

History and Physical Examination
A 4-year-old girl presented with progressive muscle wasting and bilateral pain of the lower extremities increasing in severity during a period of one year. She had a waddling gait, tendency to fatigue easily, and restless legs at night. Physical examination findings included loss of strength in her hips and knees and apparent muscle wasting. The patient had a valgus deformity of both knees with no other abnormalities on the physical examination. The girl’s birth was uncomplicated and she developed well during her early childhood years. Her basic laboratory and endocrinologic or metabolic laboratory tests were normal. There were no indications of any mental deficits. The patient’s mother experienced similar symptoms in childhood that progressed into adulthood. Her mother also had a delay in puberty and development of secondary gender characteristics. The patient’s two maternal uncles had similar symptoms with development of blindness in one maternal uncle and deafness in the other maternal uncle. The maternal grandmother was asymptomatic but had similar radiographic characteristics as the other family members.
Plain radiographs of the patient’s lower extremity were obtained. Laboratory studies including erythrocyte sedimentation rate, calcium, phosphorus, alkaline phosphatase, and parathyroid hormone were within normal limits.
Imaging Interpretation
A plain radiograph of the patient’s lower extremity revealed widening and increased density of the shafts of the femurs, tibias, and fibulas (Fig. 1). A later skeletal image confirmed her spine and skull were normal, and appropriate maturation of bone growth was observed (Fig. 2). No other abnormalities were found radiographically. The findings of the radiograph of the lower extremity were similar to those seen on a radiograph of the patient’s mother (Fig. 3).
Fig. 1.
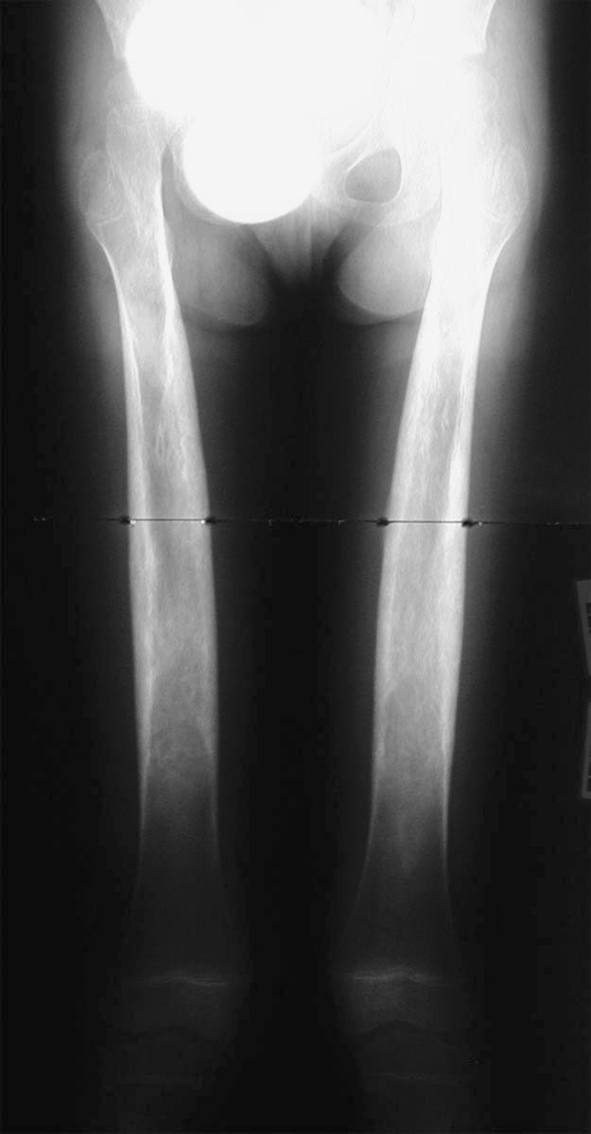
An anteroposterior radiograph of the patient at 4 years of age shows evident bilateral widening and increased density of the femurs.
Fig. 2.
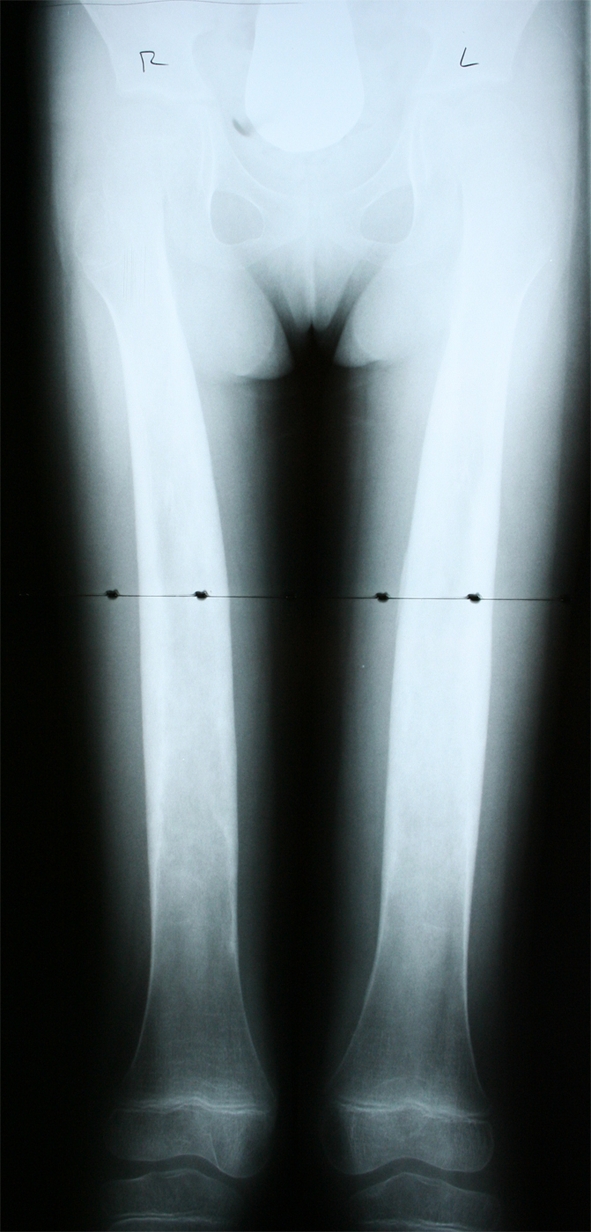
A current followup radiograph of the patient at 8 years of age shows continued widening and increased density of the femurs.
Fig. 3.
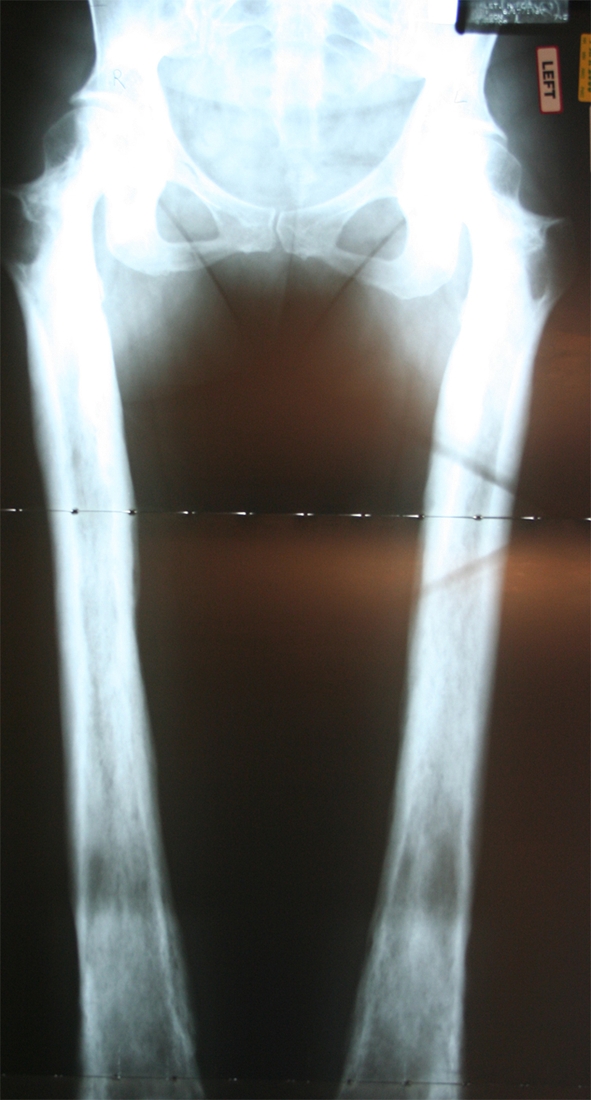
A current anteroposterior radiograph of the patient’s mother shows similar bilateral widening and increased density of the femurs.
Based on the history, physical examination, laboratory tests, and radiographic studies, what is the differential diagnosis?
Differential Diagnosis
Lenz-Majewski dysplasia
Camurati-Engelmann disease
Kenny-Caffey dysplasia
Juvenile Paget’s disease
Van Buchem dysplasia
Ribbing’s disease
Based on the history, physical examination, laboratory tests, and radiographic studies, what is the diagnosis and how should this patient be treated?
Diagnosis
Camurati-Engelmann disease
Discussion and Treatment
Camurati-Engelmann disease (CED) is a rare genetic disorder that affects the diaphysis of the long bones, skull, clavicle, or in rare cases, the facial bones. The reported incidence of the disorder (also known as progressive diaphyseal dysplasia or Engelmann’s disease) is one in 1,000,000 [9]. In 1920, Cockayne first described the disease and in 1922 Camurati proposed the genetic inheritance pattern as autosomal dominant when he treated a father and son with traditional symptoms [11]. In 1929, Engelmann reported the progressive character of the disease when he treated an 8-year-old boy, and since then there have been 199 published cases [11].
The differential diagnosis of CED is important to discuss because CED is clinically similar to other disorders. However, few disorders have clinical and radiographic findings specifically resembling those of CED [11]. Lenz-Majewski dysplasia is similar in the skeletal involvement, but it has additional congenital anomalies not found in patients with CED. These anomalies include delayed closure of the fontanel, choanal atresia, mental retardation, proximal symphalangism, joint laxity, prominent cutaneous veins, and severe growth retardation [11]. Kenny-Caffey dysplasia is characterized by dwarfism, delayed fontanel closure, craniofacial anomalies, hypocalcemia, and hypoparathyroidism [11]. As reported, CED does not have any specific laboratory abnormalities, which makes the distinction here straightforward [11]. Juvenile Paget’s disease is characterized by a predisposition to fractures, elevated alkaline phosphatase, and coarse trabeculations observed on radiographs [11]. Although patients with CED can have slightly elevated alkaline phosphatase, it is not consistent and there seems to be a resistance to fractures in these patients [11]. Van Buchem dysplasia has similar cranial features to CED; however, patients with this dysplasia do not show periosteal deposition and it is autosomal recessive rather than dominant [11]. Ribbing’s disease is a craniotubular dysplasia that is radiographically similar to CED but has milder neuromuscular symptoms, and cranial nerve deficit or other facial abnormalities are not present [9]. The differential diagnosis illustrates the complexity of diagnosing this disease; however, with the proper laboratory and radiographic testing to confirm CED, there seems to be an avenue to a straightforward diagnosis.
Camurati-Engelmann disease has vast variability in expression, making it difficult to diagnose. The variability shows up in the clinical and radiographic features of the disease [7]. The disease affects the diaphyseal of the long bones but in rare cases can affect the metaphyses and the epiphyses [7]. The reported bones of involvement in decreasing order are the femur, tibia, fibula, humerus, ulna, and radius [9]. The progression also may include the skull, facial bones, vertebrae, pelvis, and distal extremities [3]. The disease is characterized by bone growth widening of the periosteal and endosteal surfaces [12]. Symmetric bone growth is a cardinal feature of the disorder. The severity of the disease varies among patients, from those who are asymptomatic and are diagnosed only incidentally by radiographs showing bilateral increased density of the long bones to babies who are unable to walk because of severe leg pain, underdevelopment, and abnormal gait [3]. Therefore, progression of the disease is highly variable and difficult to predict.
Genetic involvement of CED has been deduced and is giving rise to developments in understanding the disease progression. A 15.1-cM segment on chromosome 19q13.1–13.3 has been shown to be the origin of the disorder [1]. Ten different mutations have been identified to date [5]. Specifically, most mutations involve the transforming growth factor-beta 1 (TGFβ1) gene, which initially is formed as a proprecursor molecule consisting of a signal peptide, latency associate peptide, and the mature peptide [5]. Posttranslational modification yields a small latent complex comprised of an association between two latency-associated peptides and two mature peptides [5]. The majority (seven of 10) of the mutations are missense mutations located in exon 4, which includes the coding region of the latency-associated peptide [5]. An arginine residue at position 218 is the most common position of mutation, making up 60% of all mutations [5]. Mutations outside exon 4 include a 9-bp duplication in exon 1 encoding the signal peptide, and two missense mutations in exon 1 and 2 at the N-terminus of the latency-associated peptide [5]. These mutations are classified into two groups depending in what exon they develop [5]. Mutations in exon 4 destabilize disulphide bridges of the latency-associated peptides and cause premature activation of the mature peptide [5]. Mutations in exon 1 affect secretion and lead to intracellular retention of the mutant protein [5].
All mutations studied to date increase the activity of TGFβ1. Receptors for TGFβ1 are expressed ubiquitously throughout the body and are vital to the development of many organ systems. How a mutation in the TGFβ1 coding sequence causes the relatively mild disease pattern of CED has not been definitely explained [5]. However, it is believed the TGFβ1 gene is directly related to the balance of osteoblast and osteoclast function, among other biologic processes. Transforming growth factor-β1 has been shown to stimulate bone formation and suppress bone resorption under physiologic conditions, which explains most of the clinical features of the disease. Furthermore, TGFβ1 is a known inhibitor of myogenesis and adipogenesis, causing reduction in fat and muscle mass, characteristic muscle wasting, and easy fatigability.
Patients with CED traditionally present with pain in their legs after walking a short distance, weakness, muscle underdevelopment, and a waddling gait [9]. The majority of cases (75%) develop during childhood, although earlier onset has been reported [7, 12]. Yet, the disorder has been reported to develop as late as the eighth decade of life and seems to worsen with age [3, 9]. The average age of patients at diagnosis is 14 years, but many previously were misdiagnosed [11]. There is no gender or ethnic predisposition, but it seems the disease shows anticipation, or increased severity of disease in subsequent generations, with the most severe inheritance pattern from mother to daughter [11]. The progression of CED is extremely variable, with some authors reporting symptom remission late into adulthood [4]. Also, patients with continued symptoms have unpredictable progression of the disease that can affect not only the long bones, but also cranial bones, vertebrae, or distal extremities [5]. The musculoskeletal involvement can cause varying abnormalities including lordosis, kyphosis, coxa valga, genu valga, pes planus, and frontal bossing [11]. There is no indication of shortened lifespan or predisposition to other chronic or life-threatening illnesses [11].
Radiographically, patients present with bilateral increased density of the cortices of long bones, most commonly affecting the femur. Other symptoms include growth retardation, dry skin, flat feet, valgus ankle, reduced subcutaneous fat, altered reflexes, exophthalmos, nystagmus, splenomegaly, and delayed puberty [3, 11]. Hearing impairment is incident in 18% of patients with CED; when the disease progresses to the facial bones, the bone growth narrows the internal auditory canal, causing encroachment on nerves and vessels [8]. When the skull is involved, symptoms such as headache, vision loss, vertigo, tinnitus, or facial paralysis can be present [11]. Patients with CED do not have an increased risk of fracture; on the contrary, there appears to be resistance to osteoporosis, and fractures in these patients usually are comminuted, representing hard bone [11]. No specific abnormalities have been seen in the laboratory tests of patients with CED to be used as a diagnostic marker [11]. As the attributes of this disease are so widespread, it is important to exclude any other possible diagnosis.
Treatment of CED has not been standardized and is used mainly to alleviate the symptoms experienced by the patient. The most commonly recommended treatment is systemic corticosteroids [4, 6, 10, 11]. Although the specific mechanism of action in CED is unknown, steroids seem to alleviate leg pain and muscle weakness, although they do not necessarily alter the course of the disease [1]. The corticosteroids also appear to stimulate osteoclast function and decrease lamellar bone deposition [2]. Some authors report patients having success with different levels of steroid treatment, although some patients experience no improvement with long-term steroid use [2]. One dose regimen included 1 to 2 mg/kg/day followed by rapid tapering to an alternate day dose when tolerated [11]. Another dose started at 0.5 mg/kg on an alternate day schedule [11]. Long-term use of corticosteroids, however, is not advised because of the unfavorable side effects.
Bisphosphonates are a class of drugs disputed in the literature for treatment of CED. Pamidronate is a bisphosphonate used experimentally in some patients, but it had no positive effects [4]. Disodium etidronate, also a bisphosphonate, was used in one case to reduce the rate of growth and mineral deposit on bone with some success [1]. Bisphosphonates reduce bone growth by reducing the growth rate of hydroxyapatite crystals. It has been suggested bisphosphonates will not provide adequate treatment of CED because of their successful use for treatment of osteoporosis to increase bone mineral density and lower fracture risk [5]. Calcitonin was described for its use to relieve bone pain in one patient, but it is also an inhibitor of bone resorption, making it unlikely to be a successful treatment [5]. Nonsteroidal antiinflammatory medications are successful only in alleviating pain symptoms but are not effective in improving bone alteration. Finally, surgery for CED is rare because the disease is progressive and will return. Medullary reaming has been used in two cases to alleviate narrowing of the medullary canal [5]. Surgical decompression of the optic nerve was used in one case to remove bone encroachment [5].
Gene therapy is another possibility in the treatment of CED. It may be possible to use molecules that sequester the mature peptide, such as decorin, biglycan, α2-macroglobulin, soluble β-glycan, α2-HS glycoprotein/fetuin, anti-TGFβ monoclonal antibodies, or a soluble inactive Type II receptor [5]. Inhibitors of downstream signaling molecules also could be successful [5]. The most difficult aspect of gene therapy in CED is controlling side effects because of the vital function TGFβ1 plays in many organ systems throughout the body [5]. Consequently, direct application to bone and muscle tissue would be the most appropriate avenue of treatment [5].
The current literature review illustrates the difficulty in treating patients with CED. Our patient was confirmed to have CED through clinical features, radiographic evidence, and family history, as her mother was diagnosed genetically. The girl has been followed for four years with observation and pain control being central to the treatment plan. Physical therapy, nonsteroidal antiinflammatory medications, and ankle orthotics have been used for strength training and pain control with success. She is being followed orthopaedically with radiographic studies to determine disease progression, and ophthalmologically to determine any visual deficits. She currently is having difficulty maintaining strength and muscle mass in her lower extremities and continues to have intermittent pain; however, her disease is tolerable with the current treatment and the support of her mother who has the same disease.
Camurati-Engelmann disease is a rare genetic disease that can provide the clinician with difficulties in diagnosing and treating patients. However, with the help of molecular genetics and the connection to clinical presentation, diagnosis of this disease may become easier. When a patient presents with painful legs, abnormal waddling gait, and fatigue, this disorder should be part of the differential diagnosis.
Footnotes
Each author certifies that he or she has no commercial associations (eg, consultancies, stock ownership, equity interest, patent/licensing arrangements, etc) that might pose a conflict of interest in connection with the submitted article.
Each author certifies that his or her institution has approved the reporting of this case report, that all investigations were conducted in conformity with ethical principles of research, and that informed consent for participation in the study was obtained.
References
- 1.Belinda A, Xavier CF, Saraiva JM, Le Merrer M, Dagoneau N, Huber C, Penet C, Munnich A, Cormier-Daire V. Genetic homogeneity of the Camurati-Engelmann disease. Clin Genet. 2000;58:150–152. [DOI] [PubMed]
- 2.Gamanagatti S, Sharma R, Goswami R, Laway BA. Camurati-Engelmann disease with metaphyseal involvement. Eur J Radiol Extra. 2004;49:93–96. [DOI]
- 3.Grey AC, Wallace R, Crone M. Engelmann’s disease: a 45-year follow-up. J Bone Joint Surg Br. 1996;78:488–491. [PubMed]
- 4.Inaoka T, Shuke N, Sato J, Ishikawa Y, Takahashi K, Aburano T, Makita Y. Scintigraphic evaluation of pamidronate and corticosteroid therapy in a patient with progressive diaphyseal dysplasia (Camurati-Engelmann disease). Clin Nucl Med. 2001;26:680–682. [DOI] [PubMed]
- 5.Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S, Guañabens N, Migone N, Wientroub S, Divizia MT, Bergmann C, Bennett C, Simsek S, Melançon S, Cundy T, Van Hul W. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006;43:1–11. [DOI] [PMC free article] [PubMed]
- 6.Naveh Y, Alon U, Kaftori JK, Berant M. Progressive diaphyseal dysplasia: evaluation of corticosteroid therapy. Pediatrics. 1985;75:321–323. [PubMed]
- 7.Saraiva JM. Anticipation in progressive diaphyseal dysplasia. J Med Genet. 2000;37:394–395. [DOI] [PMC free article] [PubMed]
- 8.Stasolla A, Magliulo G, Bellussi A, Parrotto D, Bibbolino C, Marini M. Imaging of the temporal bone in Camurati-Engelmann dysplasia with an 11-year follow-up. Otol Neurotol. 2005;26:773–777. [DOI] [PubMed]
- 9.Vanhoenacker FM, Janssens K, Van Hul W, Gershoni-Baruch R, Brik R, De Schepper AM. Camurati-Engelmann Disease: review of radioclinical features. Acta Radiol. 2003;44:430–434. [DOI] [PubMed]
- 10.Verbruggen LA, Bossuyt A, Schreurer R, Somers G. Clinical and scintigraphic evaluation of corticosteroid treatment in a case of progressive diaphyseal dysplasia. J Rheumatol. 1985;12:809–813. [PubMed]
- 11.Wallace SE, Lachman RS, Mekikian PB, Bui KK, Wilcox WR. Marked phenotypic variability in progressive diaphyseal dysplasia (Camurati-Engelmann disease): report of a four-generation pedigree, identification of a mutation in TGFB1, review. Am J Med Genet A. 2004;129A:235–247. [DOI] [PubMed]
- 12.Wright M, Miller NR, McFadzean RM, Riordan-Eva P, Lee AG, Sanders MD, McIlwaine GG. Papilloedema, a complication of progressive diaphyseal dysplasia: a series of three case reports. Br J Ophthalmol. 1998;82:1042–1048. [DOI] [PMC free article] [PubMed]