

Abstract
Background
Pancreatic cancer is a very aggressive malignancy and efficient therapeutic options are still largely lacking. The importance of interactions between tumor cells and surrounding stromal elements, e.g. mononuclear cells, for chemo-resistance has been increasingly recognized. In addition, cyclooxygenase-2 is thought to be an important mediator of chemo-resistance in several malignancies. The aim of the present study was to explore the role of mononuclear cells in pancreatic cancer chemo-resistance.
Methods
U937 cells were differentiated into macrophage-like cells. The effect of U937 conditioned medium on drug-induced pancreatic cancer cell apoptosis was measured by ELISA. The contributions of interleukin-1β and cyclooxygenase-2 were evaluated by specific receptor antagonists and inhibitors. The importance of the extracellular signal-regulated kinase (ERK1/2) pathway was also determined.
Results
U937 conditioned culture medium protected pancreatic cancer cells from drug-induced apoptosis. This protective effect was abolished by an interleukin-1 receptor antagonist and cyclooxygenase-2 inhibitor. U937 conditioned medium and interleukin-1β stimulated expression of cyclooxygenase-2 and prostaglandin E2 production in pancreatic cancer cells, which was mediated by activation of the ERK1/2 pathway. Transfection of pancreatic cancer cells with cyclooxygenase-2 increased resistance to drug-induced cell death.
Conclusions
Mononuclear cells protect pancreatic cancer cells of drug-induced apoptosis in vitro by interleukin-1β-mediated expression of cyclooxygenase-2 and production of prostaglandins. This study highlights the importance of tumor-host interactions in pancreatic cancers and may provide the basis for novel therapeutic approaches to sensitize pancreatic cancers to chemotherapeutic agents.
Introduction
Interactions between cancer cells and the surrounding host stroma are increasingly recognized as instrumental for tumor growth, survival, and spread.1 The stroma mainly comprises of various cellular elements, e.g. fibroblasts, endothelial and inflammatory cells, and deposited extracellular matrix proteins. A dense fibrous stroma, called desmoplasia, is a characteristic histological feature of pancreatic cancers (PaCa) and has recently gained recognition as an active contributor to the malignant phenotype of this disease.2 Moreover, the desmoplastic reaction is thought to be partially responsible for the notorious resistance of pancreatic cancers to common chemo- and radio-therapeutic regimens. Of the cellular components of the tumor-surrounding stroma inflammatory cells are believed to play a pivotal role in the progression and chemo-resistance of malignant tumors.3 Macrophages, neutrophils and mast cells have all been implicated in promoting tumor growth.3,4 There is an emerging concept that chronic inflammatory processes are fundamental for the development and maintenance of malignant tissues.3 Macrophages are commonly recruited into the tumor by cancer cell-secreted cytokines/chemokines.1 Although principally capable of killing tumor cells, tumor-infiltrating macrophages are often dysfunctional and lack tumoricidal activity. However, they still maintain their ability to secrete various cytokines, some of which directly promote tumor cell survival and growth.1 In PaCa inflammatory cell infiltration has been correlated with lymph node metastasis and poor prognosis with macrophages being one of the predominant leukocyte subpopulation.5,6
IL-1β is a pro-inflammatory, secreted cytokine synthesized by many cell types, including monocytes and tissue macrophages, as a 31 kDa proform, which is cleaved by IL-1β-converting enzyme or caspase-1 to generate the mature 17 kDa protein.7 IL-1β signals by binding to a high-affinity receptor aggregate of IL-1 receptor type I (IL-1RI) and IL-1 receptor accessory protein (IL-1AcP). A third receptor, IL-1 receptor type II (IL-1RII), acts as a decoy receptor and competes with IL-1RI for IL-1. The naturally occurring receptor antagonist of IL-1 (IL-1RA) has structural similarity to IL-1 and can bind to IL-1RI but does not induce any signalling response.8 Besides its major role in inflammatory and autoimmune diseases, IL-1β has also been shown to be involved in tumorigenesis, tumor growth and metastasis.9 As a proinflammatory cytokine IL-1β is capable of rapidly stimulate the expression of cyclooxygenase-2 (COX-2), the rate limiting enzyme in generating pro-inflammatory prostanoids.10 COX-2 and COX-2 generated prostanoids are implicated in the development, growth, and spread of various human tumors, including pancreatic cancers.11 Furthermore, COX-2 has been suggested to confer chemo-resistance in human cancers and preclinical cancer models.12–14 Conversely, selective inhibitors of COX-2 increased the sensitivity of cancer cells to chemotherapeutic agents.15,16 COX-2 is overexpressed in the majority of human PaCa and correlates with poor prognosis.17–19 Preclinical animal studies have clearly demonstrated that inhibiting the COX-2/prostanoid pathway attenuates the growth of PaCa and delays the progression of PaCa precursor lesions indicating that the COX-2/prostanoid pathway is an intriguing target for PaCa therapy and prevention.20,21 However, the role of COX-2 in pancreatic cancer chemo-resistance, in particular its contribution to IL-1β-mediated chemo-resistance, has not been explored. Our data provide evidence that monocyte/macrophages confer chemo-resistance to human pancreatic cancer cells in vitro by IL-1β-mediated up-regulation of COX-2 in pancreatic cancer cells.
Material and Methods
Reagents
The chemicals phorbol 12-myristate 13-acetate (PMA), lipopolysaccharide (LPS), camptothecin, genistein as well as the mouse monoclonal β-actin antibody were purchased from Sigma (Sigma Chemical Co., St. Louis, MO). Human recombinant IL-1β was obtained from Peprotech (Rocky Hill, NJ) and human recombinant IL-1 receptor antagonist (IL-1RA) was purchased from R&D Systems Inc. (Minneapolis, MN). The selective COX-2 inhibitor nimesulide and the mouse monoclonal antibody against COX-2 were from Cayman Chemicals (Ann Arbor, MI). The MEK-1 inhibitor PD98059 and the rabbit polyclonal antibodies against phospho-ERK and total-ERK were obtained from Cell Signaling Technology (Beverly, MA). The full-length human COX-2 expression vector (pBOSNeoCOX-2) was a generous gift from Y. Takahashi (Okayama, Japan).
Cell culture
Human pancreatic cancer cell lines (BxPC-3: G2; Capan-2: G1; HPAF-II: G2; MIA PaCa-2: G3)22 and the human histiocytic lymphoma cell line U937 were obtained from the American Type Culture Collection (Rockville, MD) and cultured as recommended by the vendor and described previously.20 Prior to the experiments sub-confluent cells were cultured overnight in serum-deprived (0.5%) medium. Culture medium was then replaced with serum free medium together with the indicated reagents.
Macrophage model
U937 cells were terminally differentiated with 10 nM phorbol ester (PMA) for 48 hours into macrophage-like cells and subsequently stimulated with 1 μg/ml lipopolysaccharide (LPS) for another 24 hours in serum free medium. Conditioned culture medium of differentiated and stimulated U937 cells (U937diff CM) was used for subsequent experiments. PMA differentiated and LPS stimulated U937 cells have been widely and successfully used as an in vitro model of macrophage-like cells.23,24
Cell death assay
Cells were cultured on 96-well plates in serum free culture medium or U937diff CM and exposed to the DNA topoisomerase I inhibitor camptothecin (1 μg/ml) or the isoflavonoid genistein (100 μM) for 24 hours. In some experiments cells were pre-incubated with the indicated reagents 3 hours prior to adding camptothecin or genistein. Apoptotic cell death was determined after 24 hours by the Cell Death Detection ELISA (Roche Diagnostics Corp., Indianapolis, IN), which measures the enrichment of mono- and oligonucleosomes in the cytoplasm of the apoptotic cell during DNA fragmentation, as described previously.25
Western blot analysis of COX-2
Total cell lysates were prepared using ice-cold modified radioimmunoprecipitation (RIPA) buffer containing leupeptin, aprotinin, and pepstatin A. Aliquots were fractionated on 8% SDS-PAGE and transferred to nitrocellulose membranes. Proteins were detected using specific primary antibodies and horseradish peroxidase-conjugated immunoglobulins as secondary antibodies (Pierce, Rockford, IL). Protein-antibody complexes were visualized with the SuperSignal West Pico Chemiluminescent Substrate (Pierce). Equal protein loading was confirmed by β-actin Western blot. Protein expression was quantified by laser densitometry and ImageQuant software (Amersham Biosciences Corp., Piscataway, NJ).
Activation of ERK1/2
Activation of ERK1/2 was determined as described previously.26 Briefly, pancreatic cancer cells were incubated in serum free medium with the indicated ligands and total cell lysates were separated on 8% SDS-PAGE and transferred to nitrocellulose membranes. Membranes were probed with antibodies recognizing specifically the dually phosphorylated ERK1/2. Detection of total ERK1/2 levels served as loading controls.
PGE2 and IL-1β ELISA
PGE2 and IL-1β levels in the culture medium were determined by a PGE2 (Cayman Chemical, Ann Arbor, MI) and IL-1β competitive enzyme immunoassay (Invitrogen, Carlsbad, CA), respectively, as recommended by the manufacturers and described previously.25 Protein levels were normalized to cell number.
Transient transfection
Cells were transiently transfected as described previously.27 Briefly, pancreatic cancer cells were cultured to 50% confluence and transfected with pBOSNeoCOX-2 using the Lipofectamine Plus reagent (Invitrogen) for 48 hours. The parent vector without the full length COX-2 cDNA insert was used as a control.
Statistical analysis
Data are presented as mean ± standard deviation. Comparisons of more than two groups were made by a One Way Analysis of Variance (ANOVA) with post-hoc Holm-Sidak analysis for pairwise comparisons and comparisons versus control. An alpha value of 0.01 was used to determine significant differences. All statistics were done in SigmaStat 3.1 (Systat Software, Inc.).
Results
Macrophage-like cells confer resistance to apoptotic cell death in pancreatic cancer cells through the IL-1 receptor and COX-2
As a model of mononuclear cells, we used the human histiocytic lymphoma cell line U937, which are immunological precursor cells and can differentiate into macrophage-like cells if treated with PMA.23 Compared to undifferentiated U937 cells, PMA-treated U937 cells secreted significantly more IL-1β, which was further exponentially increased after stimulation with LPS (Figure 1A). For subsequent experiments we used conditioned medium of PMA-differentiated and LPS-stimulated U937 cells (U937diff CM). To study the effect of U937diff CM on chemo-resistance of pancreatic cancer cells, apoptotic cell death in Capan-2 cells was induced by the DNA topoisomerase I inhibitor camptothecin and the isoflavonoid genistein and quantified by ELISA. U937diff CM partially protected Capan-2 cells from drug-induced apoptosis (Figure 1B). The protective effect of U937diff CM on drug-induced apoptosis was reversed by recombinant IL-1RA and a selective COX-2 inhibitor, indicating an involvement of the IL-1 receptor and COX-2 in the U937diff CM-mediated protection of pancreatic cancer cells from apoptotic cell death (Figure 1C). Recombinant IL-1β at a concentration equal to the IL-1β levels found in U937diff CM (10 ng/ml) demonstrated a similar but slightly less protective effect on drug-induced apoptosis (Figure 1C). To further test the hypothesis that U937diff CM protects pancreatic cancer cells from apoptosis through an IL-1RI and COX-2 mediated pathway, pancreatic cancer cells were incubated with U937diff CM in the absence or presence of IL-1RA and COX-2 protein expression and activity were measured. U937diff CM increased the expression of COX-2 protein and production of PGE2, a surrogate marker for COX-2 activity, in three pancreatic cancer cell lines. Pre-treatment with IL-1RA partially abrogated this effect (Figure 1D).
Figure 1. Macrophage-like cells increase the resistance of pancreatic cancer cells to apoptosis through the IL-1RI and COX-2.

A) U937 cells were terminally differentiated with phorbol 12-myristate 13-acetate (PMA) and stimulated with lipopolysaccaride (LPS). Il-1β levels in the culture medium were measured by ELISA and normalized to the cell number. B) Capan-2 cells were cultured in normal culture medium or U937diff CM and exposed to camptothecin or genistein for 24 hours. DNA fragmentation as a measure of apoptosis was determined by the Cell Death Detection ELISA and presented as fold-increase relative to the control. C) Capan-2 cells were cultured in normal culture medium with or without IL-1β or in U937diff CM and exposed to camptothecin for 24 hours. IL-1RA and nimesulide were added 3 hours prior to camptothecin. DNA fragmentation as a measure of apoptosis was determined by the Cell Death Detection ELISA and presented as fold-increase relative to the control. D) BxPC-3, Capan-2 and HPAF-II cells were cultured in normal culture medium or U937diff CM in the presence or absence of Il-1RA (500 ng/ml) for 24 hours. COX-2 protein expression was determined by Western Blotting. Beta-actin was used as a loading control. PGE2 levels in the culture medium were measured by ELISA and normalized to the cell number. *, p<0.01 vs. control; #, p<0.01 vs. U937diff CM. All experiments were done at least in duplicate using separate cultures. Data are presented as mean ± SD.
IL-1β stimulates COX-2 expression in pancreatic cancer cells by activation of ERK1/2
Next we determined whether IL-1β can up-regulate COX-2 expression in pancreatic cancer cells. IL-1β increased COX-2 expression in a dose- and time-dependent fashion in three human pancreatic cancer cell lines (Figures 2A, B). Significant up-regulation of COX-2 expression was thereby observed with IL-1β concentrations as low as 1 ng/ml. In Capan-2 cells, which have low baseline COX-2 expression,25 IL-1β at 1 ng/ml and 10 ng/ml increased COX-2 protein levels after 24 hours by about 16- and 20-fold, respectively (Figure 2A). A significant increase in COX-2 expression was seen in Capan-2 cells as early as 2 hours after adding IL-1β to the culture medium (Figure 2B). In addition, exposure of Capan-2 cells to IL-1β (10 ng/ml) for 24 hours increased PGE2 production, which was dose-dependently attenuated by IL-1RA. At a concentration of 500 ng/ml IL-1RA completely abolished the effect of IL-1β on PGE2 production (Figure 2C).
Figure 2. Il-1β dose- and time-dependently increases COX-2 expression.

A) BxPC-3, Capan-2 and HPAF-II were exposed to incremental concentrations of Il-1β (0–10 ng/ml) for 24 hours. COX-2 protein expression was determined by Western Blotting and quantified by laser densitometry; β-actin was used as a loading control. *, p<0.01 vs. control (no Il-1β). B) BxPC-3, Capan-2 and HPAF-II were exposed to vehicle or Il-1β (10 ng/ml) for various times (2–16 hours). COX-2 protein expression was determined by Western Blotting and quantified by laser densitometry; β-actin was used as a loading control. *, p<0.01 vs. vehicle. C) Capan-2 cells were exposed to Il-1β (10 ng/ml) for 24 hours in the absence or presence of IL-1RA (0–500 ng/ml). COX-2 protein expression was determined by Western Blotting and PGE2 levels in the culture medium were measured by ELISA and normalized to the cell number. *, p<0.01 vs. Il-1β (10 ng/ml) and IL-1RA (0 ng/ml). All experiments were done at least in duplicate using separate cultures. Data are presented as mean ± SD.
Having demonstrated that IL-1β up-regulates COX-2 expression through the IL-1R, we then sought to determine which signalling pathways are involved. Upon ligand binding, several adapter molecules and kinases are sequentially recruited to the IL-1R complex ultimately leading to the activation of a cytosolic multiprotein complex which can activate multiple downstream signalling pathways, including NF-κB and MAPK.28 Both NF-κB and MAPK pathways are known critical signals for transcriptional control of COX-2 expression.29 Exposure of Capan-2 cells with IL-1β (10 ng/ml) time-dependently stimulated phosphorylation of ERK1/2 (Figure 3A). A significant increase of phosphorylated ERK1/2 was already seen at 15 minutes after addition of IL-1β and was sustained for at least 24 hours. Pre-treatment of human pancreatic cancer cells with the MEK-1 inhibitor PD98059, which completely inhibited IL-1β-induced ERK1/2 phosphorylation at 10 μM (not shown), dose-dependently attenuated the IL-1β-mediated increase in COX-2 expression (Figure 3B).
Figure 3. IL-1β stimulates COX-2 expression through the ERK1/2 pathway.
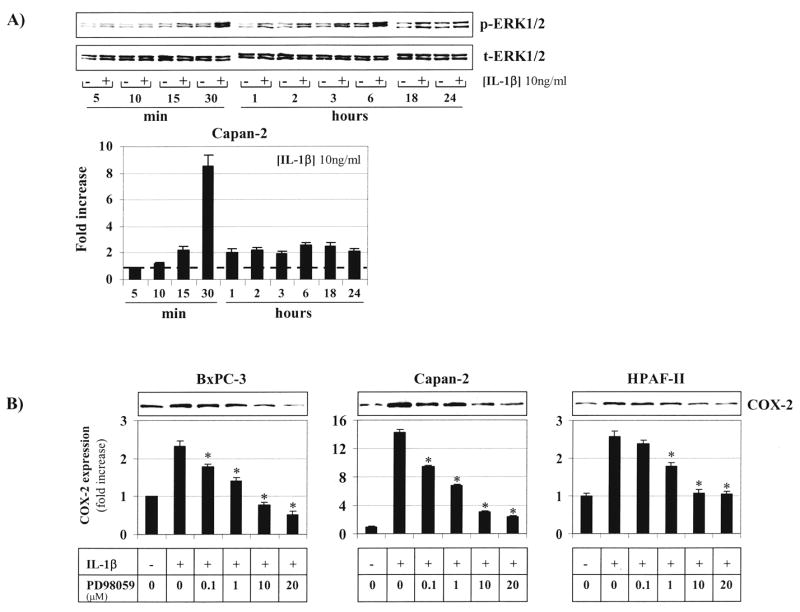
A) Capan-2 cells were exposed to vehicle or Il-1β (10 ng/ml) for different times (0–24 hours). Phosphorylation of ERK1/2 (p-ERK1/2) was determined by Western Blotting and quantified by laser densitometry; total-ERK1/2 (t-ERK1/2) was used as a loading control. B) BxPC-3, Capan-2 and HPAF-II cells were exposed to Il-1β (10 ng/ml) for 6 hours in the absence or presence of the MEK-1 inhibitor PD98059 (0–20 μM). COX-2 protein expression was determined by Western Blotting and quantified by laser densitometry. *, p<0.01 vs. Il-1β (10 ng/ml) and PD98059 (0 μM). All experiments were done at least in duplicate using separate cultures. Data are presented as mean ± SD.
COX-2 confers resistance to apoptotic cell death in pancreatic cancer cells
To further test the hypothesis that IL-1β-induced COX-2 confers resistance to apoptosis, COX-2 negative MIA PaCa-2 cells were transiently transfected with COX-2. Over-expression of COX-2 increased resistance to drug-induced apoptotic cell death of MIA PaCa-2 cells, which was reversed by the selective COX-2 inhibitor nimesulide (Figure 4A). Transfection of MIA PaCa-2 cells with the parent vector lacking the COX-2 insert had no effect on drug-induced apoptosis. Addition of exogenous PGE2 to nimesulide-treated MIA PaCa-2 cells restored the protective effect of COX-2 (Figure 4A), indicating that COX-2 generated prostanoids, e.g. PGE2, are mediating the pro-survival properties of COX-2 in transfected MIA PaCa-2 cells. Successful transfection of COX-2 and enzymatic COX-2 activity was confirmed by Western blot and PGE2 ELISA (Figures 4B, C).
Figure 4. COX-2 confers resistance to camptothecin-induced apoptosis.

A) MIA PaCa-2 cells were transfected with COX-2 or an empty vector (EV) and exposed to camptothecin (1 μg/ml) for 24 hours together with nimesulide (10 μM) or PGE2 (10 μM). DNA fragmentation as a measure of apoptosis was determined by the Cell Death Detection ELISA and presented as fold-increase relative to the control. *, p<0.01. B) PGE2 levels in the culture medium of MIA PaCa-2 cells (transfected with COX-2 or the empty vector) with or without nimesulide were measured by ELISA and normalized to the cell number. C) Successful transfection of MIA PaCa-2 cells with COX-2 was confirmed by Western Blotting; β-actin was used as a loading control. All experiments were done at least in duplicate using separate cultures. Data are presented as mean ± SD.
Discussion
The critical importance of mutual interactions between cancer cells and surrounding host stromal elements is widely accepted today. Tumor cells require the intricate cytokine network present in their microenvironment for survival, growth, and metastasis.2 Besides endothelial cells and fibroblasts, inflammatory cells have been implicated to be a main source of tumor-promoting cytokines and are commonly found in human pancreatic cancers.1,6 Our data clearly demonstrate that the pro-inflammatory cytokine IL-1β increases the resistance of pancreatic cancer cells in vitro to apoptotic cell death. Fibroblasts and mononuclear cells are major common sources of IL-1β production.10 In recent studies IL-1β production by pancreatic cancer cells in vitro was stimulated by fibroblast-secreted nitric oxide, thereby enhancing chemo-resistance.30 In our studies, we were unable to detect measurable levels of IL-1β in the culture medium of pancreatic cancer cell lines (not shown). Instead, we used differentiated and stimulated U937 cells as a monocyte/macrophage model, which secreted ample amounts of IL-1β. These cells have been widely used as a model for macrophage function.23,24 Conditioned culture medium of differentiated and stimulated U937 cells (U937diff CM) increased resistance of pancreatic cancer cells to drug-induced apoptotic cell death, which was inhibited by recombinant IL-1RA, indicating the importance of the IL-1RI and IL-1 in this process. We used the DNA topoisomerase I inhibitor camptothecin and the isoflavonoid genistein as the chemotherapeutic agent to induce apoptotic cell death in pancreatic cancer cells. DNA topoisomerase I inhibitors, which induce apoptosis by creating single strand DNA breaks with a subsequent block in cell replication, have been shown to be efficacious agents in murine models of pancreatic cancer.31,32 Although the clinical use of camptothecin is prohibitive due to its severe life threatening adverse reactions and later camptothecin-derivatives, e.g. exatecan and irinotecan, lack clinical efficacy in pancreatic cancer patients in phase III trials,33,34 our data clearly demonstrate that the DNA topoisomerase I inhibitor camptothecin can induce apoptotic cell death in pure pancreatic cancer cell cultures in vitro. However, incubation of pancreatic cancer cells with conditioned medium of macrophage-like cells, which attempts to recapitulate the intricate tumor-stroma interactions found in vivo, confers resistance to drug-induced apoptosis, which is in agreement with the clinical observation. In addition, the protective effect of macrophage-conditioned medium is not specific to camptothecin-induced apoptosis as U937diff CM also confers resistance to apoptotic cell death induced by the isoflavonoid genistein. We have previously demonstrated that genistein induced caspase-3 dependent apoptosis in pancreatic cancer cells and recent data suggest that genistein augments the anti-tumor efficacy of various chemotherapeutic agents.35–38
IL-1β is known to up-regulate the expression of COX-2 with a subsequent increase in the generation of prostaglandins; critical steps in inflammatory processes.14 Beside its mitogenic, pro-invasive, and angiogenic properties, COX-2 has also been found to increase the resistance to chemotherapeutic agents in a variety of models.14 The observation that a selective COX-2 inhibitor completely reversed the effect of U937diff CM on drug-induced cell death in pancreatic cancer cells in vitro highlights the critical role of COX-2 in mediating this effect. Furthermore, U937diff CM increased COX-2 expression in pancreatic cancer cells, which was partially inhibited by IL-1RA and exposure of pancreatic cancer cells to recombinant IL-1β dose- and time-dependently enhanced COX-2 expression. These data clearly suggest that IL-1β secreted by monocytes/macrophages enhance the expression of COX-2 and production of prostaglandins in pancreatic cancer cells thereby increasing their resistance to cell death induced by a chemotherapeutic agent. The finding that IL-1RA inhibited the effect of U937diff CM on COX-2 expression and drug-induced apoptosis only partially can be explained by the presence of other cytokines in the culture medium, e.g. tumor necrosis factor-alpha.39 This hypothesis is supported by the finding that recombinant IL-1β at the same concentration as in U937diff CM was slightly less efficacious in preventing drug-induced apoptosis. In our model system, U937 cells were differentiated with PMA and stimulated with LPS, which itself is a strong inducer of COX-2 expression in many cells.14,40 However, U937 cells stimulated with LPS but not differentiated with PMA failed to up-regulate COX-2 expression in pancreatic cancer cells after 24 hours (not shown), suggesting that U937-produced cytokines rather than LPS were responsible for the up-regulation of COX-2. It is certainly possible that LPS present in the conditioned medium increased COX-2 protein expression transiently (<24 hours) but the sustained effect on COX-2 expression at 24 hours was mediated by IL-1β.
The expression of COX-2 can be induced by various extra-cellular signals. Activation of the mitogen-activated protein kinase (MAPK) pathways is critically involved in regulating COX-2 mRNA levels at the transcriptional and post-transcriptional level.29 Our data demonstrate that the effect of IL-1β on COX-2 expression in pancreatic cancer cells in vitro was mediated by activation of the extracellular signal-regulated kinase (ERK) pathway. It has been shown that IL-1β up-regulates COX-2 expression in a variety of cell types by activation of the p38 MAPK and ERK1/2 pathways.40–43 Binding of IL-1β to the IL-1RI initiates the recruitment of MyD88 (Myeloid differentiation factor 88), which in turn recruits IL-1R-associated kinase-4 (IRAK4) into the complex inducing the further recruitment of Tollip/IRAK1 complexes. Phosphorylation of IRAK1 by IRAK4 allows transient recruitment of TRAF6 (TNF receptor-associated factor 6) which together with IRAK1 dissociates from the receptor and associates with a complex of adaptor molecules, e.g. TAK1 (TGF-activated kinase 1). Activated TAK1 phosphorylates the inhibitor of κB (IKK) complex in addition to MAPK kinases 3, 4, and 6, thereby linking IL-1β signalling to NF-κB, JNK and p38 MAPK.7,28 In addition it has been shown that the recruitment of IRAK-1 into focal adhesion complexes is required for IL-1β-induced ERK activation.44 Although we have not dissected the intracellular signalling pathways in detail, our observation that the MEK-1 inhibitor PD98059, which inhibits activation of ERK1/2, abolished the effect of IL-1β on COX-2 expression in pancreatic cancer cells, indicates the importance of the ERK1/2 pathway in this process. Our data certainly cannot rule out the involvement of NF-κB in mediating the IL-1β-induced expression of COX-2 and hence resistance to apoptosis, as NF-κB can be downstream of ERK1/2 signaling.
Our results clearly identified macrophage-secreted IL-1β with subsequent up-regulation of COX-2 expression as a mechanism to protect pancreatic cancer cells from apoptotic cell death. Since inflammatory cells, including macrophages, are commonly found in and around human pancreatic cancers, inflammatory cell-secreted cytokines, e.g. IL-1β, could create a microenvironment that provides strong survival signals, which may explain the notorious clinical resistance of pancreatic cancers to common chemotherapeutic agents. Our data strongly support the notion that inhibiting the IL-1β/IL-1RI signalling may prove an intriguing approach to sensitize pancreatic cancers to chemotherapeutic agents. Currently there are several chemicals and recombinant proteins in development, which target the IL-1β/IL-1RI signalling pathway.7 Among those, Anakinra, a recombinant human IL-1RA, has been approved for the treatment of rheumatoid arthritis. Further studies using these targeted approaches in preclinical animal models will be necessary to evaluate the efficacy of inhibiting IL-1β signalling in sensitizing pancreatic cancers to chemotherapeutic agents.
Acknowledgments
Financial support: This work was supported in part by the CURE: Digestive Diseases Research Center Pilot and Feasibility Studies Grant (Center: P30 DK041301) to G.E., the Hirshberg Foundation for Pancreatic Cancer Research (H.A.R.), and the UCLA Center for Excellence in Pancreatic Diseases (P01 AT003960). E.A. was supported by a Personal Grant from the Swiss Scientific Foundation (PBZHB-117008). G.E. is supported by the National Institutes of Health (R01 CA104027, R01 CA122042).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bronte V, Cingarlini S, Marigo I, De Santo C, Gallina G, Dolcetti L, et al. Leukocyte infiltration in cancer creates an unfavorable environment for antitumor immune responses: a novel target for therapeutic intervention. Immunol Invest. 2006;35(3–4):327–57. doi: 10.1080/08820130600754994. [DOI] [PubMed] [Google Scholar]
- 2.Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6(4):1186–97. doi: 10.1158/1535-7163.MCT-06-0686. [DOI] [PubMed] [Google Scholar]
- 3.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4(1):71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 4.Bellocq A, Antoine M, Flahault A, Philippe C, Crestani B, Bernaudin JF, et al. Neutrophil alveolitis in bronchioloalveolar carcinoma: induction by tumor-derived interleukin-8 and relation to clinical outcome. Am J Pathol. 1998;152(1):83–92. [PMC free article] [PubMed] [Google Scholar]
- 5.Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, et al. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57(6):630–6. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emmrich J, Sparmann G, Hopt U, Lohr M, Liebe S. Typing of leukocytes in pancreatic tissue surrounding human pancreatic carcinoma. Ann N Y Acad Sci. 1999;880:171–4. doi: 10.1111/j.1749-6632.1999.tb09520.x. [DOI] [PubMed] [Google Scholar]
- 7.Braddock M, Quinn A. Targeting IL-1 in inflammatory disease: new opportunities for therapeutic intervention. Nat Rev Drug Discov. 2004;3(4):330–9. doi: 10.1038/nrd1342. [DOI] [PubMed] [Google Scholar]
- 8.Martin MU, Wesche H. Summary and comparison of the signaling mechanisms of the Toll/interleukin-1 receptor family. Biochim Biophys Acta. 2002;1592(3):265–80. doi: 10.1016/s0167-4889(02)00320-8. [DOI] [PubMed] [Google Scholar]
- 9.Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25(3):387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 10.Dinarello CA. The IL-1 family and inflammatory diseases. Clin Exp Rheumatol. 2002;20(5 Suppl 27):S1–13. [PubMed] [Google Scholar]
- 11.Wang D, Dubois RN. Prostaglandins and cancer. Gut. 2006;55(1):115–22. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagai N, Tian X, Mukai K, Hirata E, Kusuda T, Shiroyama Y, et al. Overexpression of cyclooxygenase-2 protein and its relationship to apoptosis in cervical carcinoma treated with neoadjuvant chemotherapy. Int J Mol Med. 2003;12(5):709–14. [PubMed] [Google Scholar]
- 13.Saikawa Y, Sugiura T, Toriumi F, Kubota T, Suganuma K, Isshiki S, et al. Cyclooxygenase-2 gene induction causes CDDP resistance in colon cancer cell line, HCT-15. Anticancer Res. 2004;24(5A):2723–8. [PubMed] [Google Scholar]
- 14.Trifan OC, Hla T. Cyclooxygenase-2 modulates cellular growth and promotes tumorigenesis. J Cell Mol Med. 2003;7(3):207–22. doi: 10.1111/j.1582-4934.2003.tb00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau L, Hansford LM, Cheng LS, Hang M, Baruchel S, Kaplan DR, et al. Cyclooxygenase inhibitors modulate the p53/HDM2 pathway and enhance chemotherapy-induced apoptosis in neuroblastoma. Oncogene. 2007;26(13):1920–31. doi: 10.1038/sj.onc.1209981. [DOI] [PubMed] [Google Scholar]
- 16.Zatelli MC, Luchin A, Piccin D, Tagliati F, Bottoni A, Vignali C, et al. Cyclooxygenase-2 inhibitors reverse chemoresistance phenotype in medullary thyroid carcinoma by a permeability glycoprotein-mediated mechanism. J Clin Endocrinol Metab. 2005;90(10):5754–60. doi: 10.1210/jc.2005-1362. [DOI] [PubMed] [Google Scholar]
- 17.Kokawa A, Kondo H, Gotoda T, Ono H, Saito D, Nakadaira S, et al. Increased expression of cyclooxygenase-2 in human pancreatic neoplasms and potential for chemoprevention by cyclooxygenase inhibitors. Cancer. 2001;91(2):333–8. doi: 10.1002/1097-0142(20010115)91:2<333::aid-cncr1006>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 18.Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, et al. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999;59(5):987–90. [PubMed] [Google Scholar]
- 19.Juuti A, Louhimo J, Nordling S, Ristimaki A, Haglund C. Cyclooxygenase-2 expression correlates with poor prognosis in pancreatic cancer. J Clin Pathol. 2006;59(4):382–6. doi: 10.1136/jcp.2005.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eibl G, Takata Y, Boros LG, Liu J, Okada Y, Reber HA, et al. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65(3):982–90. [PubMed] [Google Scholar]
- 21.Funahashi H, Satake M, Dawson D, Huynh NA, Reber HA, Hines OJ, et al. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67(15):7068–71. doi: 10.1158/0008-5472.CAN-07-0970. [DOI] [PubMed] [Google Scholar]
- 22.Sipos B, Moser S, Kalthoff H, Torok V, Lohr M, Kloppel G. A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform. Virchows Arch. 2003;442(5):444–52. doi: 10.1007/s00428-003-0784-4. [DOI] [PubMed] [Google Scholar]
- 23.Larrick JW, Fischer DG, Anderson SJ, Koren HS. Characterization of a human macrophage-like cell line stimulated in vitro: a model of macrophage functions. J Immunol. 1980;125(1):6–12. [PubMed] [Google Scholar]
- 24.Shin YH, Lee GW, Son KN, Lee SM, Kang CJ, Kwon BS, et al. Promoter analysis of human CC chemokine CCL23 gene in U937 monocytoid cells. Biochim Biophys Acta. 2007;1769(3):204–8. doi: 10.1016/j.bbaexp.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 25.Eibl G, Bruemmer D, Okada Y, Duffy JP, Law RE, Reber HA, et al. PGE(2) is generated by specific COX-2 activity and increases VEGF production in COX-2-expressing human pancreatic cancer cells. Biochem Biophys Res Commun. 2003;306(4):887–97. doi: 10.1016/s0006-291x(03)01079-9. [DOI] [PubMed] [Google Scholar]
- 26.Okada Y, Eibl G, Guha S, Duffy JP, Reber HA, Hines OJ. Nerve growth factor stimulates MMP-2 expression and activity and increases invasion by human pancreatic cancer cells. Clin Exp Metastasis. 2004;21(4):285–92. doi: 10.1023/b:clin.0000046131.24625.54. [DOI] [PubMed] [Google Scholar]
- 27.Sawai H, Okada Y, Kazanjian K, Kim J, Hasan S, Hines OJ, et al. The G691S RET polymorphism increases glial cell line-derived neurotrophic factor-induced pancreatic cancer cell invasion by amplifying mitogen-activated protein kinase signaling. Cancer Res. 2005;65(24):11536–44. doi: 10.1158/0008-5472.CAN-05-2843. [DOI] [PubMed] [Google Scholar]
- 28.Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell. 2003;11(2):293–302. doi: 10.1016/s1097-2765(03)00053-4. [DOI] [PubMed] [Google Scholar]
- 29.Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN. Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol. 2006;38(10):1654–61. doi: 10.1016/j.biocel.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 30.Muerkoster SS, Lust J, Arlt A, Hasler R, Witt M, Sebens T, et al. Acquired chemoresistance in pancreatic carcinoma cells: induced secretion of IL-1beta and NO lead to inactivation of caspases. Oncogene. 2006;25(28):3973–81. doi: 10.1038/sj.onc.1209423. [DOI] [PubMed] [Google Scholar]
- 31.Covey JM, Jaxel C, Kohn KW, Pommier Y. Protein-linked DNA strand breaks induced in mammalian cells by camptothecin, an inhibitor of topoisomerase I. Cancer Res. 1989;49(18):5016–22. [PubMed] [Google Scholar]
- 32.Sun FX, Tohgo A, Bouvet M, Yagi S, Nassirpour R, Moossa AR, et al. Efficacy of camptothecin analog DX-8951f (Exatecan Mesylate) on human pancreatic cancer in an orthotopic metastatic model. Cancer Res. 2003;63(1):80–5. [PubMed] [Google Scholar]
- 33.Abou-Alfa GK, Letourneau R, Harker G, Modiano M, Hurwitz H, Tchekmedyian NS, et al. Randomized phase III study of exatecan and gemcitabine compared with gemcitabine alone in untreated advanced pancreatic cancer. J Clin Oncol. 2006;24(27):4441–7. doi: 10.1200/JCO.2006.07.0201. [DOI] [PubMed] [Google Scholar]
- 34.Stathopoulos GP, Syrigos K, Aravantinos G, Polyzos A, Papakotoulas P, Fountzilas G, et al. A multicenter phase III trial comparing irinotecan-gemcitabine (IG) with gemcitabine (G) monotherapy as first-line treatment in patients with locally advanced or metastatic pancreatic cancer. Br J Cancer. 2006;95(5):587–92. doi: 10.1038/sj.bjc.6603301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buchler P, Reber HA, Buchler M, Shrinkante S, Buchler MW, Friess H, et al. Hypoxia-inducible factor 1 regulates vascular endothelial growth factor expression in human pancreatic cancer. Pancreas. 2003;26(1):56–64. doi: 10.1097/00006676-200301000-00010. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee S, Zhang Y, Wang Z, Che M, Chiao PJ, Abbruzzese JL, et al. In vitro and in vivo molecular evidence of genistein action in augmenting the efficacy of cisplatin in pancreatic cancer. Int J Cancer. 2007;120(4):906–17. doi: 10.1002/ijc.22332. [DOI] [PubMed] [Google Scholar]
- 37.El-Rayes BF, Ali S, Ali IF, Philip PA, Abbruzzese J, Sarkar FH. Potentiation of the effect of erlotinib by genistein in pancreatic cancer: the role of Akt and nuclear factor-kappaB. Cancer Res. 2006;66(21):10553–9. doi: 10.1158/0008-5472.CAN-06-2333. [DOI] [PubMed] [Google Scholar]
- 38.Banerjee S, Zhang Y, Ali S, Bhuiyan M, Wang Z, Chiao PJ, et al. Molecular evidence for increased antitumor activity of gemcitabine by genistein in vitro and in vivo using an orthotopic model of pancreatic cancer. Cancer Res. 2005;65(19):9064–72. doi: 10.1158/0008-5472.CAN-05-1330. [DOI] [PubMed] [Google Scholar]
- 39.Garrelds IM, van Hal PT, Haakmat RC, Hoogsteden HC, Saxena PR, Zijlstra FJ. Time dependent production of cytokines and eicosanoids by human monocytic leukaemia U937 cells; effects of glucocorticosteroids. Mediators Inflamm. 1999;8(4–5):229–35. doi: 10.1080/09629359990397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molina-Holgado E, Ortiz S, Molina-Holgado F, Guaza C. Induction of COX-2 and PGE(2) biosynthesis by IL-1beta is mediated by PKC and mitogen-activated protein kinases in murine astrocytes. Br J Pharmacol. 2000;131(1):152–9. doi: 10.1038/sj.bjp.0703557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan XM, Wong BC, Lin MC, Cho CH, Wang WP, Kung HF, et al. Interleukin-1beta induces cyclo-oxygenase-2 expression in gastric cancer cells by the p38 and p44/42 mitogen-activated protein kinase signaling pathways. J Gastroenterol Hepatol. 2001;16(10):1098–104. doi: 10.1046/j.1440-1746.2001.02593.x. [DOI] [PubMed] [Google Scholar]
- 42.Chen P, Cai Y, Yang ZG, Zhou R, Zhang GS, Domann F, et al. Involvement of PKC, p38 MAPK and AP-2 in IL-1beta-induced expression of cyclooxygenase-2 in human pulmonary epithelial cells. Respirology. 2006;11(1):18–23. doi: 10.1111/j.1440-1843.2006.00779.x. [DOI] [PubMed] [Google Scholar]
- 43.Chen KH, Weng MS, Lin JK. Tangeretin suppresses IL-1beta-induced cyclooxygenase (COX)-2 expression through inhibition of p38 MAPK, JNK, and AKT activation in human lung carcinoma cells. Biochem Pharmacol. 2007;73(2):215–27. doi: 10.1016/j.bcp.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 44.MacGillivray MK, Cruz TF, McCulloch CA. The recruitment of the interleukin-1 (IL-1) receptor-associated kinase (IRAK) into focal adhesion complexes is required for IL-1beta -induced ERK activation. J Biol Chem. 2000;275(31):23509–15. doi: 10.1074/jbc.M003186200. [DOI] [PubMed] [Google Scholar]