of Kentucky Institutional Animal Care and Use Committee.
Table 1.
Composition of Purified Dietsa
| Constituent | g/kg diet |
|---|---|
| Casein (vitamin-free)b | 140 |
| Corn starchb | 465.7 |
| Dextrose monohydrateb | 255 |
| Cellulose fiberb | 50 |
| Corn-oil, tocopherol strippedc | 40 |
| AIN Mineral mixb | 35 |
| L-Cystineb | 1.8 |
| Choline bitartrateb | 2.5 |
| AIN Vitamin mix without vitamin Eb | 10 |
| dl-α-tocopherol acetated | 10 or 250 mg/kg |
similar to diet described by Reeves et al. (Reeves et al., 1993).
Teklad Test Diets, Madison, WI.
Acros Organics, Geel, Belgium.
Sigma Chemical, St. Louis, MO.
Preparation of whole liver homogenate (WLH)
Liver pieces were homogenized in cold 11.5 g/L KCl with 100 µmol/L EDTA, pH 7.4, using an Ultra-Turrax homogenizer (Tekmar Co., Cincinnati, OH). The protein concentration was determined for each WLH and normalized to 30 mg/ml. The homogenates were aliquoted and kept at −80°C until needed. The WLH was used to assay FAO activity and α-tocopherol levels.
Fatty acyl-CoA oxidase
The activity of peroxisomal fatty acyl-CoA oxidase in WLH was measured fluorometrically according to the method of Poosch and Yamazaki (1986).
α-Tocopherol concentrations
Tocopherols were extracted from WLH with hexane as described by Desai (1984). The samples were dried under a stream of nitrogen gas for 35–45 minutes and resuspended in 320 µL of 100% methanol. α-Tocopherol was measured using fluorometry (excitation at 205 nm and emission at 360 nm), as described by Buttriss and Diplock (1984) in an HPLC system consisting of Shimadzu Systems Controller SCL-10A, Shimadzu Liquid Chromatograph (pump) LC-10AS, Shimadzu Spectrofluorometric Detector RF-551, Shimadzu UV Detector SPD-10A, Shimadzu Auto Injector SIL-10A, Shimadzu Chromatopac CR501, and Column 25 cm × 4.6 mm packed with 5 µm particle size C18 material. A 100% methanol mobile phase at a flow rate of 1.0 ml/min was used. Standards of α-tocopherol (Lancaster Synthesis, Windham, NH) were used to create a standard curve. 50 µl of sample and standard were injected into the HPLC. α-Tocopherol was normalized to µg/g liver.
Quantitation of DNA synthesis and apoptosis
The liver tissues were kept in 10% buffered formalin for at least 24 hours for proper fixation. The tissues were paraffin embedded and sectioned at 5 µm and mounted on 3-aminopropyl-triethyloxysilane coated glass slides (Sigma Chemical Co, St. Louis, MO). The sections were immunohistochemically stained for BrdU according to the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA) manufacturer procedures. Cells that had incorporated BrdU were easily identified with brown nuclei. Apoptotic cells were quantified in 5 µm sections using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (Intergen Company, Purchase, NY.) as described by the manufacturer. At least 3000 hepatocyte nuclei from 3 liver lobes were counted randomly per slide to obtain labeling and apoptotic indexes. Other sections were stained with hematoxylin and eosin and used for histopathological analysis.
Isolation of nuclear extracts
Nuclear extracts were prepared from frozen liver tissue as described by Deryckere and Gannon (1994). 150 µg of tissue were homogenized with 5 ml of buffer A [6 ml/L IGEPAL CA-6430, 150 mmol/L NaCl, 10 mmol/L HEPES, pH 7.9, 1 mmol/L EDTA, 500 µmol/L PMSF] using a Dounce tissue homogenizer, and the nuclear pellet was resuspended with 100 µl of buffer C [250 ml/L glycerol, 20 mmol/L HEPES, pH 7.9, 420 mmol/L NaCl, 1.2 mmol/L MgCl2, 200 µmol/L EDTA, 500 µmol/L PMSF, 500 µmol/L DTT, 2 mmol/L Benzamidine, 5 mg/L aprotinin, 5 mg/L leupeptin, 5 mg/L pepstatin A]. The protein concentration was approximately 0.8 µg/µl.
Electrophoretic mobility shift assay (EMSA)
The NF-κB oligonucleotide (Promega Corporation, Madison, WI) was radiolabeled with [γ-32P] using T4-polynucleotide kinase (New England Biolabs, Beverly, MA). The labeled probe was purified by adding the components of each reaction to the center of a spin column (Bio-Rad P-30, Hercules, CA) and spinning the column at 3500 rpm in a microcentrifuge (1000 × g). The labeled probe was diluted with Tris-EDTA (TE) buffer (10 mmol/L Tris-HCl, pH 7.5; 1.0 mmol/L EDTA) to 0.5 ng/µl. The average cpm as determined in a liquid scintillation analyzer was 50,000 for 1 µl. For the gel retardation assay, the 20 µl reaction consisted of: 2 µg of nuclear extraction proteins, 1x binding buffer [10 mmol/L Hepes, pH 7.9, 50 mmol/L KCl, 0.2 mmol/L EDTA, 2.5 mmol/L DTT, 100 ml/L glycerol, 0.5 ml/L NP-40], 0.2 µg poly dI-dC, 1 ng NF-κB probe, and nuclease free water (up to 20 µl). The contents of the reaction were pre-incubated for 10 minutes in ice without the probe. 1 µg of the following antibodies from Santa Cruz (Santa Cruz, CA) were used to confirm the binding specificity of NF-κB: p50 (sc-114x), p65 (sc-372G), p52 (sc-298x), c-Rel (sc-70x), and Rel B (sc-226x). After the pre-incubation, the radiolabeled NF-κB probe was added to the reaction and incubated for 20 minutes at room temperature.
Western blotting
The liver tissues were diluted 1:7 with lysis buffer (1% Nonidet P-40, 0.1% SDS, 0.1 mg/ml PMSF, 2 µg/ml aprotinin, 2 µg/ml leupeptin, 2 µg/ml pepstatin A, and 1x PBS). The liver tissue was homogenized using an Ultra-Turrax homogenizer (Tekmar Co., Cincinnati, OH) for 90 seconds (with two 20-second rests). The cytosol was isolated as described by Schramm et al. (1985). All samples were brought to 6 mg/ml with the lysis buffer. The samples were denatured by boiling for 5 minutes in 2x gel loading buffer (173 ml/L glycerol, 1.25 mol/L β-mercaptoethanol, 52 g/L SDS, 220 mmol/L Tris pH 6.8, 1 mg bromophenol blue). For the identification of the IκBα protein, 20 µg of protein per sample were electrophoresed at 175 volts for 2 hours using a 9% separating and 4% stacking gel. The proteins in the gels were transferred to nitrocellulose membranes (Gibco Life Technologies, Grand Island, NY) at 100 volts for 1 hour. The membranes were blocked with 5% blocking buffer (10 mmol/L Tris pH 7.5, 150 mmol/L NaCl, 500 µl/L Tween-20, 50 g/L non-fat dry milk) for 1 hour at room temperature while shaken. The IκBα antibody (Santa Cruz) was diluted to 0.2 µg/ml in 5% milk blocking buffer with a total volume of 20 ml and incubated with the membrane for 1 hour with shaking at room temperature. The membrane was washed 3 times with wash buffer (10 mmol/L Tris pH 7.5, 150 mmol/L NaCl, 500 µl/L Tween-20) and incubated with anti-rabbit HRP-labeled secondary antibody (Santa Cruz) at a final concentration of 80 ng/ml in 20 ml total volume for 1 hour at room temperature with shaking. The membranes were washed 4 times and the Pierce SuperSignal Chemiluminescent Substrate Kit (Pierce, Rockford, IL) was used to detect the proteins.
Glutathione analysis
Fresh liver samples were homogenized and deproteinated in a 1:10 dilution (w/v) with 5% sulfosalicylic acid immediately in order to avoid oxidation of glutathione. Reduced and oxidized glutathione were analyzed as described by Ridnour (1999).
RNA isolation
Total RNA was isolated from frozen liver by homogenizing the tissue in TRIzol Reagent (Gibco Life Technologies) as described by the manufacturer. The purity of RNA was estimated by assessing the ratio of OD260/OD280.
Ribonuclease protection assay (RPA)
RPAs were carried out using the RiboQuant RNase Protection Assay Kit from Becton Dickinson Pharmingen (San Diego, CA), according to the protocol provided by the manufacturer. The RNA template used was the MCYC-1 RiboQuant ™ Mouse Cyclin Multi-Probe Template set (Becton Dickinson Pharmingen).
Protein assay
Protein concentrations of the WLH, nuclear extracts, and cytosol were determined using the bicinchoninic acid (BCA) method (Pierce Chemical Company, Rockford, IL) at 562 nm using bovine gamma globulin as the standard (Bio-Rad, Hercules, CA).
Statistical analyses
All statistical analyses were conducted using SYSTAT V.8 (SPSS, Inc.) software. Results were first analyzed by three-way analysis of variance (ANOVA). The results of all ANOVAs are shown in Table 2. If significant interactions were observed, individual differences between means were determined using Bonferroni’s or Tukey’s posthoc test. The results were reported as means ± standard error of mean (SEM). The level of significance was P = 0.05.
Table 2.
Results of Three-Way ANOVA for the Study Endpoints (P values)
| Study Endpoint | Main effect for vitamin E | Main effect for CIP | Main effect for p50 genotype | Vitamin E/CIP interaction | Vitamin E/p50 interaction | CIP/p50 interaction | Three-way interaction |
|---|---|---|---|---|---|---|---|
| Body weight | 0.70 | 0.28 | <0.001 | 0.030 | 0.15 | 0.17 | 0.57 |
| Liver weight | 0.50 | <0.001 | 0.028 | 0.25 | 0.12 | 0.56 | 0.70 |
| Liver weight/body weight | 0.92 | <0.001 | <0.001 | 0.97 | 0.47 | 0.45 | 0.95 |
| Vitamin E | <0.001 | 0.30 | 0.039 | 0.74 | 0.011 | 0.99 | 0.51 |
| FAO | 0.75 | <0.001 | 0.20 | 0.61 | 0.28 | 0.03 | 0.13 |
| NF-κB DNA binding activity (upper band) | 0.056 | 0.16 | 0.001 | 0.89 | 0.014 | 0.28 | 0.72 |
| NF-κB DNA binding activity (lower band) | 0.095 | 0.011 | <0.001 | 0.38 | 0.068 | 0.67 | 0.75 |
| GSH | 0.002 | 0.17 | 0.001 | 0.50 | 0.32 | 0.52 | 0.23 |
| GSSG | 0.17 | 0.17 | 0.27 | 0.45 | 0.49 | 0.22 | 0.22 |
| GSH/GSSG | 0.03 | 0.17 | 0.049 | 0.96 | 0.39 | 0.76 | 0.31 |
| Cell proliferation | 0.12 | 0.024 | <0.001 | 0.98 | 0.013 | 0.79 | 0.29 |
| Apoptosis | 0.070 | 0.65 | 0.003 | 0.69 | 0.34 | 0.91 | 0.60 |
| Cyclin A2 | 0.26 | 0.060 | 0.068 | 0.024 | 0.037 | 0.086 | 0.17 |
| Cyclin C | 0.077 | <0.001 | 0.11 | 0.38 | 0.99 | 0.041 | 0.95 |
| Cyclin D1 | 0.17 | 0.017 | 0.16 | 0.52 | 0.55 | 0.35 | 0.77 |
| Cyclin D2 | 0.13 | 0.86 | 0.028 | 0.91 | 0.42 | 0.11 | 0.67 |
| Cyclin D3 | 0.87 | <0.001 | 0.053 | 0.57 | 0.58 | 0.58 | 0.006 |
| Cyclin B2 | 0.003 | <0.001 | <0.001 | 0.009 | 0.98 | 0.98 | 0.039 |
RESULTS
In this study, we examined the ability of dietary vitamin E to alter cell proliferation, apoptosis, and oxidative stress with or without ciprofibrate-treatment in the absence or presence of the NF-κB p50 subunit. We previously observed that dietary vitamin E decreased CIP-mediated NF-κB activation (Calfee-Mason et al., 2004), and multiple studies have shown vitamin E can inhibit cell proliferation (Boscoboinik et al., 1991a; Boscoboinik et al., 1991b; Azzi et al., 1995; Azzi et al., 1998) and increase apoptosis (Barnett et al., 2002; Sakai et al., 2004). With this in mind, we sought to determine if vitamin E could decrease cell proliferation and increase apoptosis by decreasing CIP-induced NF-κB activation. By measuring cell proliferation and apoptosis in the presence and absence of the NF-κB p50 subunit, we could determine if vitamin E works through NF-κB to affect cell proliferation and apoptosis.
The final body weights of the p50−/− mice were less than their wild-type counterparts (Table 3). The liver weight and liver weight/body weight ratio were significantly greater in CIP-treated mice and p50−/− mice (Table 3). Two mice died during the study: a p50 +/+ mouse fed the low vitamin E diet plus ciprofibrate, and a p50 +/+ mouse fed the high vitamin E diet plus ciprofibrate. The control p50 −/− mice fed 10 mg/kg vitamin E all had fatty livers; only a low incidence was seen in the other groups (Table 4). Necrosis was mainly seen in p50−/− mice administered CIP. Mice fed the diet with 250 mg/kg α-tocopheryl acetate had increased hepatic α-tocopherol levels (Figure 1). The p50−/− mice fed the 250 mg/kg vitamin E diet had significantly less hepatic α-tocopherol in comparison to the p50+/+ mice fed 250 mg/kg α-tocopherol acetate. CIP administration did not affect hepatic α-tocopherol concentrations. Peroxisomal fatty acyl-CoA oxidase, which was measured as an indicator of peroxisome proliferation, was increased in CIP-treated mice, but there was no significant main effect produced by dietary vitamin E or genotype (data not shown). A significant interaction between CIP and genotype was observed, which indicated higher induction in p50−/− mice (P = 0.09 by Tukey’s test was observed for the difference between p50 −/− and +/+ mice administered CIP and 250 mg/kg vitamin E).
Table 3.
Body and Liver Weights
| Mouse | Dietary Vitamin E | Treatment | Mice per Group | Body weight (g) | Liver weight (g) | Liver wt/body wt (%) |
|---|---|---|---|---|---|---|
| p50+/+ | 10 mg/kg | Control | 4 | 25.87 ± 1.25 | 1.15 ± 0.06 | 4.46 ± 0.23 |
| CIP | 3 | 24.12 ± 1.13 | 1.96 ± 0.28a | 8.28 ± 1.51a | ||
| 250 mg/kg | Control | 4 | 25.32 ± 1.19 | 1.21 ± 0.06 | 4.78 ± 0.15 | |
| CIP | 3 | 26.73 ± 0.47 | 2.28 ± 0.14a | 8.53 ± 0.47a | ||
| p50−/− | 10 mg/kg | Control | 5 | 21.40 ± 0.62b | 1.39 ± 0.03b | 6.52 ± 0.16b |
| CIP | 5 | 21.73 ± 0.50b | 2.37 ± 0.16ab | 10.82 ± 0.51ab | ||
| 250 mg/kg | Control | 5 | 19.85 ± 0.29b | 1.25 ± 0.06b | 6.30 ± 0.26b | |
| CIP | 5 | 22.19 ± 0.50b | 2.36 ± 0.10ab | 10.61 ± 0.28ab |
Results are expressed as mean ± SEM.
Values are significantly different from respective controls.
Denotes statistical significance from p50+/+ mice which received the same treatment (p<0.05).
Table 4.
Histological Changes in the Liver
| Mouse Genotype | Dietary Vitamin E | Chemical Treatment | Fatty liver | Necrosis |
|---|---|---|---|---|
| p50+/+ | 10 mg/kg | Control | 0/4 | 0/4 |
| CIP | 1/3 | 1/3 | ||
| 250 mg/kg | Control | 0/4 | 1/4 | |
| CIP | 0/3 | 0/3 | ||
| p50−/− | 10 mg/kg | Control | 5/5 | 0/5 |
| CIP | 1/5 | 2/5 | ||
| 250 mg/kg | Control | 1/5 | 0/5 | |
| CIP | 1/5 | 3/5 |
Figure 1.
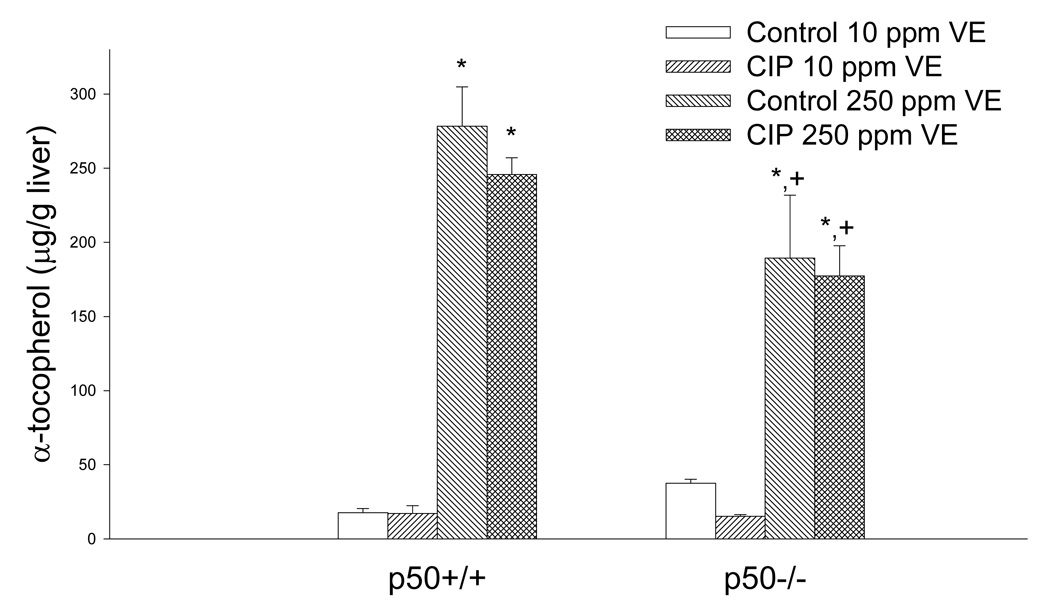
Effect of ciprofibrate (CIP), vitamin E (VE), and genotype on liver α-tocopherol levels in mice. Values represent mean ± SEM. *Values are significantly different from the group fed 10 mg/kg vitamin E receiving the same treatment. +Values are significantly different from the p50+/+ mice fed 250 mg/kg vitamin E (p<0.05).
We measured the DNA binding activity of NF-κB to verify our previous observations with ciprofibrate and vitamin E (Li et al., 1996; Calfee-Mason et al., 2002; Calfee-Mason et al., 2004). The NF-κB EMSA bands observed were comprised primarily of two moieties, which correspond to the p65:p65 homodimer and the p65:p50 heterodimer, based on supershift analysis (Figure 2). Ciprofibrate did not significantly affect the DNA binding activity of the p65:p65 homodimer (Figure 3 and Figure 4). Both vitamin E and p50−/− genotype decreased the levels of the p65:p65 homodimer, but there was a significant interaction between the two (Table 2), which indicated that there was an increase in mice fed 10 mg/kg vitamin E only in wild-type mice. For the DNA binding activity of the p50:p65 heterodimer, significant effects were observed for ciprofibrate (increased DNA binding) and genotype (decreased DNA binding in p50−/−), with no significant interactions. Vitamin E did produce a slight main effect (P = 0.095). There was also a slight interaction between vitamin E and genotype (P = 0.068), again indicating that there was a decrease in mice fed 250 mg/kg vitamin E only in wild-type mice. Since the activation and translocation of NF-κB to the nucleus requires phosphorylation and proteolytic degradation of an inhibitory subunit (IκB) (Karin and Ben-Neriah, 2000), we measured cytosolic IκB levels to determine if IκB coincided with the NF-κB data. CIP treatment qualitatively decreased cytosolic IκBα protein levels in all groups (Figure 5). Higher dietary vitamin E resulted in slightly higher IκBα protein levels in the p50+/+ mice. There was a slight qualitative decrease in the cytosolic protein levels of the NF-κB inhibitor IκBα in the p50−/− livers, which has been shown previously in p50−/− mice (DeAngelis et al., 2001).
Figure 2.
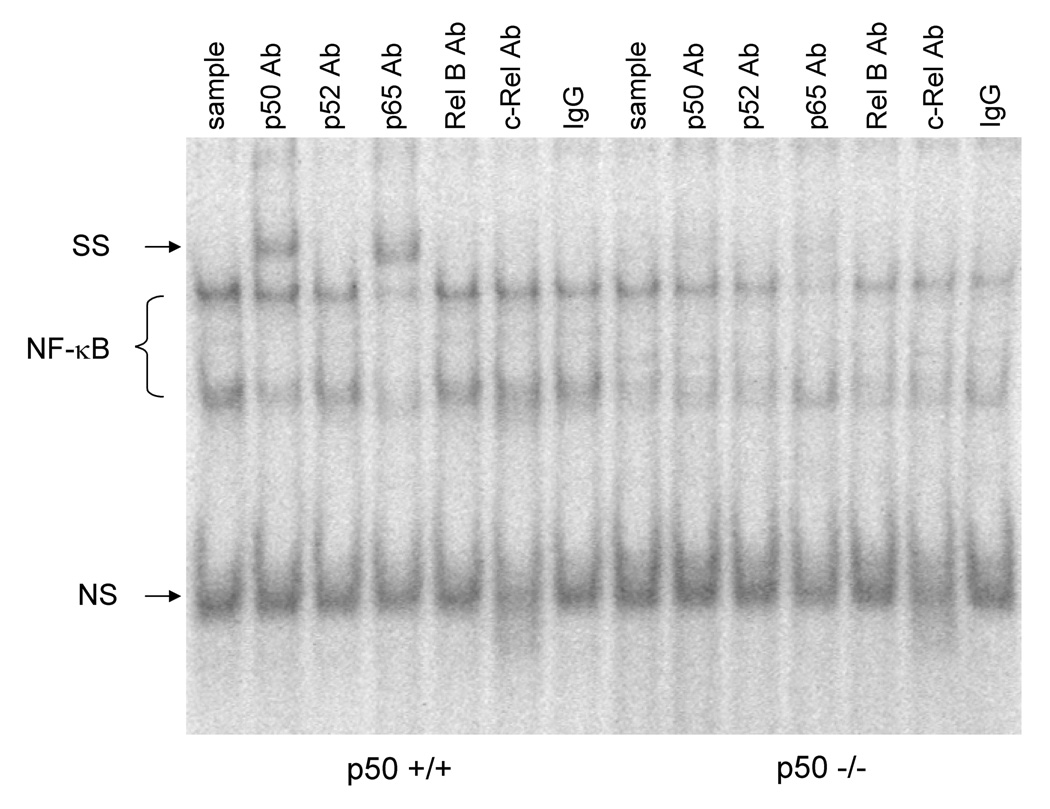
Determination of NF-κB specific binding. Antibodies specific for NF-κB subunits were added to the reaction containing a radiolabeled NF-κB oligonucleotide and liver nuclear extracts. All lanes contain 2 µg of liver nuclear extracts with the first 7 lanes from a p50 +/+ mouse fed the CIP-treated 10 mg/kg vitamin E diet and the last 7 lanes from a p50 −/− mouse fed the CIP-treated 10 mg/kg vitamin E diet. The upper NF-κB band signifies the p65:p65 moiety and the lower band is the p65:p50 moiety. SS = supershift. NS = nonspecific band. The nuclear extracts were incubated with antibodies to p50, p52, p65, RelB, c-Rel, and nonspecific preimmune serum (IgG), respectively.
Figure 3.
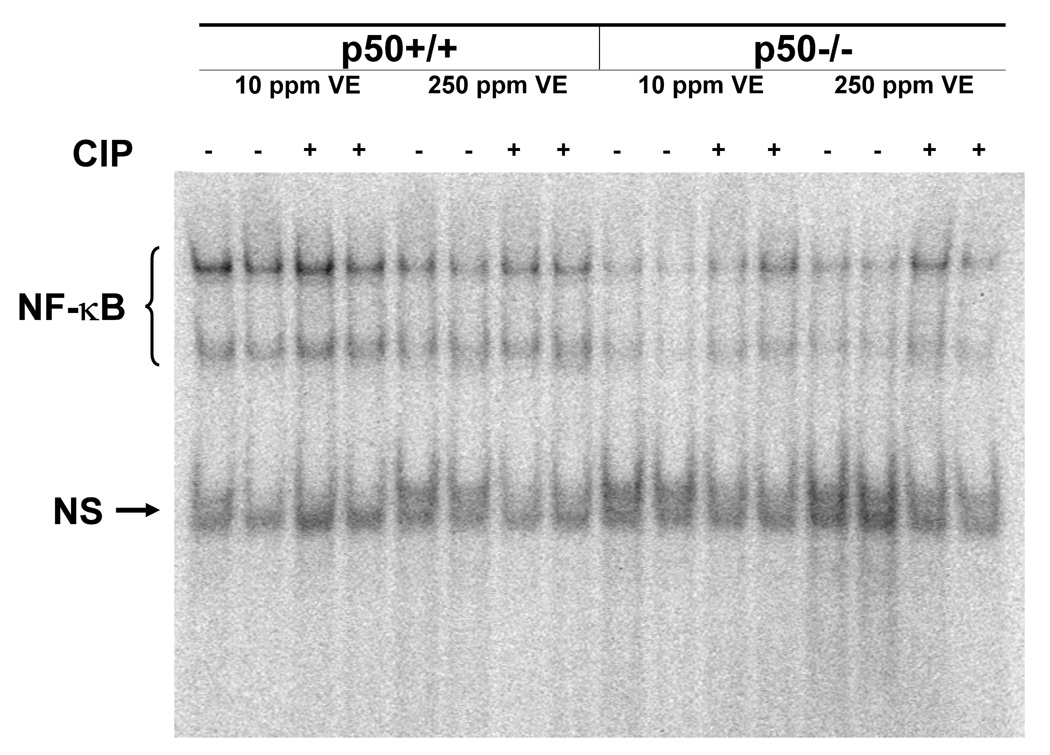
Electrophoretic mobility shift assay to detect NF-κB DNA binding in liver nuclear extracts. NS = nonspecific binding.
Figure 4.
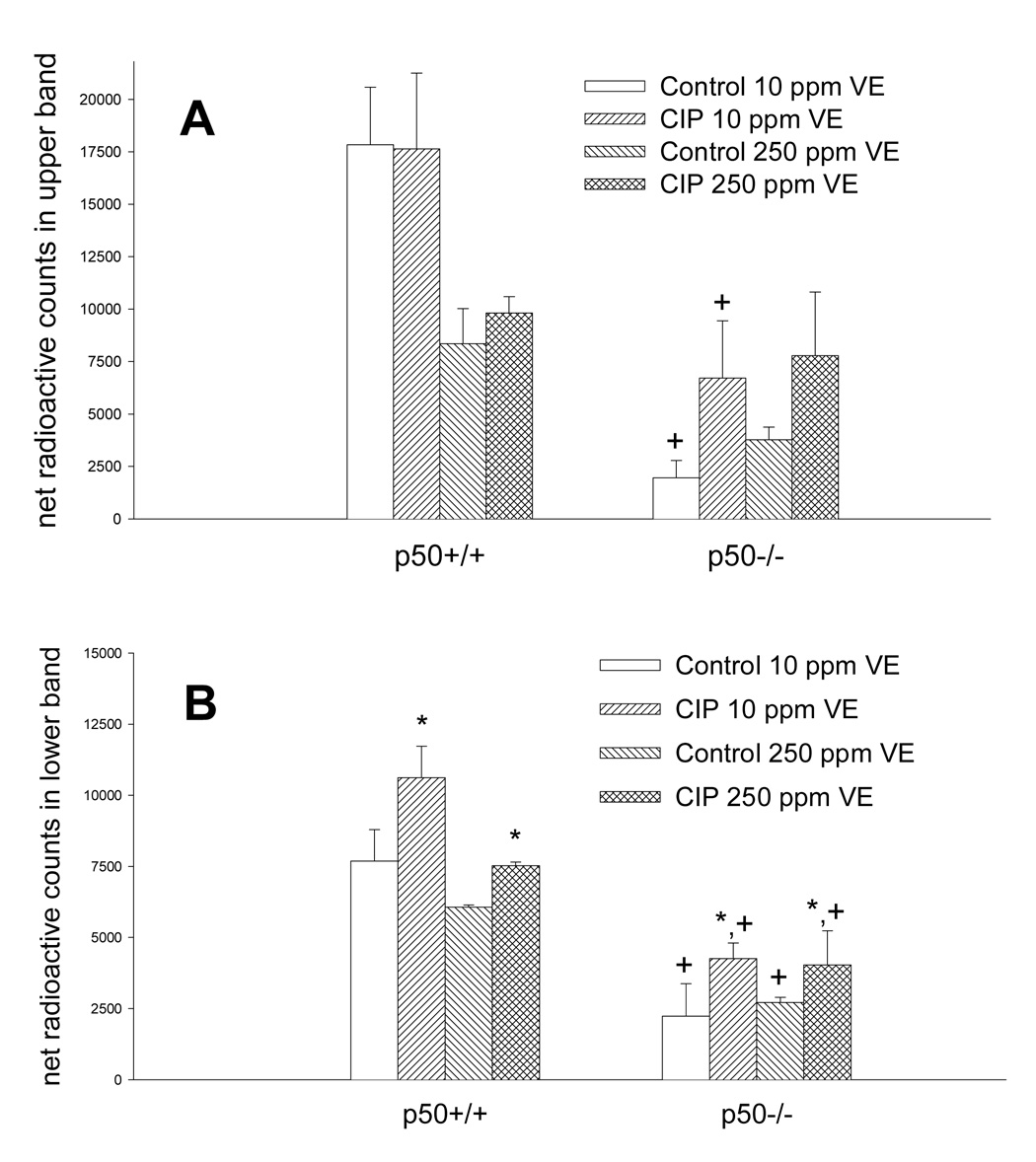
Net radioactive counts of NF-κB bands from the EMSA data (Figure 3). Quantitation of the upper NF-κB band (A) and the lower NF-κB band (B) was determined by subtracting background counts from the total counts in each NF-κB band. *Values are significantly different from respective untreated controls. +Values are significantly different from the p50+/+ group receiving the same treatment (p<0.05).
Figure 5.
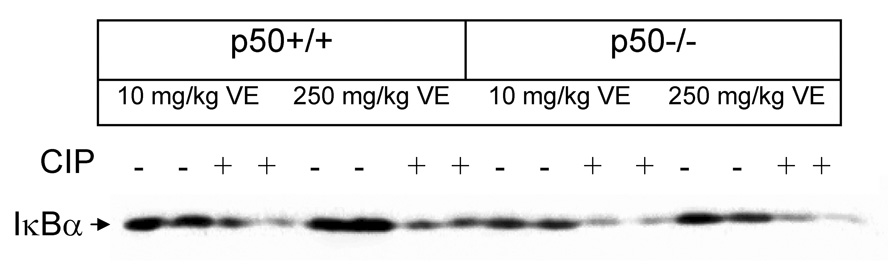
Protein levels of cytosolic IκBα as determined by Western analysis. Each lane contains 20 µg protein.
The ratio of reduced glutathione to oxidized glutathione is often used as an indicator of oxidative stress. When animals are exposed to oxidative stress, the ratio of GSH/GSSG decreases due to GSH depletion and/or GSSG accumulation. The concentration of reduced glutathione (GSH) and the ratio of reduced glutathione/oxidized glutathione (GSH/GSSG) were significantly higher in the p50+/+ mice and those fed higher dietary vitamin E (Table 5). Ciprofibrate did not have a significant effect (P = 0.17 for both endpoints). The concentration of GSSG was not significantly different among any of the groups.
Table 5.
Effects on Hepatic Glutathione Status
| Genotype | Dietary Vitamin E | Treatment | GSHa,b | GSSG | GSH/GSSGa,b |
|---|---|---|---|---|---|
| p50+/+ | 10 mg/kg | Control | 12.15 ± 0.42 | 0.84 ± 0.11 | 15.34 ± 2.28 |
| CIP | 12.11 ± 1.03 | 0.80 ± 0.08 | 15.33 ± 1.16 | ||
| 250 mg/kg | Control | 14.43 ± 0.26 | 0.69 ± 0.11 | 23.06 ± 4.13 | |
| CIP | 13.00 ± 0.42 | 0.77 ± 0.22 | 19.14 ± 4.07 | ||
| p50−/− | 10 mg/kg | Control | 11.41 ± 0.28 | 0.75 ± 0.07 | 15.63 ± 1.48 |
| CIP | 10.94 ± 0.49 | 1.35 ± 0.35 | 10.79 ± 2.66 | ||
| 250 mg/kg | Control | 12.07 ± 0.63 | 0.74 ± 0.04 | 16.51 ± 1.28 | |
| CIP | 11.99 ± 0.27 | 0.82 ± 0.07 | 15.16 ± 1.72 |
GSH and GSSG are expressed as mean (µmol/g liver) ± SEM with n = 3–5.
p50−/− mice are significantly different from wild-type mice based on ANOVA (p<0.05).
Mice fed high vitamin E are significantly different from mice fed low dietary vitamin E based on ANOVA (p<0.05).
We had hypothesized that vitamin E would decrease cell proliferation by inhibiting the activation of NF-κB. CIP-treatment and the absence of the p50 subunit increased cell proliferation in all groups, but dietary vitamin E had no significant main effect on cell proliferation (Figure 6). There was a significant interaction between vitamin E and genotype, indicating that vitamin E inhibited cell proliferation only in the p50−/− mice. Therefore it appears that the presence of the p50 subunit inhibits the ability of vitamin E to inhibit cell proliferation induced by ciprofibrate.
Figure 6.
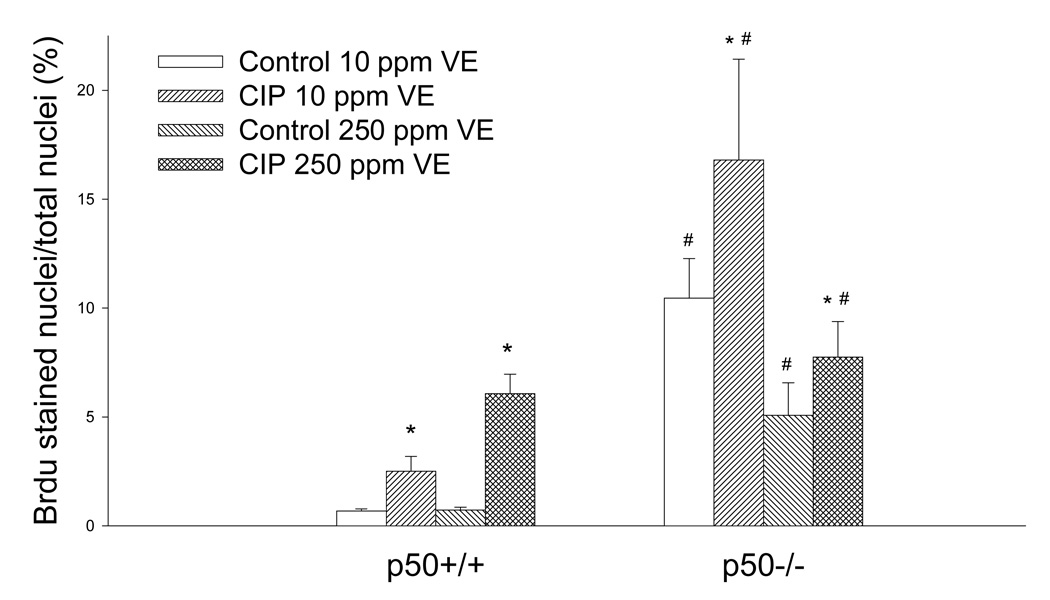
Effect of ciprofibrate (CIP), vitamin E (VE), and genotype on hepatocyte cell proliferation. The labeling index was determined by BrdU immunohistochemical staining. Values represent mean ± SEM. *Values are significantly different from their corresponding untreated controls. #Values are significantly different from the p50+/+ group that received the same treatment (p<0.05).
Although some studies have reported decreased hepatic apoptosis with PP-treatment (Bursch et al., 1984; Bayly et al., 1994; Roberts et al., 1995; Gill et al., 1998), there was no significant difference in apoptosis between control and CIP-treated mice in this study (Figure 7). Mice fed 250 mg/kg vitamin E had higher apoptosis (P = 0.070 in ANOVA) than mice fed 10 mg/kg vitamin E. Hepatic apoptosis was increased in the p50−/− mice, as has been observed previously (Tharappel et al., 2003; Lu et al., 2004). Therefore vitamin E appears to increase apoptosis independently of NF-κB, since apoptosis was increased in both p50−/− and wild-type mice fed high vitamin E diets.
Figure 7.
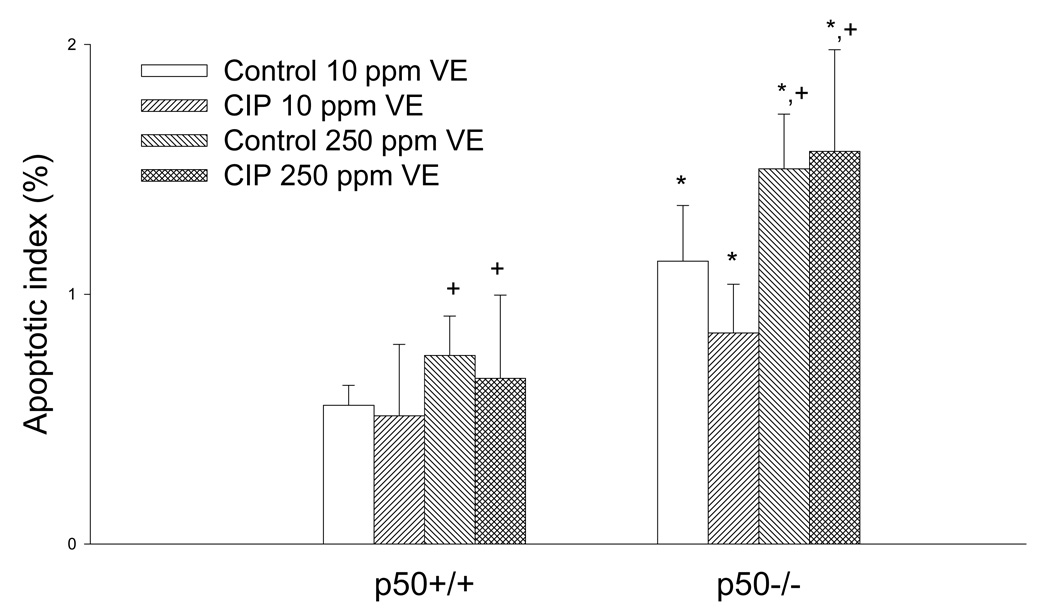
Effect of ciprofibrate (CIP), vitamin E (VE), and genotype on hepatocyte apoptotic index. Values represent mean ± SEM. *Values are significantly different from the p50+/+ mice. +Values are different from the mice fed 10 mg/kg vitamin E (p = 0.070).
Since we observed increased cell proliferation in the p50−/− mice, we measured the cyclin mRNA levels in the liver using ribonuclease protection assays (RPAs), since cyclins are regulators of the cell cycle (Table 6). All samples were normalized to the housekeeping gene, GAPDH. Based on the cell proliferation data, the p50−/− mice would be expected to have increased cyclin mRNA. Indeed, cyclins D2, D3, and B2 were increased in the p50−/− mice in comparison to the p50+/+ mice. In addition, cyclin A2 expression tended to be higher (P = 0.068), especially in mice fed 10 mg/kg of vitamin E (P = 0.037 for p50-vitamin E interaction). For vitamin E, the mRNA level of cyclin C were slightly increased in mice fed 250 mg/kg (P = 0.077), but only one difference was significant, which was an increase in cyclin B2 in the CIP-treated p50−/− mice fed 250 mg/kg vitamin E. CIP-treatment increased mRNA levels for cyclins D1, D3, and B2. However, CIP decreased cyclin C levels in all p50−/− mice, and cyclin A2 levels in p50−/− mice fed 10 mg/kg vitamin E. Thus, the increases in cell proliferation in p50−/− mice and CIP-fed mice (particularly for p50+/+) correlate with generally higher cyclin mRNA levels. For vitamin E, the inhibitory effect on cell proliferation only in the p50−/− mice is not explained by the cyclin data.
Table 6.
Effects of ciprofibrate (CIP) and dietary vitamin E on cyclin mRNA concentrations in livers from female p50+/+ and p50−/− mice
| Genotype | Dietary Vitamin E | Treatment | Cyclin A2 | Cyclin C | Cyclin D1 | Cyclin D2b | Cyclin D3 | Cyclin B2b |
|---|---|---|---|---|---|---|---|---|
| p50+/+ | 10 mg/kg | Control | 0.85 ± 0.04 | 2.24 ± 0.20 | 8.39 ± 0.84 | 2.91 ± 0.45 | 3.44 ± 0.21 | 2.58 ± 0.19 |
| CIP | 0.77 ± 0.06 | 1.93 ± 0.06 | 13.34 ± 2.11a | 3.19 ± 0.32 | 4.71 ± 0.20a | 3.10 ± 0.16 | ||
| 250 mg/kg | Control | 0.86 ± 0.01 | 2.48 ± 0.15 | 11.56 ± 2.42 | 3.36 ± 0.43 | 3.80 ± 0.15 | 2.82 ± 0.16 | |
| CIP | 0.91 ± 0.04 | 2.02 ± 0.05 | 14.50 ± 1.33a | 3.76 ± 0.37 | 4.50 ± 0.17 | 3.48 ± 0.08 | ||
| p50−/− | 10 mg/kg | Control | 1.33 ± 0.27 | 2.58 ± 0.16 | 11.94 ± 1.99 | 3.90 ± 0.34 | 4.13 ± 0.01 | 3.34 ± 0.07 |
| CIP | 0.83 ± 0.06a | 1.89 ± 0.14a | 14.22 ± 0.42a | 3.60 ± 0.13 | 4.58 ± 0.07 | 3.47 ± 0.05c | ||
| 250 mg/kg | Control | 0.86 ± 0.08 | 2.82 ± 0.17 | 13.20 ± 0.55 | 4.18 ± 0.07 | 3.68 ± 0.01 | 3.19 ± 0.13 | |
| CIP | 0.87 ± 0.06 | 1.97 ± 0.07a | 14.72 ± 0.81a | 3.65 ± 0.10 | 4.95 ± 0.09a | 4.24 ± 0.08a |
Results are expressed as the percentage of the control protein (GAPDH). Numbers are shown as the mean ± SEM with n = 2 for the controls and n = 3 for the CIP-treated. VE = vitamin E.
Values from CIP-fed mice are significantly different from respective untreated controls (p<0.05).
p50−/− values are significantly higher than p50+/+ (p<0.05).
Denotes statistical significance from the p50−/− high vitamin E group treated with CIP (p<0.05).
DISCUSSION
Reactive oxygen intermediates are proposed to be involved in the mechanism by which PPs lead to hepatic cancer. Our working hypothesis was that H2O2- and superoxide-producing enzymes are induced by PPs, thereby leading to the activation of the oxidative stress-sensitive transcription factor NF-κB. Indeed, cells overexpressing fatty acyl CoA or urate oxidase, two H2O2-producing enzymes, display signs of neoplastic transformation (Yeldandi et al., 2000). Through the activation of NF-κB, downstream target genes are induced which could result in increased cell proliferation and decreased apoptosis, both of which have been observed after PP administration in the liver. Since we have shown that dietary vitamin E can decrease NF-κB activation in vivo (Calfee-Mason et al., 2002; Calfee-Mason et al., 2004), we hypothesized that vitamin E acts on downstream events such as cell proliferation, apoptosis, and proliferation-associated gene expression through its action on NF-κB. Based on the findings from this study, the effects of vitamin E on cell growth parameters do not appear to be acting solely through decreased NF-κB activation, suggesting that vitamin E is acting by other molecular mechanisms.
Similar to what was observed in our previous studies in rats (Calfee-Mason et al., 2002; Calfee-Mason et al., 2004), higher dietary vitamin E in this study decreased NF-κB activation in CIP-treated mice, but only in wild-type mice. The basal level of NF-κB in the livers of the control wild-type mice fed 10 mg/kg vitamin E seemed high, especially since the cytosolic IκBα protein was not degraded. These B6129 mice may inherently have elevated basal NF-κB DNA binding, based on the present study and another study from our laboratory (Tharappel et al., 2003). There were two major differences between the present study and that of Calfee-Mason et al. (2004): the species and the sex of the animals. Female rats are less responsive than males to clofibrate-inducible cytochrome P-450 4A and peroxisome proliferation (Reddy and Kumar, 1979; Sundseth and Waxman, 1992), yet another study found PPs to have similar effects on peroxisomal β-oxidation and catalase activities in mice regardless of gender (Sohlenius et al., 1992). Cytochrome P450 4A2 mRNA is decreased with growth hormone, which is higher in the plasma of adult female rats (Sundseth and Waxman, 1992). This may indicate a difference between rats and mice, rather than a difference between males and females.
We observed a significant increase in both cell proliferation and apoptosis in the p50−/− mice. In the present study, basal hepatocyte proliferation was increased 10-fold in the p50−/− mice fed 10 mg/kg vitamin E and 5-fold in the p50−/− mice fed 250 mg/kg vitamin E. In previous studies in our laboratory, basal levels of hepatocyte proliferation were 4-fold higher in p50−/− mice in one study but not changed in another (Tharappel et al., 2003; Lu et al., 2004). Another study found similar cell proliferation indices in p50+/+ and p50−/− mice 5 days post-treatment with carbon tetrachloride or partial hepatectomy (2 treatments known to activate NF-κB) (DeAngelis et al., 2001). Higher vitamin E in the p50+/+ mice did not produce the expected decrease in CIP-induced cell proliferation. However, no previous studies have examined the effect of vitamin E on PP-induced cell proliferation. Therefore, the present study provides the first data examining the effect of vitamin E on PP-induced cell proliferation. It is possible that the form of vitamin E used in the study is not as effective in inhibiting cell proliferation. At least in human hepatoma cells, delta-tocopherol and vitamin E succinate have more effective anti-proliferative properties (Min et al., 2003). Increased apoptosis in the p50−/− mice has also been reported in other studies (Tharappel et al., 2003; Lu et al., 2004). In this study, we observed that the high vitamin E diet increased apoptosis (in both p50 −/− and +/+ mice), and decreased cell proliferation, but only in p50 −/− mice. Thus, the level of dietary vitamin E may play a significant role in hepatocyte proliferation and apoptosis in the p50−/− mice. Ciprofibrate increased cell proliferation, as has been observed in other studies, but surprisingly it did not affect apoptosis in either the wild-type or p50 −/− mice. Previously, ciprofibrate decreased apoptosis in p50 −/− mice (Tharappel et al., 2003). Other studies have also reported that apoptosis is inhibited by peroxisome proliferators, both in vivo and in vitro (Bursch et al., 1984; Bayly et al., 1994; Roberts et al., 1995; Gill et al., 1998).
Increased oxidative stress in the liver may be resulting from deletion of the p50 subunit as well as from the low vitamin E diet. Indeed, the ratio of GSH/GSSG, which is an indicator of oxidative stress (Gius et al., 1999), was significantly decreased in the p50−/− mice compared to the wild-type mice, regardless of dietary vitamin E. The tripeptide glutathione is present in cells primarily in its reduced form (GSH) and provides an important pool of reducing equivalents. Variations in the ratio of GSH/GSSG can affect signal transduction pathways (Gius et al., 1999). The p50−/− mice fed the 250 mg/kg vitamin E diet may have had compromised antioxidant capabilities, since the hepatic α-tocopherol levels were significantly less compared to the p50+/+ mice fed 250 mg/kg vitamin E. Indeed, changes in cellular GSH/GSSG have been shown to affect cell proliferation and apoptosis pathways likely by altering redox signaling mechanisms (Rahman et al., 2005). These mice gained comparable weight over the course of the study, so the quantity of vitamin E consumed was probably not a factor, but the lack of the NF-κB1 gene (which encodes p50) in all cells, including enterocytes, could possibly affect the absorption or transport of vitamin E.
Hepatocytes typically proliferate at a very low rate, yet increased proliferation occurs in response to physical, infectious or toxic hepatic injury (Kitamura et al., 1998). The cell cycle control system is regulated by the expression of cyclins, cyclin-dependent kinases (CDKs) and cyclin-dependent kinase inhibitors (CKIs). Nine subtypes of cyclins and eight subtypes of CDKs have been identified (Kitamura et al., 1998). The CDKs 2, 4, and 5 in conjunction with cyclins C, D, and E are associated with the passage through the G1 phase, while CDK 1 and CDK 2 and cyclins A and B are of importance to the S and M phases of the cell cycle (Sherr and Roberts, 1995; King and Cidlowski, 1998; Johnson and Walker, 1999). During liver regeneration after a partial hepatectomy, cyclin D1 mRNA is induced up to 20-fold. Changes in the cyclins/CDK complexes and the CKIs are thought to result in the loss of regulation of hepatocyte proliferation, which may account for tumor formation (Kitamura et al., 1998). The peroxisome proliferator, nafenopin, increased the protein levels of CDK2, CDK4, cyclin D1, and cyclin E in primary hepatocytes (Chevalier et al., 1999). Male rats treated with Wy-14,463 for 4 days had increased protein levels of cyclins B, D2, and D3 compared to controls (Rininger et al., 1996). Another study observed an increase in the protein levels of cyclin D1, A, and B within 48 hours after one dose of Wy-14,463 (Rininger et al., 1997). No significant differences were noted for cyclins D2 and D3 in that study. In our study, we saw an increase in cyclin D1 mRNA with CIP-treatment, and cyclins B2 and D3 were somewhat increased with CIP-treatment. The differences in the expression of cyclins among studies may result from the length of treatment, of which our study was 10 days of ongoing dietary CIP-treatment. The p50−/− mice appeared to have increased cyclin mRNA levels for multiple cyclins, which correlates with our BrdU proliferation data, and CIP-treatment resulted in increased expression of multiple cyclins, which also was in agreement with our BrdU proliferation data. Dietary vitamin E did not significantly alter cyclin mRNA levels among any groups except cyclin B2. The inhibition of cell proliferation in the p50 −/− mice by vitamin E does not correlate with the cyclin data; the reason for this is not known. NF-κB has been shown to increase cell replication by transcriptionally increasing cyclin D1 (Guttridge et al., 1999; Hinz et al., 1999), but based on our data, NF-κB activation did not coincide with the cyclin D1 mRNA data.
We have hypothesized that the antioxidant vitamin E can alleviate CIP-induced oxidative stress, thus leading to decreased NF-κB activation. Indeed, increased dietary vitamin E decreased NF-κB activation in rats (Calfee-Mason et al., 2002; Calfee-Mason et al., 2004). In this study we hypothesized that vitamin E acts through NF-κB to decrease cell proliferation and prevent the inhibition of apoptosis. If vitamin E is acting by inhibiting NF-κB activation, then vitamin E should have little or no effect on cell cycle, DNA synthesis, or apoptosis in the absence of the p50 subunit. However, there was approximately a 60% reduction in cell proliferation in the p50−/− mice fed 250 mg/kg vitamin E in comparison to the p50−/− mice fed 10 mg/kg vitamin E, whereas vitamin E did not affect cell proliferation in the wild-type mice. Higher dietary vitamin E also increased hepatic apoptosis regardless of genotype and CIP-treatment. Therefore vitamin E’s effect on cell proliferation and apoptosis appears to be independent of the p50 NF-κB subunit. This may suggest that other transcription factors are involved in the CIP-induced cell proliferation, and vitamin E is influencing their activation. Furthermore, the loss of p50 may result in an overexpression of other genes, which could lead to increased oxidative stress and hepatic cell proliferation. In fact, some long-term studies with tumor promoters in conjunction with increased dietary vitamin E have observed increased tumors and hepatic focal lesions (Glauert et al., 1990; Kolaja and Klaunig, 1997). More studies need to be performed on the effects of vitamin E on cell proliferation and apoptosis and the effects of long-term supplementation with vitamin E in order to establish if increased dietary vitamin E is beneficial in not only decreasing NF-κB activation, but also if vitamin E can be used for cancer prevention. From this study we found p50−/− mice to have increased oxidative stress, cell proliferation, and apoptosis, whereas dietary α-tocopherol increased apoptosis and decreased oxidative stress and cell proliferation, even in the absence of the p50 NF-κB subunit. Therefore, vitamin E may not be decreasing cell proliferation and increasing apoptosis solely through its action on NF-κB.
ACKNOWLEDGEMENTS
The authors thank Job Tharappel, Zijing Lu, and Michelle Twaroski for their assistance with the animal study. In addition, we thank Larry Robertson for the use of his HPLC. This work was supported by National Institutes of Health grant ES-11480 and by the Kentucky Agricultural Experimental Station. K.C.M. was supported by a National Institutes of Health training grant (CA-09509).
Abbreviations
- ANOVA
analysis of variance
- BrdU
bromodeoxyuridine
- CIP
ciprofibrate
- EMSA
electrophoretic mobility shift assay
- FAO
fatty acyl CoA oxidase
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- H2O2
hydrogen peroxide
- IκB
inhibitory κB
- IKK
IκB kinase
- NF-κB
nuclear factor κB
- p50 −/−
deficient in the p50 subunit of NF-κB
- p50 +/+
wild-type
- PPAR
peroxisome proliferator activated receptor
- PPREs
peroxisome proliferator response elements
- PPs
peroxisome proliferators
- RXR
retinoid X receptor
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
Footnotes
Conflict of Interest Statement There were no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Azzi A, Aratri E, Boscoboinik D, Clement S, Ozer NK, Ricciarelli R, Spycher S. Molecular basis of alpha-tocopherol control of smooth muscle cell proliferation. Biofactors. 1998;7:3–14. doi: 10.1002/biof.5520070102. [DOI] [PubMed] [Google Scholar]
- Azzi A, Boscoboinik D, Marilley D, Ozer NK, Stauble B, Tasinato A. Vitamin E: a sensor and an information transducer of the cell oxidation state. Am. J. Clin. Nutr. 1995;62:1337S–1346S. doi: 10.1093/ajcn/62.6.1337S. [DOI] [PubMed] [Google Scholar]
- Baichwal VR, Baeuerle PA. Apoptosis: Activate NF-kappa B or die? Curr. Biol. 1997;7:R94–R96. doi: 10.1016/s0960-9822(06)00046-7. [DOI] [PubMed] [Google Scholar]
- Barkett M, Gilmore TD. Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6910–6924. doi: 10.1038/sj.onc.1203238. [DOI] [PubMed] [Google Scholar]
- Barnett K, Fokum F, Malafa M. Vitamin E succinate inhibits colon cancer liver metastases. J. Surg. Res. 2002;106:292–298. doi: 10.1006/jsre.2002.6466. [DOI] [PubMed] [Google Scholar]
- Bayly AC, Roberts RA, Dive C. Suppression of liver cell apoptosis in vitro by the non-genotoxic hepatocarcinogen and peroxisome proliferator nafenopin. J. Cell Biol. 1994;125:197–203. doi: 10.1083/jcb.125.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscoboinik D, Szewczyk A, Azzi A. Alpha-tocopherol (vitamin E) regulates vascular smooth muscle cell proliferation and protein kinase C activity. Arch. Biochem. Biophys. 1991a;286:264–269. doi: 10.1016/0003-9861(91)90039-l. [DOI] [PubMed] [Google Scholar]
- Boscoboinik D, Szewczyk A, Hensey C, Azzi A. Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C. J. Biol. Chem. 1991b;266:6188–6194. [PubMed] [Google Scholar]
- Bursch W, Lauer B, Timmermann-Trosiener I, Barthel G, Schuppler J, Schulte-Hermann R. Controlled death (apoptosis) of normal and putative preneoplastic cells in rat liver following withdrawal of tumor promoters. Carcinogenesis. 1984;5:453–458. doi: 10.1093/carcin/5.4.453. [DOI] [PubMed] [Google Scholar]
- Buttriss JL, Diplock AT. High-performance liquid chromatography methods for vitamin E in tissues. Methods Enzymol. 1984;105:131–138. doi: 10.1016/s0076-6879(84)05018-7. [DOI] [PubMed] [Google Scholar]
- Calfee-Mason KG, Spear BT, Glauert HP. Vitamin E inhibits hepatic NF-kappa B activation in rats administered the hepatic tumor promoter, phenobarbital. J. Nutr. 2002;132:3178–3185. doi: 10.1093/jn/131.10.3178. [DOI] [PubMed] [Google Scholar]
- Calfee-Mason KG, Spear BT, Glauert HP. Effects of vitamin E on the NF-κB pathway in rats treated with the peroxisome proliferator, ciprofibrate. Toxicol. Appl. Pharmacol. 2004;199:1–9. doi: 10.1016/j.taap.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Chen E, Li CC. Association of Cdk2/cyclin E and NF-kappa B complexes at G1/S phase. Biochem. Biophys. Res. Commun. 1998;249:728–734. doi: 10.1006/bbrc.1998.9224. [DOI] [PubMed] [Google Scholar]
- Chevalier S, Macdonald N, Roberts RA. Induction of DNA replication by peroxisome proliferators is independent of both tumour necrosis factor α priming and EGF-receptor tyrosine kinase activity. J. Cell Sci. 1999;112:4785–4791. doi: 10.1242/jcs.112.24.4785. [DOI] [PubMed] [Google Scholar]
- Cimino F, Esposito F, Ammendola R, Russo T. Gene regulation by reactive oxygen species. Curr. Top. Cell. Regul. 1997;35:123–148. doi: 10.1016/s0070-2137(97)80005-2. [DOI] [PubMed] [Google Scholar]
- Corton JC, Lapinskas PJ, Gonzalez FJ. Central role of PPARalpha in the mechanism of action of hepatocarcinogenic peroxisome proliferators. Mutat. Res. 2000;448:139–151. doi: 10.1016/s0027-5107(99)00232-8. [DOI] [PubMed] [Google Scholar]
- Dalton TP, Shertzer HG, Puga A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharmacol. Toxicol. 1999;39:67–101. doi: 10.1146/annurev.pharmtox.39.1.67. [DOI] [PubMed] [Google Scholar]
- DeAngelis RA, Kovalovich K, Cressman DE, Taub R. Normal liver regeneration in p50/nuclear factor kappaB1 knockout mice. Hepatology. 2001;33:915–924. doi: 10.1053/jhep.2001.23192. [DOI] [PubMed] [Google Scholar]
- Deryckere F, Gannon F. A one-hour minipreparation technique for extraction of DNA-binding proteins from animal tissues. Biotechniques. 1994;16:405. [PubMed] [Google Scholar]
- Desai ID. Vitamin E analysis methods for animal tissues. Methods Enzymol. 1984;105:138–147. doi: 10.1016/s0076-6879(84)05019-9. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Gill JH, James NH, Roberts RA, Dive C. The non-genotoxic hepatocarcinogen nafenopin suppresses rodent hepatocyte apoptosis induced by TGFβ1, DNA damage and Fas. Carcinogenesis. 1998;19:299–304. doi: 10.1093/carcin/19.2.299. [DOI] [PubMed] [Google Scholar]
- Gius D, Botero A, Shah S, Curry HA. Intracellular oxidation/reduction status in the regulation of transcription factors NF-kappaB and AP-1. Toxicol. Lett. 1999;106:93–106. doi: 10.1016/s0378-4274(99)00024-7. [DOI] [PubMed] [Google Scholar]
- Glauert HP. Modification of the activation of NF-κB by vitamin E. In: Preedy VR, Watson RR, editors. The Encyclopedia of Vitamin E. Oxfordshire: CABI Publishing; 2007. pp. 353–364. [Google Scholar]
- Glauert HP, Beaty MM, Clark TD, Greenwell WS, Tatum V, Chen LC, Borges T, Clark TL, Srinivasan SR, Chow CK. Effect of dietary vitamin E on the development of altered hepatic foci and hepatic tumors induced by the peroxisome proliferator ciprofibrate. J. Cancer Res. Clin. Oncol. 1990;116:351–356. doi: 10.1007/BF01612917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-κB function in growth control: Regulation of cyclin D1 expression and G0/G1-to-S-Phase transition. Mol. Cell. Biol. 1999;19:2690–2698. doi: 10.1128/mcb.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- King KL, Cidlowski JA. Cell cycle regulation and apoptosis. Annu. Rev. Physiol. 1998;60:601–617. doi: 10.1146/annurev.physiol.60.1.601. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Watanabe S, Sato N. Liver regeneration, liver cancers and cyclins. J. Gastroenterol. Hepatol. 1998;13 Suppl:S96–S99. doi: 10.1111/jgh.1998.13.s1.96. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Xu Y, Bachowski S, Ketcham CA, Isenberg JS, Kolaja KL, Baker TK, Walborg EF, Jr, Stevenson DE. Oxidative stress in nongenotoxic carcinogenesis. Toxicol. Lett. 1995;82–83:683–691. doi: 10.1016/0378-4274(95)03514-1. [DOI] [PubMed] [Google Scholar]
- Kolaja KL, Klaunig JE. Vitamin E modulation of hepatic focal lesion growth in mice. Toxicol. Appl. Pharmacol. 1997;143:380–387. doi: 10.1006/taap.1996.8089. [DOI] [PubMed] [Google Scholar]
- Lander HM. An essential role for free radicals and derived species in signal transduction. FASEB J. 1997;11:118–124. [PubMed] [Google Scholar]
- Lee HH, Dadgostar H, Cheng Q, Shu J, Cheng G. NF-κB-mediated up-regulatin of Bcl-x and Bfl-1/A1 is required for CD40 survival signaling in B lymphocytes. Proc. Natl. Acad. Sci. USA. 1999;96:9136–9141. doi: 10.1073/pnas.96.16.9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Leung LK, Glauert HP, Spear BT. Treatment of rats with the peroxisome proliferator ciprofibrate results in increased liver NF-kappaB activity. Carcinogenesis. 1996;17:2305–2309. doi: 10.1093/carcin/17.11.2305. [DOI] [PubMed] [Google Scholar]
- Lock EA, Mitchell AM, Elcombe CR. Biochemical mechanisms of induction of hepatic peroxisome proliferation. Annu. Rev. Pharmacol. Toxicol. 1989;29:145–163. doi: 10.1146/annurev.pa.29.040189.001045. [DOI] [PubMed] [Google Scholar]
- Lu Z, Lee EY, Robertson LW, Glauert HP, Spear BT. Effect of 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB-153) on hepatocyte proliferation and apoptosis in mice deficient in the p50 subunit of the transcription factor NF-κB. Toxicol Sci. 2004;81:35–42. doi: 10.1093/toxsci/kfh193. [DOI] [PubMed] [Google Scholar]
- Meyer M, Schreck R, Baeuerle PA. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Guo J, Zhao F, Cai D. Effect of different vitamin E homologous analogues on human hepatoma cell HepG2 proliferation in vitro. Wei Sheng Yan Jiu. 2003;32:40–43. [PubMed] [Google Scholar]
- Nilakantan V, Spear BT, Glauert HP. Liver-specific catalase expression in transgenic mice inhibits NF- kappaB activation and DNA synthesis induced by the peroxisome proliferator ciprofibrate. Carcinogenesis. 1998;19:631–637. doi: 10.1093/carcin/19.4.631. [DOI] [PubMed] [Google Scholar]
- O'Brien ML, Spear BT, Glauert HP. Role of oxidative stress in peroxisome proliferator-mediated carcinogenesis. Crit. Rev. Toxicol. 2005;35:61–88. doi: 10.1080/10408440590905957. [DOI] [PubMed] [Google Scholar]
- Palmer HJ, Paulson KE. Reactive oxygen species and antioxidants in signal transduction and gene expression. Nutr. Rev. 1997;55:353–361. doi: 10.1111/j.1753-4887.1997.tb01561.x. [DOI] [PubMed] [Google Scholar]
- Poosch MS, Yamazaki RK. Determination of peroxisomal fatty acyl-CoA oxidase activity using a lauroyl-CoA-based fluorometric assay. Biochim. Biophys. Acta. 1986;884:585–593. doi: 10.1016/0304-4165(86)90211-4. [DOI] [PubMed] [Google Scholar]
- Rahman I, Biswas SK, Jimenez LA, Torres M, Forman HJ. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid Redox Signal. 2005;7:42–59. doi: 10.1089/ars.2005.7.42. [DOI] [PubMed] [Google Scholar]
- Rao MS, Reddy JK. An overview of peroxisome proliferator-induced hepatocarcinogenesis. Environ. Health Perspect. 1991;93:205–209. doi: 10.1289/ehp.9193205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy JK, Azarnoff DL, Svoboda DJ, Prasad JD. Nafenopin-induced hepatic microbody (peroxisome) proliferation and catalase synthesis in rats and mice. Absence of sex difference in response. J. Cell Biol. 1974;61:344–358. doi: 10.1083/jcb.61.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy JK, Kumar NS. Stimulation of catalase synthesis and increase of carnitine acetyltransferase activity in the liver of intact female rats fed clofibrate. J. Biochem. (Tokyo) 1979;85:847–856. [PubMed] [Google Scholar]
- Reddy JK, Lalwani ND. Carcinogenesis by hepatic peroxisome proliferators: evaluation of the risk of hypolipidemic drugs and industrial plasticizers to humans. Crit. Rev. Toxicol. 1983;12:1–58. doi: 10.3109/10408448309029317. [DOI] [PubMed] [Google Scholar]
- Reeves PG, Nielsen FH, Fahey GC. AIN-93 purified diets for laboratory rodents: final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A diet. J. Nutr. 1993;123:1939–1951. doi: 10.1093/jn/123.11.1939. [DOI] [PubMed] [Google Scholar]
- Ridnour LA, Winters RA, Ercal N, Spitz DR. Measurement of glutathione, glutathione disulfide, and other thiols in mammalian cell and tissue homogenates using high-performance liquid chromatography separation of N-(1-pyrenyl)maleimide derivatives. Methods Enzymol. 1999;299:258–267. doi: 10.1016/s0076-6879(99)99025-0. [DOI] [PubMed] [Google Scholar]
- Rininger JA, Goldsworthy TL, Babish JG. Time course comparison of cell-cycle protein expression following partial hepatectomy and WY14,643-induced hepatic cell proliferation in F344 rats. Carcinogenesis. 1997;18:935–941. doi: 10.1093/carcin/18.5.935. [DOI] [PubMed] [Google Scholar]
- Rininger JA, Wheelock GD, Ma X, Babish JG. Discordant expression of the cyclin-dependent kinases and cyclins in rat liver following acute administration of the hepatocarcinogen [4- chloro-6-(2,3-xylidino)-2-pyrimidinylthio] acetic acid (WY14,643) Biochem. Pharmacol. 1996;52:1749–1755. doi: 10.1016/s0006-2952(96)00596-5. [DOI] [PubMed] [Google Scholar]
- Roberts RA, James NH, Hasmall SC, Holden PR, Lambe K, Macdonald N, West D, Woodyatt NJ, Whitcome D. Apoptosis and proliferation in nongenotoxic carcinogenesis: species differences and role of PPARalpha. Toxicol. Lett. 2000;112–113:49–57. doi: 10.1016/s0378-4274(99)00243-x. [DOI] [PubMed] [Google Scholar]
- Roberts RA, Soames AR, Gill JH, James NH, Wheeldon EB. Non-genotoxic hepatocarcinogens stimulate DNA synthesis and their withdrawal induces apoptosis, but in different hepatocyte populations. Carcinogenesis. 1995;16:1693–1698. doi: 10.1093/carcin/16.8.1693. [DOI] [PubMed] [Google Scholar]
- Sakai M, Okabe M, Yamasaki M, Tachibana H, Yamada K. Induction of apoptosis by tocotrienol in rat hepatoma dRLh-84 cells. Anticancer Research. 2004;24:1683–1688. [PubMed] [Google Scholar]
- Schramm H, Robertson LW, Oesch F. Differential regulation of hepatic glutathione transferase and glutathione peroxidase activities in the rat. Biochemical Pharmacology. 1985;34:3735–3739. doi: 10.1016/0006-2952(85)90239-4. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- Sohlenius AK, Andersson K, DePierre JW. The effects of perfluoro-octanoic acid on hepatic peroxisome proliferation and related parameters show no sex-related differences in mice. Biochem. J. 1992;285:779–783. doi: 10.1042/bj2850779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundseth SS, Waxman DJ. Sex-dependent expression and clofibrate inducibility of cytochrome P450 4A fatty acid omega-hydroxylases. Male specificity of liver and kidney CYP4A2 mRNA and tissue-specific regulation by growth hormone and testosterone. J. Biol. Chem. 1992;267:3915–3921. [PubMed] [Google Scholar]
- Tharappel JC, Cunningham ML, Spear BT, Glauert HP. Differential activation of hepatic NF-kappaB in rats and hamsters by the peroxisome proliferators Wy-14,643, gemfibrozil, and dibutyl phthalate. Toxicol. Sci. 2001;62:20–27. doi: 10.1093/toxsci/62.1.20. [DOI] [PubMed] [Google Scholar]
- Tharappel JC, Nalca A, Owens AB, Ghabrial L, Konz EC, Glauert HP, Spear BT. Cell proliferation and apoptosis are altered in mice deficient in the NF-κB p50 subunit after treatment with the peroxisome proliferator ciprofibrate. Toxicol Sci. 2003;75:300–308. doi: 10.1093/toxsci/kfg201. [DOI] [PubMed] [Google Scholar]
- Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat. Res. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]