

Summary
Many enzymes in the pentein superfamily use a transient covalent intermediate in their catalytic mechanisms. Here, we use a mutant (H162G) dimethylarginine dimethylaminohydrolase from Pseudomonas aeruginosa and an alternative substrate, S-methyl-L-thiocitrulline, to trap, crystallize and determine the 2.8 Å resolution structure of a stable covalent adduct which mimics this reaction intermediate. Observed interactions between the trapped adduct and active site residues along with comparison to a previously known product-bound structure provide insight into the normal catalytic mechanism. The plane of the trapped thiouronium intermediate is angled away from that seen in the product and substrate complexes, allowing for an altered angle of attack between the nucleophiles of the first and second half reactions. The stable covalent adduct is also capable of further reaction. Addition of exogenous imidazole can rescue the original hydrolytic activity. Notably, addition of other exogenous amines can instead yield substituted arginine products. These alternative products arise from partitioning of the trapped intermediate into the evolutionarily related amidinotransferase reaction pathway. The enzyme scaffold provides both selectivity and catalysis for the amidinotransferase reaction, underscoring commonalities between different reaction pathways found in this mechanistically diverse enzyme superfamily. The promiscuous partitioning of this covalent intermediate may also help to illuminate the evolutionary history of these enzymes.
Introduction
The pentein superfamily of proteins share a β/α propeller fold that has five pseudo-symmetrical repeating ββαβ motifs (Teichmann et al., 2001). Examples of proteins in this diverse superfamily range from non-catalytic binding proteins, such as the ribosome anti-association factor IF6 (Groft et al., 2000), to a family of enzymes capable of modifying guanidino groups (Shirai et al., 2006). These guanidino modifying enzymes are capable of hydrolase reactions (arginine deiminase, peptidylarginine deiminase, dimethylarginine dimethylaminohydrolase), dihydrolase reactions (Nα-succinyl-L-arginine dihydrolase) and amidinotransferase reactions (arginine:glycine amidinotransferase, arginine:inosamine amidinotransferase). Enzymes in this superfamily are clinically relevant because of their roles in cancer, rheumatoid arthritis, multiple sclerosis, endothelial dysfunction, microbial and parasitic infections, and creatine metabolism (Shirai et al., 2006). Therefore, shared structural and mechanistic motifs are of significant interest for understanding these disease pathologies and for developing new therapeutics.
Many enzymes in the pentein superfamily use chemically similar covalent intermediates in their reaction mechanisms (Figure 1) (Shirai et al., 2001; Shirai et al., 2006). For example, one hydrolytic enzyme, dimethylarginine dimethylaminohydrolase (DDAH), uses an active-site Cys nucleophile to attack the guanidino carbon (Cζ) of Nω-methyl-L-arginine to produce methylamine and a covalent S-alkylthiouronium intermediate (Stone et al., 2005a). Its second half reaction results in hydrolysis of the intermediate to yield citrulline (Figure 1A). In contrast, the amidinotransferase enzmes bind the substrate (arginine) in an alternative conformation, holding the δ-nitrogen rather than the ω-nitrogen in the place of the leaving group. This allows conserved catalytic residues to assist in breaking the Nδ-carbon bond, rather than the Nω-carbon bond, yielding L-ornithine and the covalent S-alkylthiouronium intermediate. In contrast to the hydrolases, the intermediate of the amidinotransferases bears only a single carbon derived from arginine (Humm et al., 1997). This intermediate is stable to hydrolysis, remaining in place until attacked by the amine of the second substrate in the second half reaction (Figure 1B). Finally, a third family of enzymes, the dihydrolases, are proposed to go through a similar S-alkylthiournonium intermediate (Tocilj et al., 2005), although this species has not been detected experimentally (Figure 1C).
Figure 1.

Examples of reaction diversity in the pentein superfamily. A) Hydrolases, typified here by dimethylarginine dimethylaminohydrolase (DDAH), form a thiouronium intermediate after loss of the substrate’s Nω-substitutent, and subsequently hydrolyze the intermediate to yield a ureido-containing product. B) Amidinotransferases, typified here by Arg:Gly amidinotransferase, form a chemically similar thiouronium intermediate after loss of the Nδ-substitutent, and subsequently catalyze amidinotransfer to an acceptor amine. C) Dihydrolases, typified here by Nα-succinyl-L-arginine dihydrolase, have been proposed to catalyze their hydrolytic reactions through similar thiouronium intermediate(s), but a covalent adduct has not yet been detected experimentally.
Previous work has provided more details about the covalent intermediate formed by DDAH. A stable covalent EI complex can be formed using a mutant DDAH in which an active site His residue, which normally serves as an acid/base catalyst, is mutated to Gly (Stone et al., 2006). This H162G DDAH mutant does not react with Nω-methyl-L-arginine. However, the mutant does react with S-methyl-L-thiocitrulline, a substrate analog in which methanethiol acts as an activated leaving group in place of the methylamine found in the natural substrate. This reaction leads to formation of the same covalent thiouronium adduct that is seen during normal catalytic turnover (Stone et al., 2006; Stone et al., 2005a). The covalent adduct produced by the H162G mutant is stable to hydrolysis, presumably because the active site His is not present to deprotonate a water molecule for the second half reaction (Stone et al., 2006). Here, we have used these observations to obtain the three-dimensional structure of this covalent intermediate and shown that imidazole is capable of rescuing the normal hydrolytic reaction. We reasoned that addition of an exogenous amine to incubation mixtures of S-methyl-L-thiocitrulline and H162G DDAH might result in an amidinotransfer reaction because the related amidinotransferase enzymes are thought to use a similar covalent intermediate. Indeed, the transferase activity can be recovered. The DDAH protein scaffold both imparts selectivity for the acceptor amine and helps to catalyze the transfer. These findings provide insight into the catalytic mechanism of DDAH, experimentally demonstrate the relatedness of the hydrolase and amidinotransferase branches of this mechanistically diverse enzyme superfamily, and illustrate one way to manipulate their activities by using a combination of protein engineering, alternative substrates and chemical rescue experiments.
Results
X-ray structure determination
Co-crystallization mixtures containing the H162G DDAH protein and S-methyl-L-thiocitrulline (SMTC) produced crystals of space group P21212 with cell constants, a = 86.3, b= 127.5, c = 47.3 Å. There are two molecules per asymmetric unit, giving a Vm of 2.3 Å3/dalton. Following molecular replacement of the two molecules, a difference electron density map, with Cys249 residues omitted, clearly showed strong, continuous density for the substrate and the cysteine residue indicating a covalent bond (Figure 2A).
Figure 2.
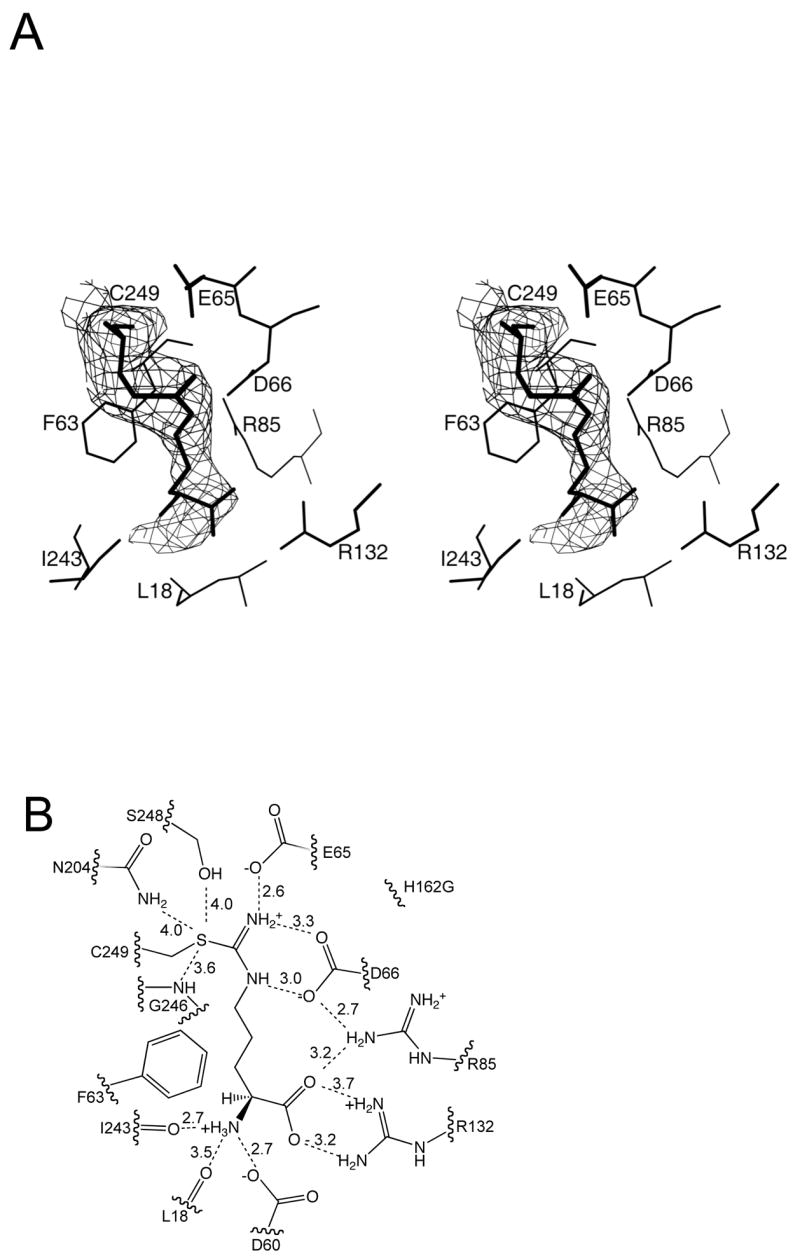
Interactions between the covalent intermediate and DDAH. A) Continuous electron density is observed between the amidino-carbon of the intermediate and the sulfur of Cys249 (wall-eyed stereoview). B) A line drawing depicting possible hydrogen bonding interactions (dashed lines). Distances between heteroatoms are given in angstroms.
Crystallographic data for the H162G DDAH-SMTC adduct are summarized in Table 1. A Ramachandran plot shows 80.2% of residues in the most favorable region, 18.4% in additional allowed space and 1.4% in generously allowed space. The refined model includes two covalent adducts and 59 solvent molecules.
Table 1.
Crystallographic Data
| H162G-SMTC | |
|---|---|
| Space group | P21212 |
| Cell constants (Å) | a=86.3, b=127.5, c=47.3 |
| Resolution (Å) (outer shell) | 20.-2.8 (2.91-2.8) |
| Rmerge (%) (outer shell) | 0.137 (0.465) |
| <I/σ|> (outer shell) | 5.4 (1.9) |
| Completeness (%) (outer shell) | 96.7 (95.0) |
| Unique reflections | 12,825 |
| Redundancy | 3.4 |
| # of residues | 508 |
| # of protein atoms | 3976 |
| # of ligand atoms | 24 |
| # of solvent atoms | 59 |
| Rworking | 0.233 |
| Rfree | 0.297 |
| Average B factor for protein atoms (Å2) | 30.1 |
| Average B factor for ligand atoms (Å2) | 31.5 |
| Average B factor for solvent atoms (Å2) | 12.0 |
| rms deviation from ideality | |
| bonds (Å) | 0.008 |
| angles (º) | 1.37 |
| Ramachandran plot | |
| % of residues in most favored region | 80.2 |
| % of residues in additional allowed region | 18.4 |
| % of residues in generously allowed region | 1.4 |
Structure of the H162G DDAH adduct
The H162G DDAH molecule is virtually identical to the previously determined P. aeruginosa C249S DDAH structure (Murray-Rust et al., 2001) with a root mean square deviation (RMSD) of 0.5 Å for equivalent Cα atoms. The largest difference comes in the loop containing the H162G mutation (residues 158–162). As described previously, DDAH is a barrel of five ββαβ units, which wrap around the active site (Murray-Rust et al., 2001). In each unit, the three β strands form a mixed β-sheet, with two parallel strands and one anti-parallel. The H162G DDAH-SMTC adduct crystallized as a dimer in the asymmetric unit. At the dimer interface, the β-sheets of the two first (N-terminal) ββαβ units are bonded together in an anti-parallel manner, resulting in a 6-strand sheet. The two monomers in the dimer are virtually identical, with an RMSD of 0.06 Å for equivalent Cα atoms, and are related by a rotation of 179º. Covalent adduct formation between SMTC and Cys249 was found in both monomers of the dimer. Similarly, a crystallographic dimer with a 180º rotation was observed in the C249S DDAH complex structure.
Binding of S-methyl-L-thiocitrulline
Reaction of SMTC with DDAH H162G results in a covalent link between the side chain of Cys249 and the remaining N5-(1-iminomethyl)-L-ornithine fragment of the substrate. The amidino-carbon (Cζ) of the adduct is covalently linked to the sulfur of Cys249, and hydrogen bonds are formed between nitrogen atoms and the side chains of Glu65 and Asp66 as shown schematically in Figure 2B. The carboxylate of the adduct makes an ion pair with the side chains of Arg132 and Arg85. The α-amino group of the adduct hydrogen bonds with the main chain carbonyl oxygen of Ile243. In the C249S DDAH-citrulline complex, the ureido and amino acid moieties of citrulline are bound by the same hydrogen bonding interactions. However, the orientation of the ureido group differs, probably due to the lack of covalent bond formed with the C249S side chain (Figure 3).
Figure 3.
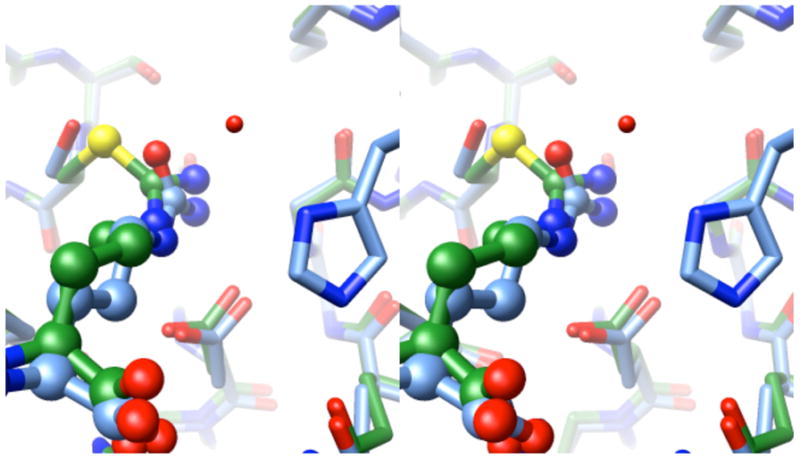
Wall-eyed stereoview of superimposed product and intermediate. The product complex structure (blue) (Murray-Rust et al., 2001) is superimposed over the covalent intermediate structure (green), with the active site ligands shown in ball and stick representations. At the bottom of the figure, Asp66 forms two hydrogen bonds with each species. An ordered water molecule (Wat143) in the product complex (missing in the intermediate structure) is within hydrogen bonding distance with the imidazole of His162 (also missing in the H162G intermediate structure). The angles of the intersecting planes formed by the urea (of the product) and the thiouronium (intermediate) groups are offset by approximately 30°. The stereoimage was prepared using UCSF Chimera (Pettersen et al., 2004).
Also, in the C249S DDAH-citrulline complex, the aliphatic backbone of citrulline is stabilized by a hydrophobic pocket with the phenyl side chain of Phe63 on one side and the side chains of Leu161 and His162 on the other. In the adduct structure with the H162G mutation, the loop of residues 158–162 has moved away from the active site by approximately 2 Å, and Leu161 no longer makes van der Waals contact with the bound compound.
Chemical Rescue Experiments
Incubation mixtures containing H162G DDAH and S-methyl-L-thiocitrulline did not show any appreciable turnover during the experimental timescale. However, in chemical rescue experiments, addition of exogenous reagents results in the catalytic disappearance of substrate. Addition of imidazole to incubation mixtures at pH 7.3 stimulated enzyme activity. Imidazole rescue displayed a concentration dependence with saturation kinetics, reaching a half-maximum rate at 95 ± 10 mM and a maximum rate of 0.03 ± 0.01 s−1 (Figure 4A). The maximum rescued rate is approximately 40-fold lower than the kcat value for S-methyl-L-thiocitrulline turnover by wild type DDAH (1.3 ± 0.1 s−1) at pH 7.5 (Stone et al., 2006). Control incubations (3 d) of imidazole (500 mM) and SMTC (500 μM) in the absence of enzyme showed a hydrolysis rate of 4 × 10−4 μM s−1, which is 900-times slower than the observed rate in the presence of enzyme. Addition of hydroxylamine (instead of imidazole) also increased the rate of substrate turnover. Control reactions in the absence of enzyme showed negligible background rates (≤ 1 %). Hydroxylamine rescue displayed a concentration dependence with sigmoidal kinetics. The data were well fit by the Hill equation with a maximum rate of 0.074 ± 0.008 s−1 and a ligand concentration at half occupancy of 250 ± 20 mM (Figure 4A). The Hill coefficient is 2.9 ± 0.4, which suggess positive cooperativity. However, further studies will be required to analyze this behavior in more detail.
Figure 4.
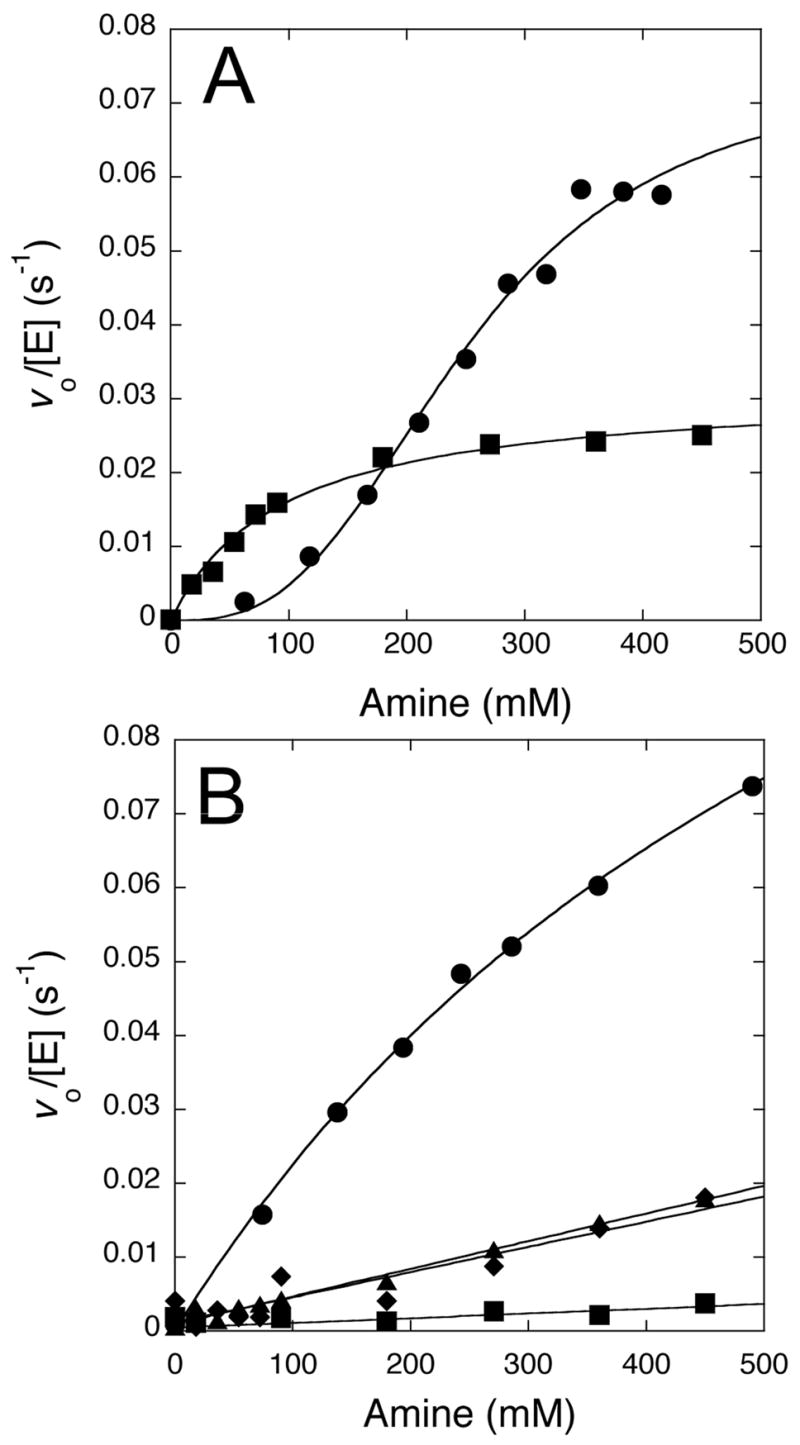
Concentration dependence of the chemical rescue reactions. A) At pH 7.3, rates of substrate disappearance increase with higher concentrations of imidazole (■, pKa 7) and hydroxylamine (●, pKa 6). Imidazole shows evidence of saturation kinetics and hydroxylamine shows sigmoidal kinetics, possibly indicative of positive cooperativity (see Results). B) At pH 9.5, rates of substrate disappearance increase with higher concentrations of methylamine (●, pKa 10.6), dimethylamine (◆, pKa 10.6), t-butylamine (▲, pKa 10.6) and 2-amino-2-methyl-1,3-propanediol (■, pKa 8.8). Methylamine shows some curvature, possibly indicative of saturation, but the remaining amines are fit by a linear concentration dependence. All experiments (except for imidazole rescue) are corrected for background rates observed in the absence of enzyme. See Experimental Details and Results for more information.
Addition of methylamine, dimethylamine, t-butylamine, and 2-amino-2-methyl-1,3-propanediol to incubation mixtures at pH 7.3 did not show appreciable turnover rates under the experimental timescale (< 10 min). Therefore, these amines were tested with incubation mixtures at a higher pH (9.5) to increase their reactivity (Figure 4B). DDAH is stable under these conditions (Stone et al., 2006). At higher pH values, there is increased background hydrolysis of S-methyl-L-thiocitrulline catalyzed by H162G DDAH alone, but the rate of this control reaction is negligible when compared to the observed rates of the rescued reactions (below). Incubations of all rescue reagents with substrate in the absence of enzyme showed low background rates (≤ 3%) with the exception of 2-amino-2-methyl-1,3-propanediol (30%). Addition of methylamine to incubations of S-methyl-L-thiocitrulline and H162G DDAH at pH 9.5 accelerated substrate turnover in a concentration dependent manner, showing a slight curvature suggestive of saturation kinetics (Figure 4B). However, the highest concentration of methylamine tested was still below the estimated concentration (690 ± 80 mM) required to reach half of the maximal (0.18 s−1) rate, so these values should be treated as resulting from extrapolations to the maximal rate. For comparison, the kcat for S-methyl-L-thiocitrulline turnover by wild type DDAH at pH 9.5 is 1.3 ± 0.1 s−1 (Stone et al., 2006). The other amines tested as rescue reagents at pH 9.5, dimethylamine, t-butylamine and 2-amino-2-methyl-1,3-propanediol, were well fit by a linear concentration dependence, with observed slopes of 3.4 ± 0.5 ×10−5, 3.7 ± 0.2 ×10−5 and 0.6 ± 0.2 ×10−5 s−1 mM−1, respectively. The observed rates qualitatively correlate with the decrease in steric bulk of the exogenous amine, but not with their pKa values.
Product Analysis
High-performance liquid chromatography (HPLC) of commercial standards for the substrate S-methyl-L-thiocitrulline (14.5 min retention time), the product L-citrulline (10.3 min) and the expected alternative product Nω-methyl-L-arginine (12.6 min) resulted in baseline separation of these compounds (Figure 5A). Derivatization and analysis of reaction mixtures previously incubated at pH 7.5 (Figure 5B) showed that the three control mixtures had only one major peak occurring at 14.6 min, matching the retention time for S-methyl-L-thiocitrulline. In contrast, the incubation mixture containing substrate, enzyme and imidazole showed a large new peak at 10.1 min, matching the retention time of the L-citrulline standard. Analysis of control incubation mixtures including substrate and enzyme also showed a minor amount of citrulline produced under these extended incubation times, but had peak heights < 5 % of those formed when imidazole was present (Figure 5). Derivatization and analysis of reaction mixtures previously incubated at pH 9.5 (Figure 5C) also showed some additional minor peaks in the control reactions, presumably due to alkaline degradation products, but the predominant peak in each control incubation occurred at 14.5 min, matching the retention time of S-methyl-L-thiocitrulline. The peak for derivatized methylamine elutes near the onset of the wash phase (22.5 min, data not shown). Notably, the incubation mixture containing substrate, enzyme and methylamine showed a large new peak at 12.7 min, matching the retention time of an Nω-methyl-L-arginine standard. Electrospray ionization mass spectrometric (ESI-MS) analysis of reaction mixtures are also consistent with assigning this product as Nω-methyl-L-arginine (Supplemental Figure S1).
Figure 5.

HPLC of Chemical Rescue Reaction Products. A) Commercial standards of S-methyl-L-thiocitrulline (S, 14.5 min), L-citrulline (10.3 min) and Nω-methyl-L-arginine (NMMA, 12.6 min) are derivatized using o-phthaldehyde, separated on a C18 analytical column and detected using fluorescence (Ex = 338 nm; Em = 445 nm). B) Reaction products of incubations (4 h) containing S, S and H162G DDAH (E), S and imidazole (Im) and S, E and Im are derivatized and separated into their components by HPLC. In the last incubation mixture, a significant peak appearing at 10.1 min is observed, consistent with citrulline production. C) Reaction products of incubations (4 h) containing S, S and E, S and methylamine (NH2CH3), and S, NH2CH3 and E are derivatized and separated into their components. In this last incubation mixture, a significant peak appearing at 12.7 min is observed, consistent with Nω-methyl-L-arginine formation. In each plot, stacked elution traces are offset by 1000, 1500 or 2000 relative fluorescence units for easier visualization.
Product analysis of incubation mixtures including S-methyl-L-thiocitrulline, H162G DDAH and each of the remaining rescue reagents, hydroxylamine, dimethylamine, t-butylamine or 2-amino-2-methyl-1,3-propanediol, can be found in supplemental data (Figure S2). Unlike methylamine, these amines can somewhat increase production of citrulline (the hydrolysis product) although to different extents. Notably, product analysis of incubation mixtures including the smaller amines, dimethylamine and hydroxylamine, showed production of new peaks with retention times that match standards of the expected amidinotransferase products, Nω,Nω-dimethyl-L-arginine and Nω-hydroxy-L-arginine, respectively. The bulkier branched amines, t-butylamine and 2-amino-2-methyl-1,3-propanediol, showed increased citrulline production, but no obvious peaks corresponding to products expected from an amidinotransferase reaction.
Discussion
In an effort to understand the similarities and differences between enzymes in the pentein superfamily, we prepared and studied a stable mimic of a covalent reaction intermediate. Previously, mass spectrometry studies have shown that incubation of Pseudomonas aeruginosa H162G DDAH with an activated substrate, S-methyl-L-thiocitrulline, can lead to formation of a stable covalent adduct (Stone et al., 2006). This stable complex presumably mimics the transient covalent reaction intermediate formed at the active-site Cys249 residue (Figure 1). Here, the covalent adduct has been crystallized under conditions similar to those reported for the non-covalent product complex formed between L-citrulline and P. aeruginosa C249S DDAH (Murray-Rust et al., 2001). Consistent with previous structural and sedimentation studies of P. aeruginosa DDAH, a crystallographic dimer of two β/α propeller monomers is clearly observed (Murray-Rust et al., 2001). Each monomer overlays quite closely with each other (0.06 Å RMSD for equivalent Cα atoms) and also matches closely the coordinates of the product complex (0.5 Å RMSD for equivalent Cα atoms) with the exception of small differences near the active site, which will be described below. The most obvious difference is the missing imidazole sidechain at position 162 due to the Gly for His substitution (Figure 3). This missing sidechain does not grossly impact the overall fold of the enzyme. However, in the mutant structure, residues 156–162 are slightly perturbed from their positions in the product complex, and have somewhat higher B values. In particular, Leu161 is moved away from the bound ligand at the active site and appears to allow increased disorder of the ligand’s carboxylate, α-amine, and α- and β-carbons. The active site residues surrounding the guanidinium binding pocket, however, are not as perturbed, and a well formed pocket remains where the His162 imidazole sidechain is positioned in the product complex (Supplemental Figure S3).
Consistent with mass spectrometry data (Stone et al., 2006), continuous electron density is observed between the sulfur atom of Cys249 and the amidino carbon of the Nδ-(iminomethyl)-L-ornithine adduct, further indicating covalent bond formation (Figure 2). Both monomers have full occupancy of the covalently bound ligand. This observation is consistent with earlier reports that the two P. aeruginosa DDAH active sites found in the dimer work independently (Plevin et al., 2004), and contrasts with the finding that a related Mycoplasma arthritidis arginine deiminase shows half-of-the-sites reactivity (Weickmann et al., 1978).
This structure of the covalent adduct provides an interesting glimpse into specific interactions that may occur with the transient covalent intermediate formed during DDAH catalyzed reactions (Figure 2). Most of the interactions with the intermediate’s α-amine, carboxylate and sidechain methylenes appear to be quite similar to those previously reported in the product and substrate complexes (Murray-Rust et al., 2001). However, the plane of the ureido group of the product, L-citrulline, appears to be significantly tilted in comparison with the S-alkylthiouronium group seen here in the covalent complex (Figure 3). The two nitrogens of the urea (in the product) and of the thiouronium (in the intermediate) each maintain hydrogen bonding distance to the two carboxylate oxygens of Asp66. This conserved interaction presumably allows the active-site ligand to hinge between the two observed angles without disrupting existing hydrogen bonds. Glu65 also maintains hydrogen bonding with one ωnitrogen of the active-site ligand in both structures. The offset angle of the intersecting planes formed by the urea (of the product) and the thiouronium (intermediate) groups is approximately 30º, and is likely larger when compared to the guanidinum of the bound substrate (although the coordinates for that complex are not available) (Murray-Rust et al., 2001). The changing angle of these planes provides an explanation for how DDAH can accommodate the different angles of attack required for each half reaction of the enzyme (Figure 1A). In the first half reaction, the thiolate nucleophile of Cys249 would be expected to attack the si-face of Nω-methyl-L-arginine, proceeding through a tetrahedral adduct and loss of methylamine to form the thiouronium intermediate. However, hydrolysis of the intermediate requires hydroxide attack on the opposite face, reflected in this change in angle. When the product and intermediate structures are overlayed, an ordered water molecule found in the product structure is appropriately placed for attack, 3.0 Å from the amidino carbon of the intermediate and at an angle of 106º (Wat-Cζ-S). This angle is close to the Bürgi Dunitz angle (105º), which describes the expected trajectory of nucleophilic attack on a carbonyl (Burgi and Dunitz, 1983). However, thiouronium electrophiles are not as well studied as carbonyls and likely have different reaction constraints. Regardless, the water molecule’s positioning is consistent with its participation in the second half reaction – formation of a second tetrahedral adduct and subsequent elimination of Cys249 to regenerate the resting enzyme. This water molecule (Wat143) is also within hydrogen bond distance of H162 (in the product structure) and is consistent with assigning this particular water as the hydrolytic water (Figure 3). An ordered water molecule is not observed at a similar position in the inactive H162G mutant.
By using this mutant enzyme and an alternative substrate to form a trapped intermediate-like structure, we can visualize how Asp66 works as a hinge to allow the conjugated guanidinium (in the substrate) or thiouronium (in the intermediate) group to flip back and forth, enabling an acceptable angle of attack for the varying nucleophiles in each half-reaction (attack of Cys249 and hydroxide, respectively), while maintaining existing hydrogen bonds. Examination of arginine deiminase and peptidylarginine deiminase structures containing similar substrate and reaction intermediates as well as computational studies of the arginine deiminase reaction suggest that this may be a common mechanistic feature in the superfamily (Arita et al., 2004; Das et al., 2004; Galkin et al., 2005; Luo et al., 2006; Thompson and Fast, 2006; Wang et al., 2007).
The conserved positions of the active-site residues in this covalent adduct structure also indicate that the observed adduct is likely trapped due to the missing imidazole sidechain and not because of an unintentional deformation of the active-site. To underscore this point, chemical rescue experiments (Toney and Kirsch, 1989) were attempted with exogenous imidazole that replaced the missing sidechain. Multiple turnovers of the activated substrate, S-methyl-L-thiocitrulline, were observed upon addition of exogenous imidazole. The rescue kinetics exhibited pseudo-first order behavior with respect to imidazole concentration, consistent with the presence of a saturatable binding site for imdiazole (95 mM) in the H162G mutant, albeit with a reduced maxiumum rate (approx 40-fold slower than wild type turnover) (Figure 4A). The slower maximum rate may be due to sub-optimal positioning or to a slowed first half reaction in the H162G mutant. The chemical rescue reaction product was identified as citrulline by o-phthaldehyde derivatization and HPLC in comparison to authentic standards (Figure 5B). Presumably, imidazole binds in the cavity created by the H162G mutation (Supplemental Figure 3) and substitutes for the missing histidine sidechain. However, we have not formally ruled out two alternative possibilities. Imidazole may bind and stabilize an alternative conformation of the enzyme that is capable of hydrolyzing the covalent intermediate. Or, Imidazole may directly attack the covalent intermediate to generate a substitution product that is rapidly hydrolyzed - Hydrolysis of p-nitrophenylacetate by imidazole has been shown to go through a covalent pathway (Bender and Turnquest, 1957).
This divergent superfamily of enzymes can be classified as a mechanistically diverse enzyme superfamily (Gerlt and Babbitt, 2001) because its members share sequence and structural motifs as well as chemically similar covalent intermediates, yet catalyze different reaction types (Shirai et al., 2006; Teichmann et al., 2001). This raises the possibility that the trapped intermediate described here might also be capable of promiscuous partitioning into evolutionarily related alternative reaction pathways. Enzyme promiscuity has been proposed to serve as one method for functional diversification during enzyme evolution (Glasner et al., 2006). Specifically, we hypothesized that addition of exogenous amines, rather than imidazole, might be able to catalyze the alternative amidinotransferase reaction found within the superfamily instead of just rescuing the original hydrolytic reaction. Indeed, addition of various amines including hydroxylamine, methylamine, dimethylamine, t-butylamine and 2-amino-2-methyl-1,3-propanediol, can trigger multiple turnovers of the activated substrate (Figure 4). Using methylamine as an example, HPLC and ESI-MS analysis of the reaction mixture clearly shows production of Nω-methylarginine as the product rather than the citrulline that was observed upon rescue by imidazole (Figure 5C and Supplemental Figure S1). The enzyme does appear to provide a degree of selectivity between acceptor amines in the rescued amidinotransferase reaction, despite their similar pKa values (Figure 4). The smaller amines, methylamine, dimethylamine and hydroxylamine, yield the expected amidinotransferase products, Nω-methyl-L-arginine, Nω,Nω-dimethyl-L-arginine and Nω-hydroxy-L-arginine, respectively (Figure 5, Supplemental Data). The larger branched amines, t-butylamine and 2-amino-2-methyl-1,3-propanediol, only appear to drive the intermediate through the hydrolytic pathway, possibly by general base catalysis (Supplemental Data). The crystal structure of H162G DDAH shows a large funnel shaped cavity, providing solvent access to the trapped intermediate (Supplemental Figure S3), which could provide access for the chemical rescue reagents. However, considerable plasticity can be seen in the DDAH active site upon ligand binding (Leiper et al., 2007), so it is difficult to predict how exogenous amines interact with the enzyme. Therefore, expanding these chemical rescue studies by using a library of structurally diverse amines may allow structure activity relationships to be determined (SAR by chemical rescue). These studies are underway.
Chemical rescue reaction rates for most of these amines were quite slow at neutral pH (data not shown), but were faster at pH 9.5, consistent with the use of their neutral forms and suggestive that their pKa values are not significantly perturbed by the H162G enzyme. To illustrate this point, we observed that hydroxylamine (pKa 6), unlike amines with higher pKa values, reacted readily at pH 7.3. The concentration dependence of methylamine appears to show some curvature, possibly reflecting a weakly saturatable binding site. However, with the exception of hydroxylamine (see Results), the other amines show only linear concentration dependencies, more consistent with reaction via encounter complexes (Figure 4B).
The protein scaffold also appears to help catalyze the rescued amidinotransfer reaction. The S-alkylthiouronium groups of the activated substrate, S-methyl-L-thiocitrulline, and the trapped covalent enzyme intermediate are chemically similar, but incubation mixtures with substrate and amines alone do not show evidence of significant amidinotransferase activity (Figure 5). However, addition of H162G DDAH to these reaction mixtures readily yields the expected amidinotransferase products, highlighting the necessity of the enzyme for catalysis. Possible explanations are that the enzyme enhances the electrophilicity of the amidino carbon by using Asp66 and Glu65 to polarize the Cζ-N bonds, and may facilitate breaking the Cζ-S bond of the intermediate by stabilizing the developing partial negative charge on the sulfur leaving group through weak hydrogen bonding interactions with Asn204, Gly246, or Ser248, as suggested by the trapped intermediate structure (Figure 2). It is possible that these interactions may also play a role in facilitating hydrolysis of the intermediate in the wild-type reaction, although further studies will be required. The evolutionary history of enzymes in this superfamily is not clear, but if the amidinotransferase reaction is more ancient, then instead of “chemical rescue,” recovery of this alternative activity in DDAH might be more accurately termed “chemical resurrection.”
Significance
Through the use of a mutant DDAH enzyme and an alternative substrate, a stable mimic of a covalent reaction intermediate was trapped and structurally characterized. This methodology provides structural insights into a normally transient reaction intermediate (Stone et al., 2005a), providing data about the catalytic mechanism of this clinically important (Knipp, 2006) enzyme. Depending on the rescue agent used, the trapped intermediate can be partitioned into its normal hydrolytic product or down an alternative amidinotransfer pathway to yield a substituted arginine product (Figure 6). The enzyme imparts both catalysis and selectivity to the transfer reaction, suggesting the possibility of using chemical rescue studies to probe active site constraints (SAR by chemical rescue). This alternative activity might also have potential applications for chemoenzymatic synthesis of substituted arginine analogs in aqueous solutions. These results also experimentally illustrate mechanistic features conserved between the hydrolyase and amidinotransferase branches of this mechanistically diverse enzyme superfamily and are consistent with the proposal that these enzymes are derived from a common ancestor with a reaction intermediate capable of promiscuous partitioning.
Figure 6.

Simplified reaction scheme for A) wild type DDAH catalyzed hydrolysis of Nω-methyl-L-arginine and B) H162G DDAH catalyzed turnover of S-methyl-L-thioctirulline. The boxed structure represents the crystallized adduct which can be partitioned into either the hydrolysis product (citrulline, upper route) or to amidinotransferase products (lower route) depending on the chemical rescue reagent.
Experimental Procedures
Unless noted otherwise, all chemicals and buffers are acquired from Sigma-Aldrich Chemical Co. (St. Louis, MO). HPLC grade acetonitrile and water were purchased from Fisher Scientific Co. (Pittsburgh, PA). The P. aeruginosa H162G DDAH enzyme was expressed in E.coli BL21(DE3) cells, induced, harvested and purified as described previously (Stone et al., 2005b). All fits were completed using Kaleidagraph software (Synergy Software, Reading, PA).
Crystallization and Data Collection
To prepare the stable covalent enzyme adduct, purified P. aeruginosa H162G DDAH was concentrated to approximately 14 mg/mL (470 mM) in Tris-HCl (50 mM), pH 8, using a Microcon YM-10 centrifugal filter device (Millipore, Billerica, MA) and the final solution was made to 5 mM DTT and 4 mM S-methyl-L-thiocitrulline.
The covalent adduct was crystallized at 25° C using the hanging drop method from 30% PEG4000, 0.1 M Tris-HCl, pH 8.5, 0.2 M sodium acetate. Prior to data collection, crystals were treated with cryoprotectant by transferring to the precipitating solution (30% PEG4000, 0.1 M Tris-HCl, pH 8.5, 0.2 M sodium acetate) for 1–5 seconds. A crystal, mounted in a cryoloop (Hampton Research, Laguna Niguel, CA), was frozen by dipping in liquid nitrogen and placed in the cold stream on the goniostat.
X-ray diffraction data were collected from the crystal at 100° K on an RAXIS IV++ image plate detector (Rigaku, The Woodlands, TX) with X-rays generated by a Rigaku RU-H3R rotating anode generator (Rigaku) operated at 50 mV, 100 mA. Diffraction images were processed and data reduced using HKL2000 (Otwinowski and Minor, 1997).
Structure determination and analysis
Crystal cell parameters indicated that the asymmetric unit likely contained two DDAH molecules. MOLREP (Vagin and Teplyakov, 1997), from the CCP4 suite (CCP4, 1994), was used to determine the molecular replacement solution for the two DDAH molecules in the asymmetric unit, using the P. aeruginosa DDAH C249S structure (PDB accession code 1H70) (Murray-Rust et al., 2001) as the model.
Model building was done on a Gateway Select SB computer (Poway, CA) using O (Jones et al., 1991). Refinement of models was performed with the Crystallography and NMR System (CNS) (Version 1.1) using the slow-cooling protocol (Brunger et al., 1998). There were several rounds of refinement followed by rebuilding of the model. To facilitate manual rebuilding of the model, a difference map and a 2Fo-Fc map, SIGMAA-weighted to eliminate bias from the model (Read, 1986), were prepared. 5% of the diffraction data were set aside throughout refinement for cross-validation (Brunger, 1993). PROCHECK (Laskowski et al., 1993) was used to determine areas of poor geometry. For the purpose of locating bound solvent molecules, CNS was used to select peaks of height 3.5 standard deviations above the mean from a difference map to eliminate those peaks that were not within 3.5 Å of a protein nitrogen or oxygen atom. O was used to manually view and accept water sites. Computations were done on an HP Pavilion a510n computer (Hewlett-Packard Co., Palo Alto, CA).
Atomic coordinates
Coordinates of the refined model of the P. aeruginosa H162G DDAH-SMTC adduct have been deposited in the Protein Data Bank with entry code XXXX.
Chemical rescue experiment
Stock solutions of imidazole (1M) and hydroxylamine (1M) were prepared in Hepes buffer (250 mM) with KCl (250 mM) and the final pH adjusted to 7.3. Stock solutions of methylamine (1M), dimethylamine (1M), t-butylamine (1M) and 2-amino-2-methyl-1,3,-propanediol (1M) were prepared in boric acid buffer (250 mM) with KCl (250 mM) and the final pH of each was adjusted to 9.5. Typically, H162G DDAH (12 μM) was mixed with saturating concentrations of the substrate, S-methyl-L-thiocitrulline (500 μM) in the appropriate reaction buffer either for rescue by imidazole or hydroxylamine [Hepes buffer (250 mM), KCl (250 mM) at pH 7.2] or for rescue by methylamine, dimethylamine, t-butylamine or 2-amino-2-methyl-1,3,-propanediol [boric acid buffer (250 mM), KCl (250 mM) at pH 9.5]. Aliquots from the amine stock solutions (final concentrations from 0 – 450 mM) were then added to initiate the reaction. Initial rates of S-methyl-L-thiocitrulline disappearance were monitored at 25° C by UV-Vis spectroscopy using a Cary 50 UV-vis spectrophotometer (Varian, Inc., Walnut Creek, CA) to follow loss of its intrinsic chromophore upon conversion to either citrulline or the substituted arginine product, as described previously (Stone et al., 2006). In all of these kinetic studies, ≤ 5% of substrate was consumed. The concentration dependence of the rescue rates for dimethylamine, t-butylamine and 2-amino-2-methyl-1,3-propanediol were fit to a linear equation. The concentration dependence of the rescue rates for methylamine and imidazole were fit to the Michaelis-Menten equation. The concentration dependence of hydroxylamine rescue rates were fit using the following form of the Hill equation, where Vo is the observed initial rate, Vmax is the maximum rate, n is the Hill coefficient, and KA is the ligand concentration that occupies half of the binding sites.
Product Analysis
Typically for product analysis, chemical rescue reactions were carried out in small volumes (100 μL) for extended times (4 h) at 25° C. For imidazole rescue reactions, various combinations of purified H162G DDAH (18 μM), S-methyl-L-thiocitrulline (6 mM) and imidazole (100 mM) were incubated in Hepes buffer (250 mM), KCl (250 mM) at pH 7.3. For methylamine rescue reactions, various combinations of purified H162G DDAH (18 μM), S-methyl-L-thiocitrulline (6 mM) and methylamine (100 mM) were incubated in boric acid buffer (250 mM), KCl (250 mM) at pH 9.5. Stock solutions of imidazole and methylamine were prepared as described above. After incubation, each sample was diluted with water (200 μL), passed through a 10,000 MWCO 96-well microtiter filter plate (Harvard Apparatus, Holliston, MA) by vacuum filtration to remove DDAH, and used immediately or frozen at −20° C for later analysis.
For separation of the reaction components, a Shimadzu Prominence analytical HPLC (Columbia, MD) equipped with two LC-20A pumps and a RF10AxL fluorescence detector was used with a 5 μm, 4.6 × 250 mm Zorbax XDB-C18 column fitted with a Zorbax XDB-C18 guard column (Phenomenex, Torrance, CA). Immediately prior to manual injections (20 μL total), aliquots (15 μL) from the incubation mixtures were mixed with an equal volume of o-phthaldehyde derivatization reagent (Sigma) and incubated for one minute. Fluorescence of the column eluate was monitored using excitation and emission wavelengths of 338 and 455 nm, respectively. Separation was achieved using the following gradient program between solvent A (aqueous, sodium phosphate (30 mM) at pH 7.5) and solvent B (acetonitrile). Solvent B was increased in a linear fashion from 8.7 % to 20 % over 20 min, to 70 % over the next 2.5 min, held at 70 % for 17.5 min, and finally decreased to 8.7 % over the next minute. A fluorescent peak found in all samples, derived from the o-phthaldehyde derivatization reagent, eluted near 22.5 min, when the organic mobile phase was increased to 70% for column washing (data not shown). All solvents were degassed daily by ultrasonication.
Supplementary Material
Supplemental data consists of three figures. ESI-MS product analysis of incubations containing methylamine. HPLC product analysis of incubation mixtures containing rescue agents hydroxylamine, dimethylamine, t-butylamine, and 2-amino-2-methyl-1,3-propanediol. Depictions of two protein cavities in the H162G DDAH protein structure. Supplemental figures are available at http://www.chembiol.com.
Acknowledgments
We thank the Analytical Instrumentation Facility Core lab (College of Pharmacy, University of Texas, Austin) for mass spectrometry studies. This work was supported in part by NIH grant GM63593 (to JDR), grant RSG-05-061-01-GMC (to WF) from the American Cancer Society, grants from the Robert A. Welch Foundation (to JDR and grant F1572 to WF), a seed grant from the Texas Institute for Drug and Diagnostic Development (to WF), and by College of Natural Sciences support to the Center for Structural Biology at the University of Texas, Austin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arita K, Hashimoto H, Shimizu T, Nakashima K, Yamada M, Sato M. Structural basis for Ca(2+)-induced activation of human PAD4. Nat Struct Mol Biol. 2004;11:777–783. doi: 10.1038/nsmb799. [DOI] [PubMed] [Google Scholar]
- Bender ML, Turnquest BW. The imidazole-catalyzed hydrolysis of p-nitrophenyl acetate. J Am Chem Soc. 1957;79:1652–1655. [Google Scholar]
- Brunger AT. Assessmet of phase accuracy by cross validation: the free R value. Methods and applications. Acta Cryst D Biol Crystallogr. 1993;49:24–36. doi: 10.1107/S0907444992007352. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Cryst D Biol Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Burgi HB, Dunitz JD. From Crystal Statics to Chemical Dynamics. Acc Chem Res. 1983;16:153–161. [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Cryst D5. 1994:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Das K, Butler GH, Kwiatkowski V, Clark AD, Jr, Yadav P, Arnold E. Crystal structures of arginine deiminase with covalent reaction intermediates; implications for catalytic mechanism. Structure (Camb) 2004;12:657–667. doi: 10.1016/j.str.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Galkin A, Lu X, Dunaway-Mariano D, Herzberg O. Crystal structures representing the Michaelis complex and the thiouronium reaction intermediate of Pseudomonas aeruginosa arginine deiminase. J Biol Chem. 2005;280:34080–34087. doi: 10.1074/jbc.M505471200. [DOI] [PubMed] [Google Scholar]
- Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- Glasner ME, Gerlt JA, Babbitt PC. Evolution of enzyme superfamilies. Curr Opin Chem Biol. 2006;10:492–497. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Groft CM, Beckmann R, Sali A, Burley SK. Crystal structures of ribosome anti-association factor IF6. Nat Struct Biol. 2000;7:1156–1164. doi: 10.1038/82017. [DOI] [PubMed] [Google Scholar]
- Humm A, Fritsche E, Steinbacher S, Huber R. Crystal structure and mechanism of human L-arginine:glycine amidinotransferase: a mitochondrial enzyme involved in creatine biosynthesis. Embo J. 1997;16:3373–3385. doi: 10.1093/emboj/16.12.3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, M K. Improved methods for building protein models in electron density maps and the locatin of errors in these models. Acta Cryst A. 1991;47(Pt 2) doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Knipp M. How to control NO production in cells: N(omega), N(omega)-dimethyl-L-arginine dimethylaminohydrolase as a novel drug target. Chembiochem. 2006;7:879–889. doi: 10.1002/cbic.200500527. [DOI] [PubMed] [Google Scholar]
- Knipp M, Vasak M. A colorimetric 96-well microtiter plate assay for the determination of enzymatically formed citrulline. Anal Biochem. 2000;286:257–264. doi: 10.1006/abio.2000.4805. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- Luo Y, Arita K, Bhatia M, Knuckley B, Lee YH, Stallcup MR, Sato M, Thompson PR. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry. 2006;45:11727–11736. doi: 10.1021/bi061180d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray-Rust J, Leiper J, McAlister M, Phelan J, Tilley S, Santa Maria J, Vallance P, McDonald N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat Struct Biol. 2001;8:679–683. doi: 10.1038/90387. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;27:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Plevin MJ, Magalhaes BS, Harris R, Sankar A, Perkins SJ, Driscoll PC. Characterization and manipulation of the Pseudomonas aeruginosa dimethylarginine dimethylaminohydrolase monomer--dimer equilibrium. J Mol Biol. 2004;341:171–184. doi: 10.1016/j.jmb.2004.05.057. [DOI] [PubMed] [Google Scholar]
- Read RJ. Improved Fourier coefficients for maps using phases from partial structures with errors. Acta Cryst Sect A. 1986;42:140–149. [Google Scholar]
- Shirai H, Blundell TL, Mizuguchi K. A novel superfamily of enzymes that catalyze the modification of guanidino groups. Trends Biochem Sci. 2001;26:465–468. doi: 10.1016/s0968-0004(01)01906-5. [DOI] [PubMed] [Google Scholar]
- Shirai H, Mokrab Y, Mizuguchi K. The guanidino-group modifying enzymes: structural basis for their diversity and commonality. Proteins. 2006;64:1010–1023. doi: 10.1002/prot.20863. [DOI] [PubMed] [Google Scholar]
- Stone EM, Costello AL, Tierney DL, Fast W. Substrate-assisted cysteine deprotonation in the mechanism of dimethylargininase (DDAH) from Pseudomonas aeruginosa. Biochemistry. 2006;45:5618–5630. doi: 10.1021/bi052595m. [DOI] [PubMed] [Google Scholar]
- Stone EM, Person MD, Costello NJ, Fast W. Characterization of a Transient Covalent Adduct Formed during Dimethylarginine Dimethylaminohydrolase Catalysis. Biochemistry. 2005a;44:7069–7078. doi: 10.1021/bi047407r. [DOI] [PubMed] [Google Scholar]
- Stone EM, Schaller TH, Bianchi H, Person MD, Fast W. Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: dimethylargininase and peptidylarginine deiminase. Biochemistry. 2005b;44:13744–13752. doi: 10.1021/bi051341y. [DOI] [PubMed] [Google Scholar]
- Teichmann SA, Murzin AG, Chothia C. Determination of protein function, evolution and interactions by structural genomics. Curr Opin Struct Biol. 2001;11:354–363. doi: 10.1016/s0959-440x(00)00215-3. [DOI] [PubMed] [Google Scholar]
- Thompson PR, Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol. 2006;1:433–441. doi: 10.1021/cb6002306. [DOI] [PubMed] [Google Scholar]
- Tocilj A, Schrag JD, Li Y, Schneider BL, Reitzer L, Matte A, Cygler M. Crystal structure of N-succinylarginine dihydrolase AstB, bound to substrate and product, an enzyme from the arginine catabolic pathway of Escherichia coli. J Biol Chem. 2005;280:15800–15808. doi: 10.1074/jbc.M413833200. [DOI] [PubMed] [Google Scholar]
- Toney MD, Kirsch JF. Direct Bronsted analysis of the restoration of activity to a mutant enzyme by exogenous amines. Science. 1989;243:1485–1488. doi: 10.1126/science.2538921. [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Cryst. 1997;30:1022–1025. [Google Scholar]
- Wang C, Xu D, Zhang L, Xie D, Guo H. Molecular dynamics and density functional studies of substrate binding and catalysis of arginine deiminase. J Phys Chem B. 2007;111:3267–3273. doi: 10.1021/jp067541g. [DOI] [PubMed] [Google Scholar]
- Weickmann JL, Himmel ME, Smith DW, Fahrney DE. Arginine deiminase: demonstration of two active sites and possible half-of-the-sites reactivity. Biochem Biophys Res Commun. 1978;83:107–113. doi: 10.1016/0006-291x(78)90404-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental data consists of three figures. ESI-MS product analysis of incubations containing methylamine. HPLC product analysis of incubation mixtures containing rescue agents hydroxylamine, dimethylamine, t-butylamine, and 2-amino-2-methyl-1,3-propanediol. Depictions of two protein cavities in the H162G DDAH protein structure. Supplemental figures are available at http://www.chembiol.com.