

Abstract
Fibrodysplasia ossificans progressiva (FOP) is a rare human genetic disorder of extensive and debilitating extra-skeletal bone formation. While the challenges of investigating a rare condition are many, the potential benefits are also great – not only for the specific disease under investigation, but also for the unique perspective on how cells normally function and the mechanisms that underlie more common disorders. This review will illustrate some of the many insights that we have gained by studying FOP.
Keywords: ACVR1, ALK2, BMP type I receptor, fibrodysplasia ossificans progressiva (FOP), bone morphogenetic protein (BMP)
Introduction
William Harvey, the seventeenth century physician who discovered blood circulation, wrote [1]: “Nature is nowhere accustomed more openly to display her secret mysteries than in cases where she shows traces of her workings apart from the beaten path; nor is there any better way to advance the proper practice of medicine than to give our minds to the discovery of the usual law of Nature by careful investigation of cases of rare forms of disease.”
The primary goal of biomedical research is “to advance the proper practice of medicine” by developing reliable diagnostics, efficient treatments, and effective preventions. As William Harvey’s quotation illustrates, the value of rare disorders to help us understand common diseases, as well as understand the normal patterns of development and function, has long been recognized.
An increasingly valuable approach to determining the causes of many commonly occurring human diseases is through identifying common genetic variants (polymorphisms) that cause or pre-dispose an individual to a particular disease in combination with other common genetic variants and/or environmental influences [2]. Such an approach has great potential to understand many human disorders while also providing an opportunity to elucidate the genes and cellular pathways that regulate normal function. However, special opportunities for insight into how our bodies and our cells function are provided by rare genetic diseases that are caused by mutations in single genes. These conditions are rare because such genes are likely of such critical functional importance that mutations in these genes are rarely tolerated. By identifying the genes that cause these genetic disorders, it becomes possible to reveal key components of cellular function that can be targeted for therapeutic intervention – not only for the rare disease under examination, but also for more common conditions that utilize the same cellular pathways.
In this review, we focus on the rare human genetic disease fibrodysplasia ossificans progressiva (FOP; MIM #135100). While our understanding of FOP remains incomplete, we already have gained tremendous insight into cellular mechanisms that regulate bone formation and that, when mis-regulated, cause this disease.
FOP - Overview
FOP is a rare condition, occurring at a population frequency of about 1 per 2 million [3, 4]. Most cases of FOP are sporadic, with a single affected person in a family. FOP can be inherited in an autosomal dominant pattern, however due to the severe disability of FOP, only a few cases of inheritance from one generation of a family to the next are known to have occurred [4, 5].
The main characteristic clinical feature of FOP is the formation of extra-skeletal, or heterotopic, bone. In addition, specific skeletal malformations that occur during embryonic development frequently are noted in patients [6]. These clinical observations tell us that the underlying genetic mutation in FOP affects a gene that is important during both pre- and post-natal bone formation.
The clinical features of FOP have been discussed in detail in recent reviews [6, 7], and only a brief contextual overview will be described here.
Clinical description and diagnostic criteria for classic FOP
Two major characteristics define “classic” FOP [6, 8]. Congenital malformation of the great toes (Figure 1) is the earliest phenotypic feature of FOP [9–11]. This is the most recognizable skeletal feature of FOP, although other subtle skeletal changes (such as cervical spine fusions, short/broad femoral necks, and osteochondromas) also commonly occur [12–14].
Figure 1. The classic FOP phenotype.

Classic fibrodysplasia ossificans progressiva (FOP) is defined by two characteristic clinical features. (a) Extensive heterotopic bone formation is seen in this 3-dimensional reconstructed computed tomography (CT) scan of the back of a twelve-year-old child. Flareups of FOP arise and progress in a well-defined spatial pattern that result in ribbons, sheets, and plates of bone that fuse the joints of the axial and appendicular skeleton, entombing the patient in a “second skeleton” of heterotopic bone. (b) An anteroposterior radiograph of the feet of a three-year-old child shows symmetrical great toe malformations of metatarsals and proximal phalanges along with microdactyly, fused interphalangeal joints, and hallux valgus deviations at the metatarsophalangeal joints.
The second classic feature is extra-skeletal bone formation (Figure 1) that begins during childhood; formation of heterotopic bone during embryonic development has not been observed [9, 10, 15]. Heterotopic ossification forms in soft tissues such as skeletal muscle, tendon, and ligaments. The bone occurs independently of the normal skeleton and forms discrete skeletal elements that can fuse with normal skeletal bone. Early stage lesions, particularly those occurring during childhood, are associated with pre-osseous soft tissue swelling and inflammation [9, 16, 17].
Heterotopic bone formation is episodic with ossification sites generally forming in predictable temporal and spatial patterns, with the first episodes typically occurring along the upper back and neck [9, 15, 18]. However, the natural progression of the disease can be altered by environmental factors, such as soft tissue injury. Tissue damage and/or the wound healing response that follows are often associated with induction of heterotopic bone formation at the site of injury [10, 19–23].
FOP histology
Since surgical trauma to tissues of FOP patients induces additional bone formation, biopsy specimens of developing FOP lesions are extremely rare and have only been obtained before a diagnosis of FOP has been made. The opportunities to observe the stages of lesion formation have therefore been limited, but extremely informative.
FOP heterotopic bone characteristically forms through an endochondral pathway [18, 24]. We have recognized several stages of FOP lesion formation leading to mature FOP heterotopic bone: lymphocytic infiltration (inflammation), degradation of muscle tissue, fibroproliferative and highly angiogenic stages, cartilage and finally bone [25, 26].
In addition to histological evaluation that revealed tissue changes consistent with endochondral bone formation, extensive analyses determined that the heterotopic bone formed in FOP is normal by several other criteria such as biochemistry, metabolism, radiography, and biomechanics [27, 28]. FOP heterotopic bone can form a marrow cavity and also heals normally following fracture [24, 29].
POH – A second rare genetic disease of extra-skeletal bone formation
One of the first insights that FOP provided was that all cases of genetic heterotopic ossification do not have the same underlying cause. Clinical referrals of patients with a preliminary diagnosis of FOP soon revealed that these patients segregated into two clearly distinct patterns of type and location of bone formation, leading us to characterize and identify of a second genetic disorder, progressive osseous heteroplasia (POH) [30].
FOP and POH share significant clinical similarities [31, 32]. In each condition, patients develop extensive bone formation within the soft connective tissues. The bone formation occurs episodically and is cumulative over time. In both FOP and POH, the progression and stages of heterotopic bone formation appear to be normal, but the induction of bone formation is regulated inappropriately. In other words, normal bone forms in the wrong place at the wrong time, suggesting that these diseases are caused by changes in critical regulators of chondro/osteo-progenitor cells and/or cell fate decisions.
Despite these similarities, POH can be distinguished from FOP by several clinical criteria [31, 32]. A key difference is that POH patients characteristically develop ossification within the superficial dermal layer of the skin while this tissue is unaffected in FOP. A diagnostic clinical characteristic of POH is the progression of heterotopic ossification from the dermis to the underlying deep connective tissues, with bone forming through the skeletal muscle and in some cases extending to and fusing with skeletal bone. Unlike FOP, POH is not associated with congenital toe malformations, inflammatory tumor-like swellings, or predictable regional patterns of heterotopic ossification. Also distinct from FOP, POH heterotopic bone forms in an asymmetric mosaic distribution and shows a predominance of membranous rather than endochondral bone formation.
Both FOP and POH are rare conditions, with most cases apparently the result of a de novo (spontaneous) genetic mutation. However, examples of inheritance of FOP and POH from one generation to the next occasionally occur, and both diseases are transmitted through an autosomal dominant inheritance pattern. The low frequency of spontaneous mutation cases and the simple Mendelian inheritance pattern suggested to us that each of these diseases is caused by mutations in single genes. Heterozygous inactivating mutations in the GNAS gene, which encodes alpha subunit of the stimulatory G protein (Gsα) of adenylyl cyclase, have been identified in POH patients [33].
FOP experimental approaches
A standard approach to investigating Mendelian genetic diseases is to identify the causative gene through genetic linkage and positional cloning approaches. Although FOP is an inherited disease, through the early stages of our investigations to understand its cause, we were limited in using these genetic strategies by the small numbers of families in which FOP is inherited from one generation to the next.
An alternate approach to studying a rare genetic disease is to identify differences in cell function that reflect the altered cellular activities of the mutated genes. For FOP, we quickly focused on the bone morphogenetic protein (BMP) signaling pathway [34–37]. BMPs are extra-cellular signaling proteins that regulate specific target gene transcription by binding to complexes of type I and type II serine/threonine kinase receptors in the cell membrane. Ligand-bound activated receptors mediate signaling through BMP pathway specific signal transduction factors (Smad1, Smad5, Smad8) and through MAPK signaling pathways.
Bone morphogenetic proteins (BMPs) are members of the TGF-beta family, proteins that regulate cell fate decisions in many different cellular and developmental contexts [38–40]. Many members of the BMP family can specifically induce the complete program of endochondral bone formation [41, 42] and BMPs are active both pre- and post-natally in tissues that are affected in FOP.
Identifying a safely obtainable source of cells for gene expression and cell signaling assays was a particular challenge since heterotopic ossification in FOP patients can be triggered by trauma from tissue biopsies. Trauma to deep connective tissues is avoided in peripheral blood sampling, however, and we routinely used circulating lymphocytes from blood to establish lymphoblast cell lines (LCLs), a common source of DNA for genetic analyses. When we initiated our studies there was little precedent for using LCLs for gene expression analyses, however, these cells have proven to be useful for this purpose [43, 44].
Although lymphocytes are present in early FOP lesions, no experimental evidence supports a specific role for these cells in FOP bone formation. Therefore, while LCLs have been an important and valuable tool for gene expression studies by revealing differences that reflect the underlying genetic mutation, these studies do not lead us to conclusions regarding the in vivo functional relevance of these cells in FOP.
More recently, we have isolated multi-potent progenitor cells from the tooth pulp of discarded primary teeth of FOP children (SHED cells: stem cells from human exfoliated deciduous teeth). These highly proliferative and adherent cells are an important cell system to investigate changes in cell differentiation potential of FOP cells [45, 46].
BMP signaling in FOP
We examined several aspects of BMP pathway signaling in FOP cells in order to identify differences in expression of pathway components and pathway activation. Through an extensive series of experiments, we demonstrated significant changes affecting the BMP pathway in FOP cells, suggesting to us that increased BMP pathway signaling plays a role in FOP pathogenesis.
Early studies used several approaches to examine the expression of BMP family members in cells from FOP patients. One of our first discoveries was that BMP4 mRNA and protein are specifically over-expressed [47–50]. Although over-expression was not observed in cell lines from every patient, subsequent experiments revealed a consistent difference among FOP and control cells: while control cell lines regulate BMP4 mRNA levels at relatively steady amounts in culture over time, FOP cells appear unable to regulate and maintain an even level of BMP4 mRNA instead accumulating steadily increasing levels of BMP4 [51]. Further support for a loss of normal feedback mechanisms in FOP cells was found in studies showing that FOP cells do not appropriately up-regulate mRNA expression of BMP antagonists in response to BMP ligand stimulation [52].
Additional studies further implicated a mis-regulated BMP pathway in FOP, such as impaired receptor internalization and higher levels of cell surface BMPRIA protein (a BMP type I receptor) [51, 53] and dysregulated cell surface heparan sulfate proteoglycan (HSPG) modulation of BMP signaling [54] in FOP cells. Increased levels of BMP receptors and altered expression of HSPGs could plausibly lead to increased BMP signaling activity that trigger abnormal bone formation.
BMP receptor signaling is mediated through at least two known pathways: Smad 1/5/8 and p38MAPK. We determined that while LCLs lack BMP-specific Smad mRNA, BMP signaling is activated through ligand-stimulated p38MAPK phosphorylation in control cells [51, 53]. FOP LCLs, however, show p38MAPK phosphorylation in the absence of BMP ligand, and in response to ligand, FOP cells show a hyper-phosphorylation of p38MAPK compared to controls cells. Consistent with constitutive signaling in FOP cells that was suggested by the increased levels of basal p38MAPK activation, we found that expression levels of known BMP transcriptional targets are also increased [51, 53]. Analyses using SHED cells were consistent with these LCL studies, and also demonstrated ligand-independent BMP signaling and ligand-dependent hyper-responsiveness to BMP in cells from FOP patients [45]. SHED cells, which are amenable to osteogenic differentiation assays, additionally showed that FOP SHED cells expressed higher levels of alkaline phosphatase (ALP) and Runx2 mRNAs and more rapidly differentiated to an osteogenic phenotype.
These data consistently showed enhanced BMP-induced activity in FOP cells, leading us to conclude that increased BMP pathway signaling plays a role in FOP pathogenesis.
Genetics of FOP
Concurrently with investigations of the BMP signaling pathway in FOP cells, we continued to work with colleagues world-wide to identify families with inherited FOP in order to conduct an informative whole-genome linkage analysis. An initial linkage study had supported linkage of FOP to a small number of chromosomal regions with the strongest linkage to a region of chromosome 4 [55], containing the Smad1 gene. However, DNA sequence analysis of Smad1 and other candidate genes within the interval failed to reveal FOP-associated mutations.
As additional families with inherited FOP were identified, a second genome-wide linkage analysis using five two-generation families with classic FOP (malformed toes and patterns of heterotopic ossification) was conducted and identified specific linkage to the 2q23-24 region of chromosome 2 [8]. This relatively large linkage interval contained several candidate genes including ACVR1/Alk2 (Activin A type I receptor) which encodes a receptor for bone morphogenetic proteins (BMPs).
Classic FOP mutations
We examined the ACVR1 gene for mutations in both inherited and sporadic FOP patients with the classic features of FOP by DNA sequencing analysis of ACVR1 coding regions (exons and splice junctions). We discovered that all people with classic features of FOP contain a unique and very specific mutation - the identical heterozygous single nucleotide change at nucleotide position 617 of the cDNA [8] (Figure 2).
Figure 2. ACVR1 mutation in classic FOP.
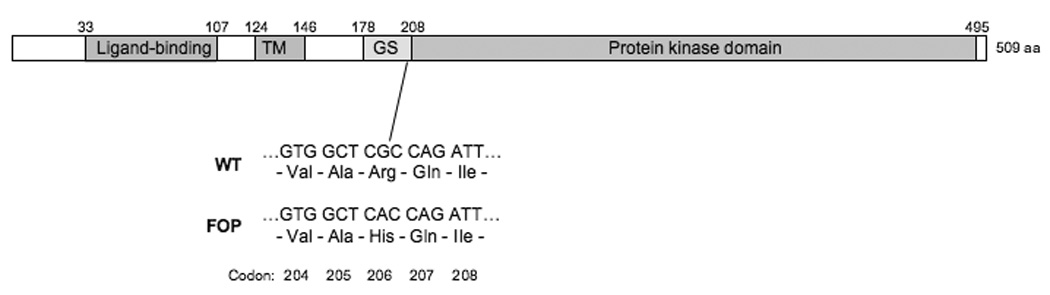
A schematic of the ACVR1 protein is shown, with key functional domains indicated: the extracellular BMP ligand-binding domain, the single trans-membrane (TM) domain, the glycine-serine (GS) activation domain, and the protein kinase signaling domain. Amino acid positions of each domain boundary are indicated above the schematic. All patients who show the classic characteristic features of FOP have the identical mutation in codon 206 within the GS domain. The nucleotide and amino acid sequences of codons 204–208 are shown for the wild-type and FOP mutation. The classic FOP mutation (a G to A substitution at nucleotide position 617 of the cDNA) causes a substitution of histidine for arginine (Arg206His).
This recurrent mutation changes amino acid 206 from arginine to histidine. Amino acid 206 is a highly conserved amino acid in the ACVR1 suggesting its functional importance. Codon 206 occurs within the GS domain of ACVR1, an important functional region of the receptor since phosphorylation of the GS domain is required for downstream signaling [56, 57]. The GS domain binds the inhibitory protein FKBP12 to prevent leaky type I receptor activation in the absence of ligand [58–60] and to regulate receptor endocytosis and ubiquitination [61, 62].
Although both arginine and histidine are positively-charged amino acids, in silico protein homology modeling of the ACVR1 protein predicts that the shorter histidine side chain at amino acid 206 changes the protein structure of the receptor at the GS domain [8, 63] and could disrupt intramolecular interactions that stabilize ACVR1 or that alter interactions between the GS domain and other signaling pathway molecules. Modeling of the ACVR1 structure also predicts that a molecular salt bridge that forms between arginine at residue 206 forms an intramolecular salt bridge with an invariant aspartate in the L45 Smad specificity loop of the protein kinase domain, however histidine at amino acid 206 (as occurs in FOP) would form this interaction only at decreased intracellular pH, effectively acting as a pH-sensitive switch that leads to ligand-independent ACVR1 activation [63].
Atypical and Variant FOP
Following our analysis of individuals with “classic” FOP features, we next turned our attention to more unusual presentations of patients with FOP-like heterotopic ossification [64]. We classified these patients into two groups (Table 1). “Atypical FOP” patients have the characteristic classic FOP features of extensive extra-skeletal ossification and toe malformations, but additionally have one or more features that are not typically associated with FOP. “FOP variants” have heterotopic ossification, but show variations of the classic FOP features, most notably more or less severe malformations of the first digits of the toes and thumbs.
Table 1.
FOP Ossification Syndromes
| Classic | Atypical | Variant | |
|---|---|---|---|
| Progressive heterotopic ossification | 100% | 100% | 100% |
| Malformed great toe (hallux valgus) | 100% | 100% | - |
| Severe or mild digit reduction deficits | no | no | 100% |
| Unique/unusual FOP-associated clinical feature(s) | no | 100% | variable |
| ACVR1 mutation | |||
| c.617G>A; R206H | yes | yes | no |
| other ACVR1 mutation | no | yes | yes |
Most atypical FOP patients (those with features not typically associated with FOP) have the classic FOP mutation (c.617G>A; R206H) in the GS domain of ACVR1. This finding suggests that the additional atypical features are independent of FOP and the R206H ACVR1 mutation, however we cannot exclude that these features are unusual variations caused by the ACVR1 mutation. This may be the case for two atypical FOP patients with non-classic ACVR1 mutations. One patient is heterozygous for a mutation within the ACVR1 GS domain, in codon 207, and the second occurs in codon 356 within the ACVR1 protein kinase domain [64].
None of the FOP variant patients had the classic FOP mutation (c.617G>A; R206H) in ACVR1. However, mutations in other amino acid codons of ACVR1 were found in each patient. With one exception (an in-frame 3 base pair deletion in the GS domain), all of the FOP variants had mutations in the protein kinase domain of ACVR1 [64]. This suggested that the classic R206H mutation causes a relatively predictable effect on embryonic skeletal development, at least for first digit formation, but that some ACVR1 mutations induce a greater or lesser signaling defect during skeletal development.
Of particular interest is ACVR1 codon 328 which is mutated in several FOP variants and shows evidence of genotype-phenotype correlations. In some patients, specific codon 328 mutations (c.982G>T; G328W or c.983G>A; G328E) are associated with a more severe developmental phenotype, with nearly complete truncation of the large toes and thumbs, while other mutations (c.982G>A or c.982G>C; G328R) are associated with a significantly milder skeletal phenotype and little or no effects on the development of the thumbs or toes [64].
ACVR1 mutations in FOP - Summary
All patients with FOP-type heterotopic ossification that we examined contain heterozygous mutations in ACVR1 [8, 64]. These dominant ACVR1 mutant alleles, affecting the GS or protein kinase domains of the receptor protein, appear to have high penetrance: thus far, the mutations have not been found in any individuals without heterotopic ossification and conversely every person with FOP-like heterotopic ossification has an identified ACVR1 mutation. One immediate benefit of identifying ACVR1 mutation in FOP has been our ability to develop a DNA diagnostic test [65], preventing harmful interventions that frequently have occurred prior to a correct diagnosis.
All of the ACVR1 mutations identified in FOP patients occur in evolutionarily conserved amino acids that are predicted to alter protein conformation by in silico structure modeling. Our data further suggest that the severity of developmental skeletal defects (such as observed in FOP variants) may depend on the specific ACVR1 mutant allele.
ACVR1/ALK2
ACVR1/ALK2 is one of four type I receptors that mediate signaling through BMPs [66–69]. In response to BMP ligand binding, these receptors phosphorylate cytoplasmic signal transduction molecules to regulate target gene transcriptional activation or repression. In vivo, specific functional roles for each of the three type I receptors are likely determined by varying efficiencies of ligand binding and by non-overlapping receptor expression patterns in specific tissues and during development [37, 38, 40].
The TGF-β/BMP family is among the classes of extracellular signaling proteins that are key mediators of cell differentiation, with essential roles during embryonic development as well as in maintaining and directing cell fate decisions at later stages [38]. Mutations in the ACVR1 type I BMP receptor in patients with FOP confirms that this receptor mediates critical BMP pathway signals that regulate embryonic skeletal bone formation as well as heterotopic ossification in FOP.
Homozygous null mutation of ACVR1/ALK2 in the mouse demonstrated a requirement for ACVR1/ALK2 in extra-embryonic tissues around the time of gastrulation for normal mesoderm formation and showed that ACVR1/ALK2 signaling is essential for later stage embryonic development [70, 71]. Conditional ACVR1/ALK2 knockout mouse studies have identified roles for ACVR1 in specific tissues and developmental processes. In order to investigate effects of ACVR1 gain of function during embryonic development in specific tissues, a conditionally regulated mouse model to express a constitutively active form of ACVR1 (caALK2) has also been developed [72].
ACVR1 is expressed in several tissues, including skeletal muscle and cartilage, consistent with the sites of heterotopic ossification in FOP. In vitro and in vivo assays have shown that ACVR1 induces BMP signaling activities and chick limb culture experiments demonstrated that constitutive activation of ACVR1 signaling expands cartilage elements and stimulates joint fusions [73–76].
Identification of ACVR1 as the mutated gene in FOP provides the foundation for examining the mechanism through which BMP signaling directs cell differentiation leading to cartilage and bone formation. Key importance is the identification of ACVR1 and the BMP signaling pathway as clear targets for developing treatments for FOP as well as for other more common conditions of too much or too little cartilage and bone.
Ongoing and future FOP studies
We are currently examining the functional effects of FOP ACVR1 mutations through both cell signaling studies and animal models in order to understand the mechanism of the altered BMP signaling and to identify the wide range of effects on tissues and cell systems.
In vitro studies [75] using ACVR1 expression constructs for the classic FOP mutation (c.617G>A; R206H) showed ligand-independent activation of BMP pathway signaling through Smad phosphorylation and promoter reporter assays. We further observed reduced binding of the inhibitory factor FKBP12 to mutant ACVR1, suggesting that increased BMP pathway activity in cells with mutant ACVR1 could be due at least in part to decreased binding of FKBP12 (Figure 3).
Figure 3. A hypothetical model for ligand-independent activation of BMP signaling in FOP.
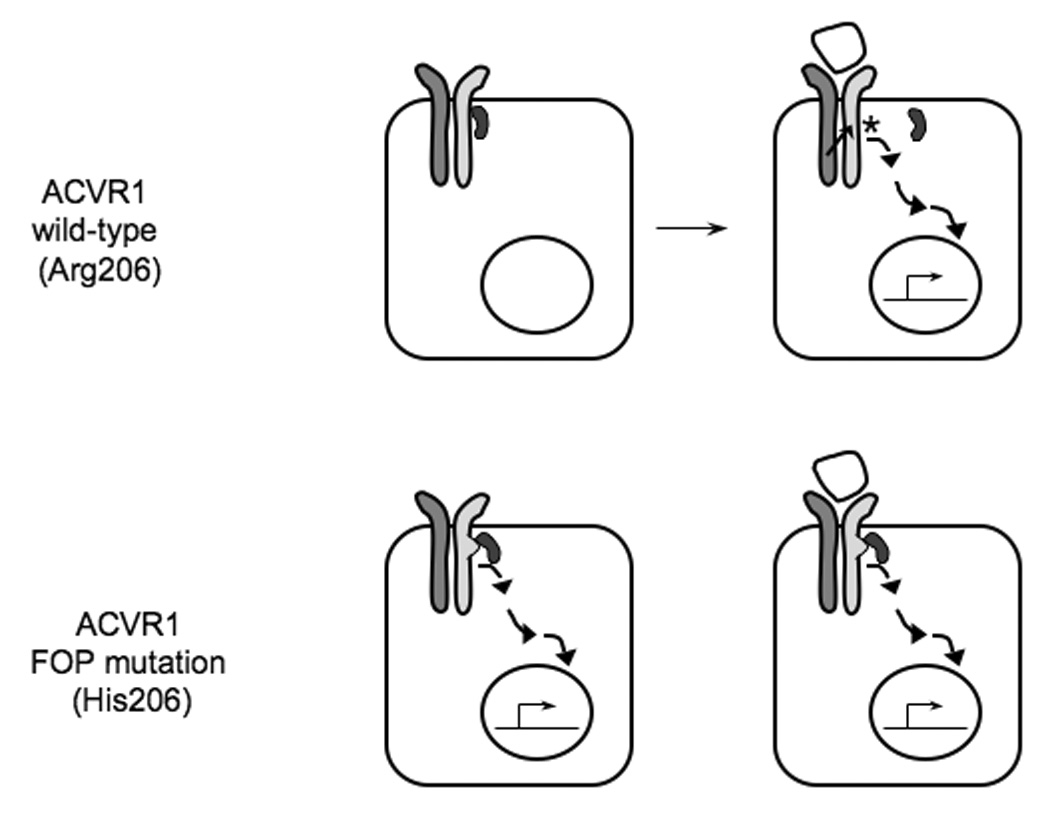
In the absence of BMP ligand, FKBP12 binds the wild-type type I BMP receptor (with arginine at codon 206; Arg206) and prevents “leaky” activation of downstream signaling in the absence of ligand. Upon BMP binding to the type I/type II BMP receptor complex, the type II receptor phosphorylates (asterisk) the type I receptor within the GS domain and induces the release of FKBP12 from the receptor and activation of downstream signaling. We hypothesize that FKBP12 does not correctly bind to the mutant FOP ACVR1 type I receptor (with histidine at codon 206; His206) either in the presence or the absence of BMP-receptor binding, allowing increased activation of BMP signaling.
In vivo functional studies [75] are examining the ability of ACVR1 to rescue the zebrafish Alk8 (homolog of human ACVR1) knockout, which shows a dorsalized phenotype (lost-a-fin) [77]. Transient expression of control ACVR1 RNA in zebrafish embryos showed rescue of the lost-a-fin phenotype. However, mutant ACVR1 induced a ventralized phenotype in the embryos, demonstrating an over-compensation of the knockout phenotype and indicating hyper-activation of BMP signaling.
These and additional preliminary investigations using both in vitro and in vivo assays support that the FOP mutations are activating mutations that enhance BMP signaling pathways and that the mutant receptor stimulates ligand-independent signaling. These data are consistent with our previous observations of increased BMP signaling activity in cells from FOP patients.
Conclusions
FOP is a rare genetic disorder that has given us, and continues to provide, important insight into the cellular and genetic regulation of skeletal and extra-skeletal bone formation. However, the beneficial lessons that FOP has taught us extend far beyond what the relatively small numbers of affected patients might initially cause one to expect. The heterotopic bone that forms in FOP is normal bone by all evaluated criteria; its aberration lies in the lost regulation of cell fate determination that allows inappropriate formation of bone and cartilage in soft connective tissues. Identification of activating mutation of a specific BMP receptor as the cause of FOP reveals a critical regulator of chondro-osseous differentiation potential. However, this receptor and its signaling pathway are not only therapeutic targets for patients with FOP, but also provide an important new focus for developing many wider applications preventing more common nonhereditary forms of heterotopic ossification and for developing tissue engineering strategies for skeletal bone and cartilage repair.
Acknowledgements
We thank members of our research laboratory and our many collaborators for their contributions. We also thank the NIH/NIAMS-supported Penn Center for Musculoskeletal Disorders (AR050950). This work was supported in part by the Center for Research in FOP and Related Disorders, the International FOP Association (IFOPA), the Ian Cali Endowment, the Weldon Family Endowment, the Isaac and Rose Nassau Professorship of Orthopaedic Molecular Medicine, and by a grant from the National Institutes of Health (R01-AR40196).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Harvey W. Letters. In: Alfred Fishman M, editor. The Works of William Harvey. Philadelphia: University of Pennsylvania Press; 1989. pp. 616–617. [Google Scholar]
- 2.Kruglyak The road to genome-wide association studies. Nature Reviews Genetics. 2008;9 doi: 10.1038/nrg2316. [DOI] [PubMed] [Google Scholar]
- 3.Connor JM, Evans DA. Genetic aspects of fibrodysplasia ossificans progressiva. Journal of Medical Genetics. 1982;19:35–39. doi: 10.1136/jmg.19.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shore EM, Feldman GJ, Xu M, Kaplan FS. The genetics of fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:201–204. [Google Scholar]
- 5.McKusick VA. Heritable Disorders of Connective Tissue. 4th ed. St. Louis, MO: C.V. Mosby; 1972. [Google Scholar]
- 6.Kaplan FS, Glaser DL, Shore EM, Deirmengian G, Gupta R, Delai P, Morhart R, Smith R, Le Merrer M, Rogers JG, Connor JM, Kitterman JA. The phenotype of fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:183–188. [Google Scholar]
- 7.Kaplan FS, Le Merrer M, Glaser DL, Pignolo RJ, Goldsby RE, Kitterman JA, Groppe J, Shore EM. Fibrodysplasia ossificans progressiva. Best Practice and Research Clinical Rheumatology. 2008;22:191–205. doi: 10.1016/j.berh.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shore EM, Xu MQ, Feldman GJ, Fenstermacher DA, Cho T-J, Choi IH, Connor JM, Delai P, Glaser DL, Le Merrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genetics. 2006;38:525–527. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 9.Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. Journal of Bone & Joint Surgery - American Volume. 1993;75:215–219. doi: 10.2106/00004623-199302000-00008. [DOI] [PubMed] [Google Scholar]
- 10.Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. Journal of Bone & Joint Surgery - British Volume. 1982;64:76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- 11.Smith R. Fibrodysplasia (myositis) ossificans progressiva. Clinical lessons from a rare disease. Clinical Orthopaedics & Related Research. 1998:7–14. [PubMed] [Google Scholar]
- 12.Deirmengian GK, Hebela NM, O'Connell M, Glaser DL, Shore EM, Kaplan FS. Proximal tibial osteochondromas in patients with fibrodysplasia ossificans progressiva. Journal of Bone & Joint Surgery - American Volume. 2008;90:366–374. doi: 10.2106/JBJS.G.00774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahboubi S, Glaser DL, Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva. Pediatric Radiology. 2001;31:307–314. doi: 10.1007/s002470100447. [DOI] [PubMed] [Google Scholar]
- 14.Schaffer AA, Kaplan FS, Tracy MR, O'Brien ML, Dormans JP, Shore EM, Harland RM, Kusumi K. Developmental anomalies of the cervical spine in patients with fibrodysplasia ossificans progressiva are distinctly different from those in patients with Klippel-Feil syndrome - Clues from the BMP signaling pathway. Spine. 2005;30:1379–1385. doi: 10.1097/01.brs.0000166619.22832.2c. [DOI] [PubMed] [Google Scholar]
- 15.Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age- and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clinical Orthopaedics & Related Research. 1994;301:243–248. [PubMed] [Google Scholar]
- 16.Kaplan FS, Shore EM, Gupta R, Billings PC, Glaser DL, Pignolo RJ, D G, Kamoun M. Immunological features of fibrodysplasia ossificans progessiva and the dysregulated BMP4 pathway. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:189–193. [Google Scholar]
- 17.Moriatis JM, Gannon FH, Shore EM, Bilker W, Zasloff MA, Kaplan FS. Limb swelling in patients who have fibrodysplasia ossificans progressiva. Clinical Orthopaedics and Related Research. 1997;336:247–253. doi: 10.1097/00003086-199703000-00033. [DOI] [PubMed] [Google Scholar]
- 18.Pignolo RJ, Suda RK, Kaplan FS. The fibrodysplasia ossificans progressiva lesion. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:195–200. [Google Scholar]
- 19.Glaser DL, Rocke DM, Kaplan FS. Catastrophic falls in patients who have fibrodysplasia ossificans progressiva. Clinical Orthopaedics & Related Research. 1998;346:110–116. [PubMed] [Google Scholar]
- 20.Hebela N, Shore EM, Kaplan FS. Three pairs of monozygotic twins with fibrodysplasia ossificans progressiva. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:205–208. [Google Scholar]
- 21.Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS. Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. Journal of Pediatrics. 1995;126:762–764. doi: 10.1016/s0022-3476(95)70408-6. [DOI] [PubMed] [Google Scholar]
- 22.Luchetti W, Cohen RB, Hahn GV, Rocke DM, Helpin M, Zasloff M, Kaplan FS. Severe restriction in jaw movement after routine injection of local anesthetic in patients who have fibrodysplasia ossificans progressiva. Oral Surgery Oral Medicine Oral Pathology Oral Radiology & Endodontics. 1996;81:21–25. doi: 10.1016/s1079-2104(96)80141-7. [DOI] [PubMed] [Google Scholar]
- 23.Scarlett RF, Rocke DM, Kantanie S, Patel JB, Shore EM, Kaplan FS. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva. Clinical Orthopaedics and Related Research. 2004;423:275–279. doi: 10.1097/01.blo.0000129557.38803.26. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan FS, Tabas JA, Gannon FH, Finkel G, Hahn GV, Zasloff MA. The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. Journal of Bone & Joint Surgery - American Volume. 1993;75:220–230. doi: 10.2106/00004623-199302000-00009. [DOI] [PubMed] [Google Scholar]
- 25.Gannon FH, Valentine BA, Shore EM, Zasloff MA, Kaplan FS. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clinical Orthopaedics and Related Research. 1998;346:19–25. [PubMed] [Google Scholar]
- 26.Glaser DL, Economides AN, Wang LL, Liu X, Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS, Shore EM. In vivo somatic cell gene transfer of an engineered noggin mutein prevents BMP4-induced heterotopic ossification. Journal of Bone and Joint Surgery-American Volume. 2003;85A:2332–2342. doi: 10.2106/00004623-200312000-00010. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan FS, Strear CM, Zasloff MA. Radiographic and scintigraphic features of modeling and remodeling in the heterotopic skeleton of patients who have fibrodysplasia ossificans progressiva. Clinical Orthopaedics ' Related Research. 1994;304:238–247. [PubMed] [Google Scholar]
- 28.Lutwak L. Myositis Ossificans Progressiva. Mineral, Metabolic and Radioactive Calcium Studies of the Effects of Hormones. American Journal of Medicine. 1964;37:269–293. doi: 10.1016/0002-9343(64)90011-7. [DOI] [PubMed] [Google Scholar]
- 29.Einhorn TA, Kaplan FS. Traumatic fractures of heterotopic bone in patients who have fibrodysplasia ossificans progressiva. A report of 2 cases. Clinical Orthopaedics & Related Research. 1994;308:173–177. [PubMed] [Google Scholar]
- 30.Kaplan FS, Craver R, MacEwen GD, Gannon FH, Finkel G, Hahn G, Tabas J, Gardner RJ, Zasloff MA. Progressive osseous heteroplasia: a distinct developmental disorder of heterotopic ossification. Two new case reports and follow-up of three previously reported cases. Journal of Bone & Joint Surgery - American Volume. 1994;76:425–436. [PubMed] [Google Scholar]
- 31.Kaplan FS, Shore EM. Progressive osseous heteroplasia. Journal of Bone and Mineral Research. 2000;15:2084–2094. doi: 10.1359/jbmr.2000.15.11.2084. [DOI] [PubMed] [Google Scholar]
- 32.Shore EM, Kaplan FS. Fibrodysplasia ossificans progressiva and progressive osseous heteroplasia: Two genetic disorders of heterotopic ossification. Clinical Reviews in Bone and Mineral Metabolism. 2005;3:257–259. [Google Scholar]
- 33.Shore EM, Ahn J, de Beur SJ, Li M, Xu MQ, Gardner RJM, Zasloff MA, Whyte MP, Levine MA, Kaplan FS. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. New England Journal of Medicine. 2002;346:99–106. doi: 10.1056/NEJMoa011262. [DOI] [PubMed] [Google Scholar]
- 34.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 35.Herpin A, Cunningham C. Cross-talk between the bone morphogenetic protein pathway and other major signaling pathways results in tightly regulated cell-specific outcomes. Febs Journal. 2007;274:2977–2985. doi: 10.1111/j.1742-4658.2007.05840.x. [DOI] [PubMed] [Google Scholar]
- 36.Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine & Growth Factor Reviews. 2005;16:251–263. doi: 10.1016/j.cytogfr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 37.Schmierer B, Hill CS. TGFbeta-SMAD signal transduction: molecular specificity and functional flexibility. Nature Reviews Molecular Cell Biology. 2007;8:970–982. doi: 10.1038/nrm2297. [DOI] [PubMed] [Google Scholar]
- 38.Derynck R, Akhurst RJ. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nature Cell Biology. 2007;9:1000–1004. doi: 10.1038/ncb434. [DOI] [PubMed] [Google Scholar]
- 39.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 40.ten Dijke P, Korchynskyi O, Valdimarsdottir G, Goumans M-J. Controlling cell fate by bone morphogenetic protein receptors. Molecular & Cellular Endocrinology. 2003;211:105–113. doi: 10.1016/j.mce.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 41.Urist MR. Bone: formation by autoinduction. Science. 1965;150:893–899. doi: 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- 42.Wozney JM, Rosen V, Celeste AJ, Mitsock LM, Whitters MJ, Kriz RW, Hewick RM, Wang EA. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 43.Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen K-Y, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nature Genetics. 2003;33:422–425. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- 44.Goring HHH, Curran JE, Johnson MP, Dyer TD, Charlesworth J, Cole SA, Jowett JBM, Abraham LJ, Rainwater DL, Comuzzie AG, Mahaney MC, Almasy L, MacCluer JW, Kissebah AH, Collier GR, Moses EK, Blangero J. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nature Genetics. 2007;39:1208–1216. doi: 10.1038/ng2119. [DOI] [PubMed] [Google Scholar]
- 45.Billings PC, Fiori JL, Bentwood JL, O'Connell MP, Jiao X, Nussbaum B, Caron RJ, Shore EM, Kaplan FS. Dysregulated BMP signaling and enhanced osteogenic differentiation of connective tissue progenitor cells from patients with fibrodysplasia ossificans progressiva (FOP) Journal of Bone and Mineral Research. 2008;23:305–313. doi: 10.1359/JBMR.071030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, Shi S. SHED: stem cells from human exfoliated deciduous teeth. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5807–5812. doi: 10.1073/pnas.0937635100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gannon FH, Kaplan FS, Olmsted E, Finkel GC, Zasloff MA, Shore E. Bone morphogenetic protein 2/4 in early fibromatous lesions of fibrodysplasia ossificans progressiva. Human Pathology. 1997;28:339–343. doi: 10.1016/s0046-8177(97)90133-7. [DOI] [PubMed] [Google Scholar]
- 48.Lanchoney TF, Olmsted EA, Shore EM, Gannon FA, Rosen V, Zasloff MA, Kaplan FS. Characterization of bone morphogenetic protein 4 receptor in fibrodysplasia ossificans progressiva. Clinical Orthopaedics and Related Research. 1998;346:38–45. [PubMed] [Google Scholar]
- 49.Olmsted EA, Kaplan FS, Shore EM. Bone morphogenetic protein-4 regulation in fibrodysplasia ossificans progressiva. Clinical Orthopaedics and Related Research. 2003;408:331–343. doi: 10.1097/00003086-200303000-00044. [DOI] [PubMed] [Google Scholar]
- 50.Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, Kaplan FS. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. New England Journal of Medicine. 1996;335:555–561. doi: 10.1056/NEJM199608223350804. [DOI] [PubMed] [Google Scholar]
- 51.Serrano de la Pena LS, Billings PC, Fiori JL, Ahn J, Kaplan FS, Shore EM. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. Journal of Bone and Mineral Research. 2005;20:1168–1176. doi: 10.1359/JBMR.050305. [DOI] [PubMed] [Google Scholar]
- 52.Ahn J, de la Pena LS, Shore EM, Kaplan FS. Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. Journal of Bone and Joint Surgery-American Volume. 2003;85A:667–674. doi: 10.2106/00004623-200304000-00013. [DOI] [PubMed] [Google Scholar]
- 53.Fiori JL, Billings PC, Serrano de la Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) Journal of Bone and Mineral Research. 2006;21:902–909. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- 54.O'Connell MP, Billings PC, Fiori JL, Deirmengian G, Roach HI, Shore EM, Kaplan FS. HSPG modulation of BMP signaling in fibrodysplasia ossificans progressiva cells. Journal of Cellular Biochemistry. 2007;102:1493–1503. doi: 10.1002/jcb.21370. [DOI] [PubMed] [Google Scholar]
- 55.Feldman G, Li M, Martin S, Urbanek M, Urtizberea JA, Fardeau M, LeMerrer M, Connor JM, Triffitt J, Smith R, Muenke M, Kaplan FS, Shore EM. Fibrodysplasia ossificans progressiva, a heritable disorder of severe heterotopic ossification, maps to human chromosome 4q27-31. American Journal of Human Genetics. 2000;66:128–135. doi: 10.1086/302724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wieser R, Wrana JL, Massague J. GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO Journal. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 58.Chen YG, Liu F, Massague J. Mechanism of TGFbeta receptor inhibition by FKBP12. EMBO Journal. 1997;16:3866–3876. doi: 10.1093/emboj/16.13.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell. 1999;96:425–436. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 60.Huse M, Muir TW, Xu L, Chen YG, Kuriyan J, Massague J. The TGF beta receptor activation process: an inhibitor- to substrate-binding switch. Molecular Cell. 2001;8:671–682. doi: 10.1016/s1097-2765(01)00332-x. [DOI] [PubMed] [Google Scholar]
- 61.Yamaguchi T, Kurisaki A, Yamakawa N, Minakuchi K, Sugino H. FKBP12 functions as an adaptor of the Smad7-Smurf1 complex on activin type I receptor. Journal of Molecular Endocrinology. 2006;36:569–579. doi: 10.1677/jme.1.01966. [DOI] [PubMed] [Google Scholar]
- 62.Yao D, Dore JJ, Jr, Leof EB. FKBP12 is a negative regulator of transforming growth factor-beta receptor internalization. Journal of Biological Chemistry. 2000;275:13149–13154. doi: 10.1074/jbc.275.17.13149. [DOI] [PubMed] [Google Scholar]
- 63.Groppe JC, Shore EM, Kaplan FS. Functional Modeling of the ACVR1 (R206H) mutation in FOP. Clinical Orthopaedics and Related Research. 2007;462:87–92. doi: 10.1097/BLO.0b013e318126c049. [DOI] [PubMed] [Google Scholar]
- 64.Shore EM, Xu M, Connor JM, Kaplan FS. Mutations in the BMP type I receptor ACVR1 in patients with fibrodysplasia ossificans progressiva (FOP) Journal of Bone and Mineral Research. 2006;21:S75–S75. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- 65.Kaplan FS, Xu MQ, Glaser D, Collins F, Connor JM, Kitterman JA, Sillence D, Zackai E, Ravitsky V, Zasloff M, Ganguly A, Shore EM. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics. 2008 doi: 10.1542/peds.2007-1980. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen YG, Hata A, Lo RS, Wotton D, Shi Y, Pavletich N, Massague J. Determinants of specificity in TGF-beta signal transduction. Genes & Development. 1998;12:2144–2152. doi: 10.1101/gad.12.14.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.David L, Mallet C, Mazerbourg S, Feige J-J, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- 68.Macias-Silva M, Hoodless PA, Tang SJ, Buchwald M, Wrana JL. Specific activation of Smad1 signaling pathways by the BMP7 type I receptor, ALK2. Journal of Biological Chemistry. 1998;273:25628–25636. doi: 10.1074/jbc.273.40.25628. [DOI] [PubMed] [Google Scholar]
- 69.ten Dijke P, Yamashita H, Ichijo H, Franzen P, Laiho M, Miyazono K, Heldin CH. Characterization of type I receptors for transforming growth factor-beta and activin. Science. 1994;264:101–104. doi: 10.1126/science.8140412. [DOI] [PubMed] [Google Scholar]
- 70.Gu Z, Reynolds EM, Song J, Lei H, Feijen A, Yu L, He W, MacLaughlin DT, van den Eijnden-van Raaij J, Donahoe PK, Li E. The type I serine/threonine kinase receptor ActRIA (ALK2) is required for gastrulation of the mouse embryo. Development. 1999;126:2551–2561. doi: 10.1242/dev.126.11.2551. [DOI] [PubMed] [Google Scholar]
- 71.Mishina Y, Crombie R, Bradley A, Behringer RR. Multiple roles for activin-like kinase-2 signaling during mouse embryogenesis. Developmental Biology. 1999;213:314–326. doi: 10.1006/dbio.1999.9378. [DOI] [PubMed] [Google Scholar]
- 72.Fukuda T, Scott G, Komatsu Y, Araya R, Kawano M, Ray MK, Yamada M, Mishina Y. Generation of a mouse with conditionally activated signaling through the BMP receptor, ALK2. Genesis: the Journal of Genetics & Development. 2006;44:159–167. doi: 10.1002/dvg.20201. [DOI] [PubMed] [Google Scholar]
- 73.Fukuda T. A constitutively activated BMP receptor, ALK2, induces heterotopic bone formation in patients with Fibrodysplasia Ossificans Progressiva (FOP) Journal of Bone and Mineral Research. 2007;22:S10–S10. [Google Scholar]
- 74.Payne TL, Postlethwait JH, Yelick PC. Functional characterization and genetic mapping of alk8. Mechanisms of Development. 2001;100:275–289. doi: 10.1016/s0925-4773(00)00541-4. [DOI] [PubMed] [Google Scholar]
- 75.Shen Q, Xu M, Little SC, Kaplan FS, Mullins MC, Shore EM. Activation of BMP signaling by the FOP ACVR1 R206H mutation. Journal of Bone and Mineral Research. 2007;22:S43–S43. [Google Scholar]
- 76.Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O'Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. Journal of Bone & Mineral Research. 2003;18:1593–1604. doi: 10.1359/jbmr.2003.18.9.1593. [DOI] [PubMed] [Google Scholar]
- 77.Bauer H, Lele Z, Rauch GJ, Geisler R, Hammerschmidt M. The type I serine/threonine kinase receptor Alk8/Lost-a-fin is required for Bmp2b/7 signal transduction during dorsoventral patterning of the zebrafish embryo. Development. 2001;128:849–858. doi: 10.1242/dev.128.6.849. [DOI] [PubMed] [Google Scholar]