

Summary
During the last 25 years, neuropathological, biochemical, genetic, cell biological and even therapeutic studies in humans have all supported the hypothesis that the gradual cerebral accumulation of soluble and insoluble assemblies of the amyloid β-protein (Aβ) in limbic and association cortices triggers a cascade of biochemical and cellular alterations that produce the clinical phenotype of Alzheimer’s disease (AD). The reasons for elevated cortical Aβ42 levels in most patients with typical, late-onset AD are unknown, but based on recent work, these could turn out to include augmented neuronal release of Aβ during some kinds of synaptic activity. Elevated levels of soluble Aβ42 monomers enable formation of soluble oligomers that can diffuse into synaptic clefts. We have identified certain APP-expressing cultured cell lines that form low-n oligomers intracellularly and release a portion of them into the medium. We find that these naturally secreted soluble oligomers -- at picomolar concentrations -- can disrupt hippocampal LTP in slices and in vivo and can also impair the memory of a complex learned behavior in rats. Aβ trimers appear to be more potent in disrupting LTP than are dimers. The cell-derived oligomers also decrease dendritic spine density in organotypic hippocampal slice cultures, and this decrease can be prevented by administration of Aβ antibodies or small-molecule modulators of Aβ aggregation. This therapeutic progress has been accompanied by advances in imaging the Aβ deposits non-invasively in humans. A new diagnostic-therapeutic paradigm to successfully address AD and its harbinger, mild cognitive impairment-amnestic type, is emerging.
Introduction
During most of the 20th century, neurodegenerative diseases remained among the most enigmatic disorders of medicine. The scientific study of these conditions was descriptive in nature, detailing the clinical and neuropathological phenotypes associated with various diseases, but etiologies and pathogenic mechanisms remained obscure. Beginning in the 1970s, advances in two principal areas – biochemical pathology and molecular genetics –combined to yield powerful clues to the molecular underpinnings of several previously “idiopathic” brain disorders. Among the classical neurodegenerative diseases, perhaps the most rapid progress occurred in research on Alzheimer’s disease (AD). In disorders like Huntington’s disease, amyotrophic lateral sclerosis and even Parkinson’s disease, unbiased genetic screens, linkage analysis and positional cloning have identified causative genes that subsequently allowed the formulation of specific biochemical hypotheses. In sharp contrast, modern research on AD developed in the opposite order: the identification of the protein subunits of the classical brain lesions guided geneticists to disease-inducing genes, for example, APP, apolipoprotein E and tau. Thus, a biochemical hypothesis of disease - that AD is a progressive cerebral amyloidosis caused by the aggregation of the amyloid β-protein (Aβ) - preceded and enabled the discovery of etiologies.
As progress in deciphering genotype-to-phenotype relationships in AD accelerated during the last two decades, it became apparent that the key challenge for understanding and ultimately treating AD was to focus not on what was killing neurons over the course of the disease but rather on what was interfering subtly and intermittently with episodic declarative memory well before widespread neurodegeneration had occurred [53]. In other words, one wishes to understand the factors underlying early synaptic dysfunction in the hippocampus and then attempt to neutralize these as soon as feasible, perhaps even before a definitive diagnosis of AD can be made. This steady movement of the field toward ever-earlier stages of the disorder is exemplified by the recognition and intensive study of minimal cognitive impairment–amnestic type (MCI; [46]). And yet patients who die with a diagnosis of MCI have been found to already have a histopathology essentially indistinguishable from classical AD [48]. Therefore, even earlier phases of this continuum are likely to become recognized, and these might show milder histopathology and might have biochemically, but not yet microscopically, detectable Aβ species that mediate synaptic dysfunction.
The IPSEN symposium for which this volume serves as a record focused on bringing together investigators at the forefront of elucidating the structure and function of hippocampal synapses with investigators focused on understanding how early assemblies of Aβ may compromise some of these synapses. This chapter will summarize some of the observations and discoveries made by the author and his colleagues over several years that have the goal of identifying the earliest synaptotoxic molecules in Alzheimer’s disease – and neutralizing them.
Moving from synthetic Aβ peptides to naturally secreted Aβ assemblies
A wealth of data from many laboratories now supports the once controversial hypothesis that the accumulation and aggregation of Aβ initiates a complex cascade of molecular and cellular changes that gradually leads to the clinical features of MCI-amnestic type and then frank Alzheimer’s disease.[20, 21, 52]. As a result, understanding precisely how Aβ accumulation and assembly compromise synaptic structure and function has become the centerpiece of therapeutically oriented research on the disease.
A great many studies have been conducted using synthetic Aβ peptides of either 40 or 42 amino acids, mimicking the two most common lengths of Aβ found in normal human brain and in the cortical and vascular amyloid deposits of AD patients. But naturally generated Aβ peptides in brain, cerebrospinal fluid (CSF) or the media of cultured cells are considerably more heterogeneous in length (see, e.g [5, 19, 25, 47, 50, 54, 56, 64, 70]. More importantly, almost all studies of synthetic Aβ peptides have employed concentrations upwards of 1 uM, often 10–40 uM, because the critical concentration allowing relatively rapid assembly of synthetic Aβ40 (the most commonly used peptide) into amyloid fibrils is in the high nanomolar range or greater. Also, synthetic Aβ peptides are often solublized, at least initially, in highly non-physiological solvents (acetonitrile, trifluoroacetic acid, sodium hydroxide, etc.), since these allow the hydrophobic, relatively insoluble peptide to be dissolved and used experimentally.
Many different types of assembly forms of synthetic Aβ, including amyloid fibrils, protofibrils (PF), annular structures, paranuclei, Aβ-derived diffusible ligands and globulomers, have been described over the last two decades (for reviews, see [6, 60]). Even some of the smaller synthetic aggregates do not fulfill the definition of soluble oligomers: Aβ assemblies that are not pelleted from physiological fluids by high-speed centrifugation. For example, protofibrils are intermediates that were observed in the course of studying the fibrillization of synthetic Aβ [22, 23, 66]. They are flexible structures that can continue to polymerize in vitro to form amyloid fibrils or can de-polymerize to lower-order species. Protofibrils are narrower than bona-fide amyloid fibrils (~4–5 nm versus 8–10 nm). Ultrastructural analyses of synthetic protofibril preparations by electron microscopy (EM) and atomic force microscopy (AFM) have revealed both straight and curved assemblies up to 150 nm in length. Synthetic Aβ protofibrils have been shown to contain substantial β-sheet structure, as they are able to bind Congo red or Thioflavin T in ordered fashion.
Annular assemblies of synthetic Aβ are donut-like structures with an outer diameter of 8–12 nm and an inner diameter of 2.0–2.5 nm that are distinct from protofibrils by AFM and EM [2, 32]. Smaller oligomeric species of synthetic Aβ than protofibrils and annuli have been observed, depending on how synthetic Aβ is prepared and incubated, and some have been designated Aβ-derived diffusible ligands (ADDLs; [31]). Apparent ADDL-like oligomeric assemblies have been isolated from postmortem AD brains and their presence correlated with memory loss [17]. In separate work, chemical stabilization of synthetic Aβ42 assembly intermediates has revealed an apparent hexamer periodicity, with hexamer, dodecamer, and octadecamer structures observed [1]
In striking contrast to the properties of these various synthetic assemblies, Aβ peptides that are generated in vivo by humans and lower mammals or by cultured cells are diverse with regard to their N- and C-termini, occur naturally in extracellular fluids at low to sub-nanomolar concentrations [18, 43, 51, 65] and can begin to assemble into metastable dimers, trimers and higher oligomers while still at low nanomolar levels.[47, 67]. Dimeric, trimeric and apparently tetrameric soluble oligomers have been described in cultured cells [47, 67], and SDS-stable oligomers of varying sizes have also been detected by Western blotting in APP transgenic mouse brain and human brain [14, 16, 27, 33, 40, 49]. Such natural (i.e., non-synthetic) Aβ oligomers can be resistant not only to SDS but also to chaotropic salts like guanidine hydrochloride and to the Aβ-degrading protease IDE (insulin-degrading enzyme), which can only efficiently digest monomeric Aβ [65]. Aβ oligomers produced by cultured cells could be related to the recently described Aβ*56 species, which represents a soluble, SDS-stable dodecamer found in the brains of at least some APP transgenic mouse lines[33]. Like the Aβ oligomers produced from cultured cells [65], Aβ*56 can disrupt synaptic function and thus affect memory [33]. Whether Aβ*56, the cell-derived dimers and trimers [47] and other soluble oligomers observed in biological systems represent stable assemblies of solely Aβ under native conditions or whether such small oligomeric assemblies stably associate with one or more “carrier” proteins in vivo is currently unclear. Upon further study, Aβ*56 and Aβ trimers secreted by cultured cells could turn out to share common synaptotoxic properties.
Naturally secreted Aβ oligomers abrogate hippocampal synaptic plasticity
In collaboration with the laboratory of Michael Rowan, we have taken advantage of our discovery that Chinese hamster ovary (CHO) cells stably expressing the AD-causing Val717Phe mutation in APP secrete soluble oligomers detectable on SDS gels as dimers, trimers and tetramers [47] to conduct a series of studies defining the electrophysiological activities of these assemblies. As mentioned above, we view these cell-derived, low-n oligomers as having several advantages over synthetic Aβ aggregates, most notably their natural production by these and other cells at low- to sub-nanomolar concentrations. Importantly, the CHO-derived oligomers are entirely soluble: centrifuging the conditioned medium at >100,000 g for >1 hour still leaves all of the monomers and oligomers in the supernatant, with retained neurobiological activity. We have confirmed the identity of these species as bona fide Ab oligomers using radiosequencing, immunoprecipitation by both N-and C-terminal-specific Aβ monoclonal antibodies, and non-denaturing size exclusion chromatography (SEC). The oligomers can be readily detect in CHO cells stably expressing wild-type human APP [47], and their disease relevance is supported by our finding that expressing either an APP mutation, a presenilin 1 mutation or a presenilin 2 mutation in CHO cells elevates Aβ42 levels in the media and increases the levels of the SDS-stable dimers and trimers [71]. We are currently attempting to determine the atomic masses of the oligomers by mass spectrometry to establish their bonding structure; we have already confirmed the mass of the secreted 4 kDa species in the medium as human Aβ monomer, but larger quantities are being prepared to try to obtain the masses of the dimers and trimers.
Dominic Walsh (now at University College Dublin), who led many of our studies characterizing the biochemical and neurobiological properties of the cell-derived oligomers while he was a fellow in my laboratory, established our very productive collaboration with Michael Rowan and his fellow, Igor Kluybin, at Trinity College Dublin. We conducted experiments together to determine whether the monomers and oligomers present in the conditioned medium of the APP V717F-expressing CHO cells (called 7PA2 cells) can interfere with basal synaptic transmission and/or long-term potentiation (LTP) in the hippocampus in vivo. Through a cannula implanted in the lateral ventricle of adult rats, we administered tiny amounts (usually 1.5 – 5.0 ul) of the straight conditioned medium collected from the 7PA2 cells. Basal synaptic transmission was unaffected by the medium, but CA1 field recordings after high-frequency stimulation (HFS) of the CA3 Schaeffer collateral-to-CA1 pathway revealed a highly consistent failure to maintain an LTP response in the presence of the 7PA2 medium but not with the medium of untransfected CHO cells (called CHO-cells;[65]). In the presence of the 7PA2 medium, synaptic potentiation would initially be induced by the HFS but waned over the next 1–3 hours, so that LTP was not maintained. We next performed a number of experiments that indicated that the soluble Aβ monomers in the 7PA2 medium were not responsible for the failure to maintain LTP but the soluble oligomers were. For example, we took advantage of the ability of IDE to degrade the monomers but leave the oligomers (which are surprisingly stable) behind, and this preparation still inhibited hippocampal LTP in vivo. Conversely, injecting the medium after treating the 7PA2 cells with a γ-secretase inhibitor at low doses that reduced the monomers by just ~40% but thereby decreased dimer/trimer levels by ~90% led to a normal LTP response. We concluded that secreted, low-n oligomers of human Aβ at picomolar concentrations and in the absence of monomers as well as higher order aggregates (protofibrils, fibrils) could potently inhibit the maintenance of LTP in the mammalian hippocampus.
In recent work, our laboratory has extended our electrophysiological findings to hippocampal slices in vitro. We confirmed the effect of the 7PA2 medium in inhibiting LTP in slices from wild-type mice [62]. Further, the cell-derived oligomers blocked a chemically induced form of LTP caused by stimulating dissociated mouse hippocampal neurons with picrotoxin and glycine in the absence of added magnesium [63]. The use of the chemically induced LTP paradigm (chem-LTP) on neuronal cultures allowed us to measure the biochemical effects of the soluble oligomers on activation of several different signaling kinases. We found that of five kinases we evaluated, three (ERK MAPK CaMKII and Akt/PKB) had clearly reduced activation upon chem-LTP, whereas two others (PKA and PKC) were unaffected. These data begin to reveal some of the signaling pathways that can be interrupted by naturally secreted oligomers, indicating that there is specificity to the hippocampal response and suggesting that there are discrete receptor-mediated pathways through which soluble oligomers act.
We have also developed protocols for separating the secreted oligomers by SEC under non-denaturing conditions [29, 62, 69]. When as many as 100 SEC fractions of the 7PA2 conditioned medium were obtained and then tested separately on mouse hippocampal slices, we observed that fractions rich in trimers inhibited LTP even more potently than did fractions enriched principally in dimers; agina, monomers were without effect [62]. These results suggest again the existence of a level of molecular specificity in the interactions of Aβ oligomers with neuronal targets. With Dominic Walsh and his colleagues, we are now attempting to purifiy the oligomers from the CHO cell medium to homogeneity so that they might be labeled and used in ligand binding studies (e.g., by autoradiography) to try to identify the cognate receptors for oligomers but not monomers.
Cell-derived oligomers interfere with the memory of a complex learned behavior
In collaboration with James Cleary and Karen Ashe at the University of Minnesota, we have been able to demonstrate significant cognitive deficits in a complex lever pressing task in adult rats that are directly attributable to a naturally secreted assembly form of Aβ [9]. The active Aβ species were the soluble oligomers, not monomers and in the absence of protofibrils and fibrils, and the oligomer effects were characterized by rapid onset, high potency and transience. These combined biochemical and cognitive analyses provided the first direct behavioral data supporting the emerging hypothesis that diffusible oligomers of Aβ are responsible for important components of neuronal dysfunction leading up to or associated with AD (reviewed in [28, 53, 68]. The rapid onset and transience of the soluble oligomer effects we saw in the rats are indicative of a pathophysiological action, similar to that seen with amnestic drugs such as scopolamine. Such pathophysiological activities can occur independently of structural neuronal injury and, indeed, our experimental paradigm does not suggest a role for any neurodegenerative processes in the deleterious cognitive effects produced transiently by low picomolar quantities of soluble Aβ oligomers.
In studies reported prior to the Cleary et al. [9] paper, intracerebral injections of synthetic Aβ that included mixtures of Aβ fibrils, protofibrils, oligomers and monomers in indeterminate proportions exerted deleterious effects on learned behavior in rats [8, 15, 45]. However, the deficits were reported to develop over weeks and, once present, they persisted or worsened even without further injections. Because intracerebral injections are usually made during stereotaxic surgery (rather than long after such surgery, as in our paradigm), any immediate acute effects of the injectate on learning or memory typically go unstudied. Two studies attempted to address potential acute effects of very high concentrations of synthetic Aβ species. In one study, increased maze entry errors were observed after i.c.v. injections of 2.0 μl of 1 mM synthetic Aβ (1–40) that were given immediately before maze testing [59]. In another study, no effect on intrahippocampal injections of 0.5 μlof 1 mM synthetic Aβ (1–40) that were administered 30 minutes before testing were observed in a radial arm maze test [38]. The concentrations and total amounts of synthetic Aβ administered were several orders of magnitude higher than those of the natural, cell-secreted oligomers used in our study [9]. Also, the duration between administration and testing was different. Because the assembly sizes of the synthetic Aβ that was injected were not defined in either study, the specific forms of Aβ being tested could not be deduced, in contrast to the present work.
Our work may provide an explanation for the observation that subtle brain dysfunction can be detected in certain individuals who are genetically at risk for AD but remain neurologically stable for many years before the expected onset of disease [4, 57]. In addition, our linkage of acute behavioral deficits with soluble oligomers provides a mechanistic explanation for the rapid restoration of cognitive function that followed administration of anti-Aβ antibodies in behaviorally impaired APP transgenic mice [12, 30].
Cell-derived oligomers decrease dendritic spine density in hippocampus by an NMDA-dependent signaling pathway
Our next approach to deciphering the synaptic effects of natural Aβ oligomers was to ask whether they can induce structural alterations of synapses in association with the clear functional deficits described in the previous two sections above. We exposed organotypic rat hippocampal slice cultures that had been biolistically transfected with EGFP to subnanomolar concentrations of SEC-separated 7PA2 cell Aβ monomers or dimers/trimers for periods varying from 1 to 15 days. We observed a marked decrease in dendritic spine density in EGFP-positive pyramidal neurons that reached ~60% loss after 15 days of exposure [55]. Electrophysiological experiments confirmed that the decreased spine density reflected a loss of excitatory synapses. Miniature EPSCs were measured from neurons in hippocampal slices treated for 10–15 days. mEPSC amplitude, which reflects the postsynaptic AMPA-type glutamate receptor (AMPAR)-mediated response to the release of a single vesicle of glutamate, was slightly but significantly reduced in oligomer-treated neurons compared to control-medium treated neurons. Moreover, as expected from the loss of dendritic spines, the inter-mEPSC interval was significantly increased (195 +/− 5 ms vs. 85 +/− 3 ms), corresponding to a ~50% reduction in mEPSC frequency [55].
The strong decrease in dendritic spine density was reversible when the Aβ oligomer-rich medium (which had been applied for 10 days) was exchanged for normal slice medium for 5 days; spine density returned to almost normal levels [55]. Spine loss was also fully prevented by co-application to the slices of an N-terminal monoclonal antibody to Aβ (6E10) together with the soluble oligomers; denatured (boiled) 6E10 had no rescue effect. We also examined scyllo-inositol (AZD-103), a myo-inositol steroisomer shown by Joanne McLaurin and colleagues to decrease Aβ plaque number and size and prevent behavioral deficits in an APP transgenic mouse line [39]. At 5 uM concentrations, scyllo-inositol co-administered with the soluble oligomers fully prevented the spine loss, whereas a known inactive stereoisomer, chiro-inositol, did nothing.
Acute application of synthetic Aβ can trigger a rapid reduction of NMDAR-dependent currents in dissociated cortical neurons that is thought to result from Aβ-mediated activation of nicotinic acetylcholine receptors (nAchRs; [58]). To determine if the same signaling pathway was responsible for the Aβ oligomer-mediated spine loss, we examined the ability of NMDAR and nAchR antagonists to mimic or block the effects of secreted Aβ. Chronic blockade of nAchRs by 10-day exposure to the irreversible antagonist α-Bungarotoxin had no effect on the spine density of control neurons and did not affect the degree of spine loss produced by oligomer application. Conversely, although 10 days of application of the NMDAR antagonist CPP (20 μM) alone had no effect on spine density, it completely prevented the spine loss normally seen with Aβ oligomer incubation, indicating that NMDAR-activity is required for Aβ-mediated spine loss [55].
To determine if Aβ oligomers reduce NMDAR-mediated synaptic Ca influx in hippocampal pyramidal neurons, 2-photon laser photoactivation (2PLP) of caged (MNI)-glutamate was used to stimulate the postsynaptic terminal on a visualized spine while evoked calcium transients were measured in the spine head with 2P laser scanning microscopy [3]. Whole-cell recordings were obtained from CA1 pyramidal neurons in acute hippocampal slices, and cells were filled through the patch pipette with the Ca-sensitive, green-fluorescing fluorophore Fluo-5F and the Ca-independent, red-fluorescing fluorophore Alexa Fluor-594. Red fluorescence was used to visualize morphology and to select spines within the proximal 150 μm of an apical dendrite for analysis. Uncaging-evoked fluorescence transients were monitored in the spine head and adjacent dendrite while uncaging-evoked postsynaptic potentials (uEPSPs) were recorded at the soma. Fluorescence transients were quantified relative to maximal green fluorescence at saturating levels of Ca (ΔGuEPSP/Gsat), a measure that, under our recording conditions, is linearly proportional to evoked changes in Ca (Δ[Ca]uEPSP; [3]). We found that Δ[Ca]uEPSP was smaller in the presence of Aβ oligomers (ΔGuEPSP/Gsat=7.4 ± 0.5 %, 21/5 spines/cells) than in control conditions (ΔGuEPSP/Gsat= 10.2 ± 1.1%, 20/7 spines/cells, p<0.05). In contrast, Δ[Ca]uEPSP in the presence of Aβ monomers (ΔGuEPSP/Gsat= 9.9 ± 1.0%, 19 spines/6 cells) was the same as in control conditions. Thus, exposure to picomolar levels of soluble Aβ oligomers acutely reduces but does not abolish Δ[Ca]uEPSP, consistent with a partial reduction of NMDAR-mediated Ca influx into active spines.
Partial blockade of NMDARs and reduced Ca2+ influx through NMDARs favor the induction of long-term depression (LTD) via a calcineurin-dependent pathway [10, 41]. LTD induction is accompanied by a shrinkage of dendritic spines that is mediated by regulation of the actin-depolymerization factor cofilin [55, 72] at a conserved serine (position 3) that can be phosphorylated by LIM-kinase. We found that neurons transfected with a plasmid encoding cofilin S3D, in which serine 3 is replaced by the phosphomimetic amino acid aspartate, rendering cofilin constitutively inactive, displayed a net increase in dendritic spine density. Expression of this construct also prevented the loss of dendritic spines normally seen following a 10-day exposure to Aβ oligomers. To examine whether Aβ oligomers mediate spine loss through activation of calcineurin, we examined the ability of FK506 to block Aβ oligomer-induced spine loss. Incubation in FK506 (1 uM) alone for 24 hr had no effect on spine density. However, co-administration of FK506 with Aβ oligomers fully prevented spine loss [55].
Conclusions
Through a series of systematic studies of soluble oligomers of human Aβ secreted by cultured cells, we have documented that low-n oligomers – but not monomers from the same source and at higher concentrations – can inhibit LTP without affecting basal synaptic transmission, can reversibly alter the structure of excitatory synapses by decreasing spines, and can interfere with the memory of a learned behavior in healthy adult rats. We interpret these data to signify that small diffusible oligomers impair both synaptic function and synaptic structure on glutamatergic neurons in the hippocampus. Therapeutically, we show that oligomer-mediated spine loss is prevented by antibodies to Aβ and a small molecule inhibitor of Aβ aggregation. We had previously reported that both active and passive immunotherapy of normal adult rats protect these animals form the LTP-inhibiting effects of soluble Aβ oligomers administered intracerebroventricularly [29]. Mechanistically, we find that dendritic spine reduction requires activity of a signaling cascade involving NMDARs, calcineurin, and cofilin. These results suggest that Aβ oligomers shift the activation of NMDAR-dependent signaling cascades towards pathways involved in the induction of LTD. We propose that chronic activation of these pathways by soluble Aβ oligomers found in the brains of patients with AD contributes to the pathogenesis of the disease and may underlie the synapse loss in hippocampus that occurs early in the disease process and is a key quantitative correlate of degree of cognitive impairment in AD patients [11, 36, 61].
Overall, our studies with soluble natural oligomers suggest a model (Fig. 1) in which exposure to Aβ oligomers mimics a state of partial NMDAR blockade, either by reducing NMDAR activation, reducing NMDAR-dependent Ca influx, or enhancing NMDAR-dependent activation of calcineurin. We propose that these effects promote the LTD-inducing mode and inhibit the LTP-inducing mode of NMDAR-dependent signaling. Since LTP induction promotes spine enlargement and the growth of new spines whereas LTD induction promotes spine shrinkage and retraction [13, 35, 37, 42, 72], this oligomer-induced imbalance would promote the progressive loss of dendritic spines and glutamatergic synapses. Furthermore, a previous study reported that FK506 prevented the ability of synthetic Aβ to inhibit LTP in hippocampus [7], suggesting that the inhibition of LTP by Aβ oligomers reflects a shift in the LTP/LTD balance rather than a direct blockade of LTP-inducing pathways.
Figure 1.
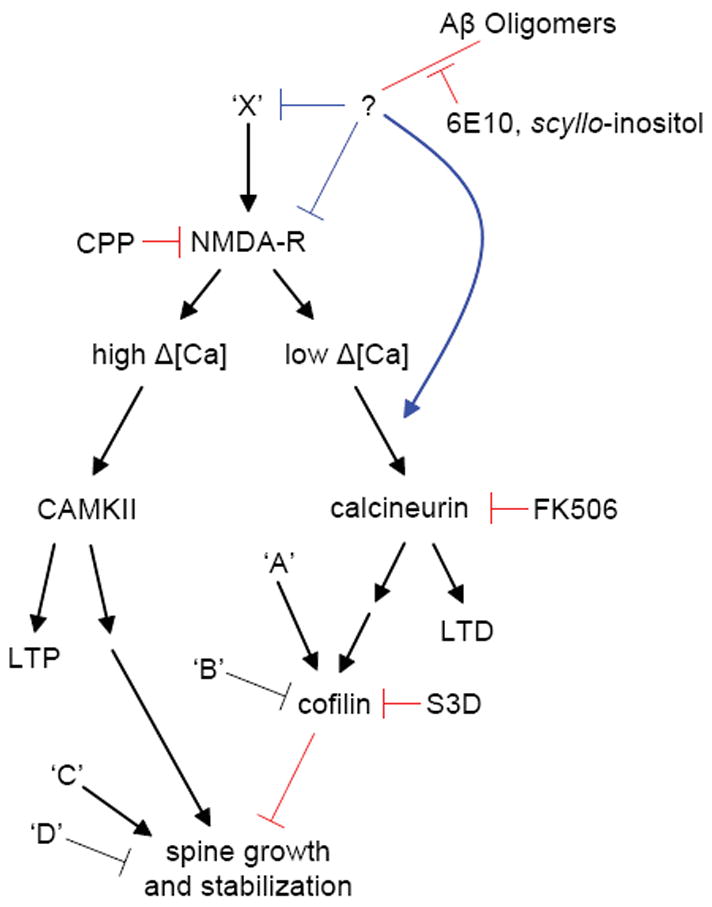
Proposed pathways that regulate spine density and that are affected by Aβ oligomers, based on the results of our results. Ca2+ influx through synaptic NMDARs can activate at least two pathways that regulate spine density. On the left-hand side, high levels of Ca2+ accumulation, such as those reached during tetanic or suprathreshold synaptic stimulation, induce LTP via a CAMKII-dependent pathway (reviewed in [44]). LTP-inducing stimuli also trigger the enlargement of dendritic spines and growth of new spines in a NMDAR- and CAMKII-dependent manner [13, 26, 35, 37, 42]. Introduction of active CAMKII in neurons is sufficient to induce new spine growth [26]. In the right-hand side pathway, low levels of Ca2+ accumulation, such as those reached during low-frequency subthreshold stimulation, induce LTD through a calcineurin-dependent pathway (reviewed in [34]. LTD-inducing stimuli also lead to spine shrinkage via an NMDAR/calcineurin/cofilin-dependent pathway and spine retraction through an NMDAR-dependent pathway [42, 72]. The calcineurin and cofilin dependence of LTD-associated spine retraction have not been examined. In this model, full block of NMDARs interrupts both pathways, leading to no net spine loss. Partial block of NMDARs favors activation of the right-hand pathway, LTD induction, and loss of spines. In addition, multiple factors (A, B, C, and D) act independently of NMDARs, CAMKII, and calcineurin to regulate cofilin and spine density. We find that soluble Aβ oligomers decrease spine density in an NMDAR/calcineurin/cofilin-dependent manner, consistent with activation of the pathway shown on the right. Aβ oligomers reduce NMDAR-dependent Ca2+ transients, possibly shifting stimuli that normally activate the left-hand pathway to instead activate those on the right. This activation might occur through direct interaction of Aβ with NMDARs or by first activating unknown factors (X) that may lead to inhibition of NMDAR-mediated synaptic Ca2+ influx. Aβ may also facilitate NMDAR-dependent activation of calcineurin via additional pathways. Blue lines indicate levels at which soluble Aβ oligomers may modulate the pathway and red lines indicate elements of the pathway tested in this study.
Our proposed model is largely consistent with a recent report that also concludes that Aβ decreases dendritic spine density and alters glutamatergic signaling in CA1 pyramidal neurons in rat hippocampal organotypic slices [24]. Hsieh et al. found that neurons overexpressing human APP have decreased dendritic spine density compared to control neurons. Furthermore, transduction with a C-terminal fragment of APP (β-CTF) decreased AMPAR-mediated synaptic currents. This effect mimicked and partially occluded metabotropic glutamate receptor-induced LTD, involving endocytosis of GluR2-containing AMPARs. Important differences between the two studies may explain why we found no decrease in AMPAR-mediated mEPSCs whereas Hsieh et al. found a ~ 30% reduction in AMPAR EPSCs. For example, Hsieh et al. performed recordings ~ 24 hours after transduction with β-CTF and therefore described effects of short-term exposure to Aβ that may be mechanistically different from the longer-term effects described here. Furthermore, Hsieh et al. found alterations in synaptic transmission in neurons expressing β-CTF but not in neighboring cells, suggesting cell-autonomous or autocrine effects of APP or Aβ. Aβ in their system is generated intracellularly and secreted, so at least some of the observed effects could be attributable to Aβ acting intracellularly, whereas our system involves extracellular application of secreted Aβ. The APP transduction paradigm used by Hsieh et al. does not allow a distinction between the effects of Aβ oligomers and monomers, whereas we are able to ascribe the synaptic changes specifically to soluble oligomers of Aβ. However, despite the differences in the design and analysis performed in the two studies, both support a model in which Aβ perturbs excitatory synapses by enhancing LTD in an activity- and calcineurin-dependent manner.
The next step in this line of research is to translate this approach and the resultant findings to the human disease. We must determine whether soluble Aβ monomers and oligomers isolated directly from the cerebral cortex of typical, late-onset AD cases can induce the same structural and functional deficits and whether they do so by similar signaling mechanisms as summarized above. If this can be achieved, we may be able to conclude that we have identified the “smoking gun” that induces synaptic impairment in the brains of Alzheimer’s patients. In a sense, this next step would be attempting to fulfil one of Koch’s postulates for identifying the causative agent of an idiopathic disease: isolating the putative agent, transmitting it to normal brain tissue (of a rodent, as this cannot be done in humans), and showing that the agent reproduces the key phenotypic features of the disorder. If such experiments are successful, they will strongly validate and encourage the current intensive effort to treat, and perhaps ultimately prevent, AD using agents that lower Aβ in the brain.
Footnotes
This article has also been published in D.J. Selkoe and Y. Christen (eds.), Synaptic plasticity and the mechanism of bAlzheimer’s disease, Heidelberg, Springer Verlag, 2008. It is published in the special issue of Behavioural Brain Research with Springer’s permission.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB. Amyloid β--protein (Aβ) assembly: Aβ 40 and Aβ 42 oligomerize through distinct pathways. Proc Natl Acad Sci USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bitan G, Teplow DB. Preparation of aggregate-free, low molecular weight amyloid-beta for assembly and toxicity assays. Methods Mol Biol. 2005;299:3–9. doi: 10.1385/1-59259-874-9:003. [DOI] [PubMed] [Google Scholar]
- 3.Bloodgood BL, Sabatini BL. Nonlinear regulation of unitary synaptic signals by CaV(2.3) voltage-sensitive calcium channels located in dendritic spines. Neuron. 2007;53:249–260. doi: 10.1016/j.neuron.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 4.Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of β-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caughey B, Lansbur PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 7.Chen F, Gu Y, Hasegawa H, Ruan X, Arawaka S, Fraser P, Westaway D, Mount H, St George-Hyslop P. Presenilin 1 mutations activate gamma 42-secretase but reciprocally inhibit epsilon-secretase cleavage of amyloid precursor protein (APP) and S3-cleavage of notch. Biol Chem. 2002;277:36521–36526. doi: 10.1074/jbc.M205093200. [DOI] [PubMed] [Google Scholar]
- 8.Cleary J, Hittner JM, Semotuk M, Mantyh P, O’Hare E. Beta-amyloid(1–40) effects on behavior and memory. Brain Res. 1995;682:69–74. doi: 10.1016/0006-8993(95)00323-i. [DOI] [PubMed] [Google Scholar]
- 9.Cleary JP, Walsh D, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid β-protein specifically disrupt cognitive function. Nature Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 10.Cummings BJ, Pike CJ, Shankle R, Cotman CW. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging. 1996;17:921–933. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- 11.Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer’s disease. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- 12.Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nature Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 13.Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- 14.Enya M, Morishima-Kawashima M, Yoshimura M, Shinkai Y, Kusui K, Khan K, Games D, Schenk D, Sugihara S, Yamaguchi H, Ihara Y. Appearance of sodium dodecyl sulfate-stable amyloid beta-protein (Abeta) dimer in the cortex during aging. Am J Pathol. 1999;154:271–279. doi: 10.1016/s0002-9440(10)65273-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frautschy SA, Hu W, Kim P, Miller SA, Chu T, Harris-White ME, Cole GM. Phenolic anti-inflammatory antioxidant reversal of Abeta-induced cognitive deficits and neuropathology. Neurobiol Aging. 2001;22:993–1005. doi: 10.1016/s0197-4580(01)00300-1. [DOI] [PubMed] [Google Scholar]
- 16.Funato H, Enya M, Yoshimura M, Morishima-Kawashima M, Ihara Y. Presence of sodium dodecyl sulfate-stable amyloid beta-protein dimers in the hippocampus CA1 not exhibiting neurofibrillary tangle formation. Am J Pathol. 1999;155:23–28. doi: 10.1016/s0002-9440(10)65094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer’s disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gravina SA, Ho L, Eckman CB, Long KE, Otvos LJ, Younkin LH, Suzuki N, Younkin SG. Amyloid β protein (Aβ) in Alzheimer’s disease brain. J Biol Chem. 1995;270:7013–7016. doi: 10.1074/jbc.270.13.7013. [DOI] [PubMed] [Google Scholar]
- 19.Haass C, Schlossmacher M, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski B, Lieberburg I, Koo EH, Schenk D, Teplow D, Selkoe D. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 20.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 21.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 22.Harper JD, Wong SS, Lieber CM, Lansbury PT., Jr Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- 23.Hartley D, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina H, Ihara Y. Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- 26.Jourdain P, Fukunaga K, Muller D. Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J Neurosci. 2003;23:10645–10649. doi: 10.1523/JNEUROSCI.23-33-10645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein WL, Krafft GA, Finch CE. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 29.Klyubin I, Walsh DM, Lemere CA, Cullen WK, Shankar GM, Betts V, Spooner ET, Jiang L, Anwyl R, Selkoe DJ, Rowan MJ. Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo. Nature Med. 2005;11:556–561. doi: 10.1038/nm1234. [DOI] [PubMed] [Google Scholar]
- 30.Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH. Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci. 2002;22:6331–6335. doi: 10.1523/JNEUROSCI.22-15-06331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Iosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfribrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 33.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 34.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 35.Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- 36.Masliah E, Mallory M, Alford M, DeTeresa R, Hansen LA, McKeel DW, Jr, Morris JC. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology. 2001;56:127–129. doi: 10.1212/wnl.56.1.127. [DOI] [PubMed] [Google Scholar]
- 37.Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald MP, Dahl EE, Overmier JB, Mantyh P, Cleary J. Effects of an exogenous beta-amyloid peptide on retention for spatial learning. Behav Neural Biol. 1994;62:60–67. doi: 10.1016/s0163-1047(05)80059-7. [DOI] [PubMed] [Google Scholar]
- 39.McLaurin J, Kierstead ME, Brown ME, Hawkes CA, Lambermon MH, Phinney AL, Darabie AA, Cousins JE, French JE, Lan MF, Chen F, Wong SS, Mount HT, Fraser PE, Westaway D, St George-Hyslop P. Cyclohexanehexol inhibitors of Abeta aggregation prevent and reverse Alzheimer phenotype in a mouse model. Nature Med. 2006;12:801–808. doi: 10.1038/nm1423. [DOI] [PubMed] [Google Scholar]
- 40.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 41.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 42.Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 43.Näslund J, Haroutunian V, Mohs R, Davis K, Davies P, Greengard P, Buxbaum J. Correlation between elevated levels of amyloid β-peptides in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 44.Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann NY Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- 45.O’Hare E, Weldon DT, Mantyh PW, Ghilardi JR, Finke MP, Kuskowski MA, Maggio JE, Shephard RA, Cleary J. Delayed behavioral effects following intrahippocampal injection of aggregated A beta (1–42) Brain Res. 1999;815:1–10. doi: 10.1016/s0006-8993(98)01002-6. [DOI] [PubMed] [Google Scholar]
- 46.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Arch Neurol. 1999;56:303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 47.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydel RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid β-protein into SDS-stable oligomers in cell culture. J Biol Chem. 1995;270:9564–9570. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 48.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 49.Roher AE, Chaney MO, Kuo Y-M, Webster SD, Stine WB, Haverkamp LJ, Woods AS, Cotter RJ, Tuohy JM, Krafft GA, Bonnell BS, Emmerling MR. Morphology and toxicity of Aβ-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J Biol Chem. 1996;271:20631–20635. doi: 10.1074/jbc.271.34.20631. [DOI] [PubMed] [Google Scholar]
- 50.Saido TC, Iwatsubo T, Mann DMA, Shimada H, Ihara Y, Kawashima S. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(p3), in senile plaques. Neuron. 1995;14:457–466. doi: 10.1016/0896-6273(95)90301-1. [DOI] [PubMed] [Google Scholar]
- 51.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid b-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 52.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 53.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 54.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmache MG, Whaley J, Swindlehurst C, McCormack R, Wolfert R, Selkoe DJ, Lieberburg I, Schenk D. Isolation and quantitation of soluble Alzheimer’s β-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 55.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai X, McKay DM, Tintner R, Frangione B, Younkin SG. Production of the Alzheimer amyloid β protein by normal proteolytic processing. Science. 1992;258:126–129. doi: 10.1126/science.1439760. [DOI] [PubMed] [Google Scholar]
- 57.Small GW, Ercoli LM, Silverman DH, Huang SC, Komo S, Bookheimer SY, Lavretsky H, Miller K, Siddarth P, Rasgon NL, Mazziotta JC, Saxena S, Wu HM, Mega MS, Cummings JL, Saunders AM, Pericak-Vance MA, Roses AD, Barrio JR, Phelps ME. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc Natl Acad Sci USA. 2000;97:6037–6042. doi: 10.1073/pnas.090106797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nature Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 59.Sweeney WA, Luedtke J, McDonald MP, Overmier JB. Intrahippocampal injections of exogenous beta-amyloid induce postdelay errors in an eight-arm radial maze. Neurobiol Learn Mem. 1997;68:97–101. doi: 10.1006/nlme.1997.3770. [DOI] [PubMed] [Google Scholar]
- 60.Teplow DB. Structural and kinetic features of amyloid beta-protein fibrillogenesis. Amyloid. 1998;5:121–142. doi: 10.3109/13506129808995290. [DOI] [PubMed] [Google Scholar]
- 61.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzma R. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 62.Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Townsend M, Mehta T, Selkoe DJ. Soluble amyloid β-protein inhibits specific signal transduction cascds common to the insulin reception pathway. J Biol Chem. 2007 doi: 10.1074/jbc.M610390200. in press. [DOI] [PubMed] [Google Scholar]
- 64.Vigo-Pelfrey C, Lee D, Keim PS, Lieberburg I, Schenk D. Characterization of β-amyloid peptide from human cerebrospinal fluid. J Neurochem. 1993;61:1965–1968. doi: 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- 65.Walsh D, Klyubin I, Fadeeva J, Cullen WK, Anwyl R, Wolfe M, Rowan M, Selkoe D. Naturally secreted oligomers of the Alzheimer amyloid β-protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 66.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J Biol Chem. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 67.Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ. Detection of intracellular oligomers of amyloid β-protein in cells derived from human brain. Biochemistry. 2000;39:10831–10839. doi: 10.1021/bi001048s. [DOI] [PubMed] [Google Scholar]
- 68.Walsh DM, Hartley DM, Selkoe DJ. The many faces of Aβ: Structures and activity. Curr Med Chem - Immun Endoc Metab Agents. 2003;3:277–291. [Google Scholar]
- 69.Walsh DM, Townsend TM, Podlisny MB, Shankar GM, Fadeeva J, El-Agnaf O, Hartley DM, Selkoe DJ. Certain inhibitors of synthetic Aβ fibrillogenesis block oligomerization of natural Aβ and thereby rescue long term potentiation. J Neurosci. 2005;25:2455–2462. doi: 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang R, Sweeney D, Gandy SE, Sisodia SS. The profile of soluble amyloid b protein in cultured cell media. Detection and quantificaiton of amyloid β protein and variants by immunoprecipitation-mass spectrometry. J Biol Chem. 1996;271:31894–31902. doi: 10.1074/jbc.271.50.31894. [DOI] [PubMed] [Google Scholar]
- 71.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ. Enhanced production and oligomerization of the 42-residue amyloid b-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem. 1997;272:7977–7982. doi: 10.1074/jbc.272.12.7977. [DOI] [PubMed] [Google Scholar]
- 72.Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]