

Introduction
Various drug formulations of hydrophilic nanogel carriers consisted of the cross-linked network of neutral polymers, e.g. poly(ethylene glycol) (PEG), and charged polymers, e.g. polyethylenimine (PEI), have been described recently [1–5]. Potential nanogel applications include delivery of oppositely charged drugs, such as oligonucleotides, plasmid DNA, small interfering RNA, nucleoside 5′-triphosphates, otherwise degraded in biological media. These formulations were also found to significantly enhance biological activity of encapsulated drugs. In addition to passive drug release, we recently confirmed a triggered release mechanism mediated by Nanogel interaction with cellular membranes [6]. Phospholipids are capable of interacting with the charged nanogel network, when nanogel particles get into a close contact with cellular membrane, easily substituting nanogel-formulated drugs. As a result, the drug is rapidly released into the cytosol.
The goal of this work was chemical engineering of nanogel structure in order to increase bioavailability of nanogels through modification of polymeric components of the carrier and surface modification with short peptides for specific targeting and modulation of Nanogel biodistribution. Preparation of nanogels with a biodegradable PEI in the structure [9] can provide a significantly less toxic alternative to the regular carriers. Finally, a novel micellar approach to the environmentally clean (‘green’) synthesis of nanogels with an enhanced cellular accumulation was proposed [7].
Experimental
Materials
Reagents were used with the highest available purity. Solvents were stored over molecular sieves 4A. Branched PEI (MW 2 kDa), PEG (MW 8 kDa), rhodamine isothiocyanate (RITC), thiazolyl blue tetrazolium bromide (MTT) and 1,1′-carbonyldiimidazole (CDI) were purchased from Aldrich Chemical Co. Pluronic® F127, P85 and P123 block copolymers were kindly provided by BASF Co. N-Hydroxysuccinimide [3H]-propionate was from Moravek Biochemicals. Maleimido-PEG-N-hydroxysuccinimide (M-PEG-NHS, MW 5 kDa) was purchased from Nektar Therapeutics. Dimethyl 3,3′-dithiobispropionimidate (DTBP) was purchased from Pierce. Custom C-amides of brain-specific NAFTPDYC (BP) and EGFR-specific MYIEALDSYAC (EP) peptides were synthesized by SynPep Co. and purified by reverse phase HPLC. Human MCF-7 and murine CL-66 breast carcinoma cells were obtained from the ATCC collection.
Instrumentation
Pharmacia FPLC system was used to purify polymer samples by gel permeation chromatography (GPC) with refractive index detector. Particle size was measured using a Brookhaven Instruments Zetasizer equipped with multiangle option. Fluorescent samples were analyzed using a BioTek FLx-800 microplate reader. Cytotoxicity was determined using MTT reagent. Protein content was calculated based on Pierce BCA protein assay.
Micellar Synthesis of Nanogel Carriers
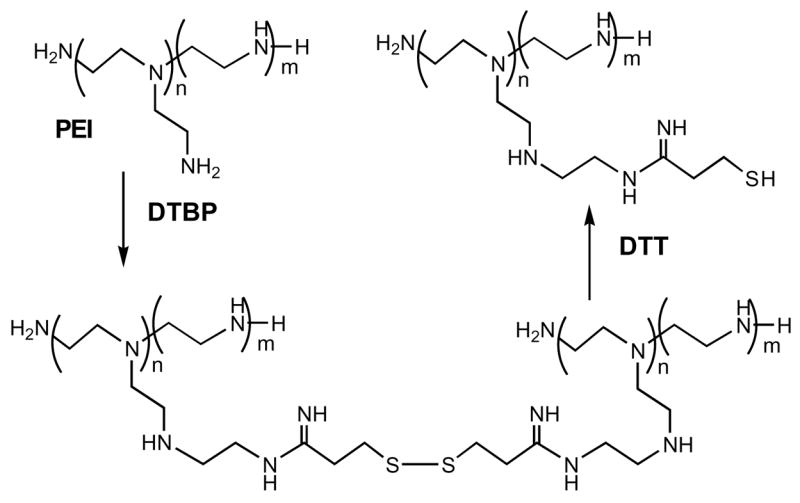
PEG (MW 8 kDa) and Pluronic® (F127, P85 or P123) were dried in vacuo over phosphorus pentoxide and activated by excess of CDI in anhydrous acetonitrile (25°C, 4 h). Activated polymers were dialyzed from the excess of reagent and directly used in the following synthetic steps. To prepare biodegradable PEI (Scheme 1), the branched PEI (MW 2 kDa) was treated overnight with equimolar amount of DTBP [8] in buffered aqueous solution and the obtained oligomeric product was isolated by GPC using Sephadex G-25 column. Fraction of PEI with high MW was used in nanogel synthesis. Synthesis of nanogel NG(PEG) was performed as described previously [3].
Scheme 1.
Other nanogels NG(F127), NG(P85) and NG(P123) were synthesized as following (Figure 1). Aqueous 0.5% (w/v) PEI solution was added dropwise into an equal volume of aqueous 1% solution of the freshly activated Pluronic® (A) under a vigorous stirring to form polymer micelles (B). Reaction of PEI with activated ends of polymer micelles was continued for 48h at 25°C (C). The same volume of aqueous 1% solution of the activated PEG was added next to reaction mixture to cross-link the PEI layer surrounding the polymer micelles (D). The stirring was continued for another 48 h at 25°C. The formed nanogel dispersions were purified by dialysis (MWCO 50 kDa) twice during 24 h against 10% ethanol containing 0.02% aqueous ammonia and lyophilized. Elemental analysis (M-H-W Laboratories), proton NMR, transmission electron microscopy (TEM) and Ellman’s reaction for analysis of thiol content were used to characterize nanogel samples (data not shown).
Figure 1.
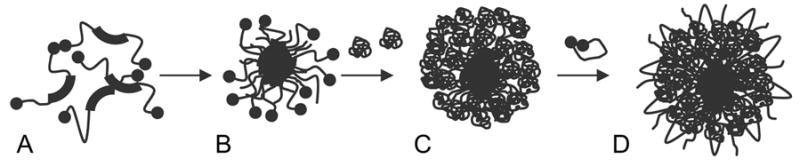
Micellar synthesis of Pluronic®-based nanogels.
Synthesis of Peptide-Nanogel Conjugates
Amino groups of nanogels (80 mg) were modified with M-PEG-NHS (10 mg) in the phosphate-buffered saline (PBS) for 30 min at 25°C. Nanogel-PEG-linker was treated overnight with an excess of thiol-peptide (20 mg) and, then, peptide-modified nanogel was separated by gel-filtration on NAP-20 column. 4–7% of covalently bound peptide was found in these peptide-nanogel conjugates.
Cellular accumulation
Human breast carcinoma MCF-7 cells were grown in 96-well plates, incubated with 0.01 mg/ml of rhodamine-labeled nanogels for 2–4 hrs at 37°C and washed. Fluorescence intensity of the cell-accumulated nanogels was measured and normalized by protein content in the samples.
Biodistribution
Female Balb/c mice were s.c. inoculated with murine mammary carcinoma CL-66 cells and maintained until tumor reached size of ca. 0.5 cm. Then, 2 uCi of tritium-labeled nanogels were injected through the tail vein, and animals were sacrificed in 4h. Radioactivity counts were measured, normalized by organ/tumor weight and expressed as % injected dose per gram (%ID/g).
Results and discussion
The synthesis of the biodegradable PEI with disulfide bonds is shown in Scheme 1. Oligomerized product (PEIss) with MW 30 kDa was isolated as determined by capillary viscosimetry using commercial PEI samples as standards. Pluronic® block copolymers formed micelles in solution at critical micellar concentration (CMC) and higher (usually, 0.005–0.5%). This property was used for preparation of nanogel network by novel micellar approach (Figure 1). Nanogels were obtained from the PEIss the same way as from regular PEI with very good yields (>70% return by polymer material). No organic solvents were used in this protocol making the whole procedure an example of “green chemistry”. Particle size of the products was usually between 100 and 200 nm, which place them in the range applicable for sterilization by filtration through 0.22 um filters (Table 1).
Table 1.
Particle size and cytotoxicity of nanogels
| Nanogel | Diameter, nm | IC50, ug/ml* |
|---|---|---|
| NG(PEG) | 152 +/− 6 | 70 |
| NG(PEGss) | 100 +/− 8 | 250 |
| NG(F127) | 132 +/− 4 | 80 |
| NG(F127ss) | 187 +/− 6 | 190 |
| NG(P85) | 270 +/− 3 | 25 |
| NG(P85ss) | 99 +/− 7 | 170 |
| NG(P123) | 102 +/− 1 | 30 |
| NG(P123ss) | 75 +/− 8 | 130 |
| PEI 25kDa | n/a | 5 |
| PEIss | n/a | 360 |
Measured after 24h-incubation with MCF-7 cells.
Degradation of urethane bonds in regular nanogels was slow showing the half-life 8–9 days (Figure 2A). While more than 2/3 of the biodegradable nanogel network was readily converted into lower MW products following short treatment with reducing agent (DTT) (Figure 2B). These products wereshown to be significantly less cytotoxic than PEI or nanogels and are expected to have a better renal clearance (MWCO 40 kDa) than nanogels. Biodegradable PEIss and nanogels NG(ss) demonstrated significantly decreased cytotoxicity in human breast carcinoma MCF-7 cell line compared to regular nanogels (Table 1).
Figure 2.
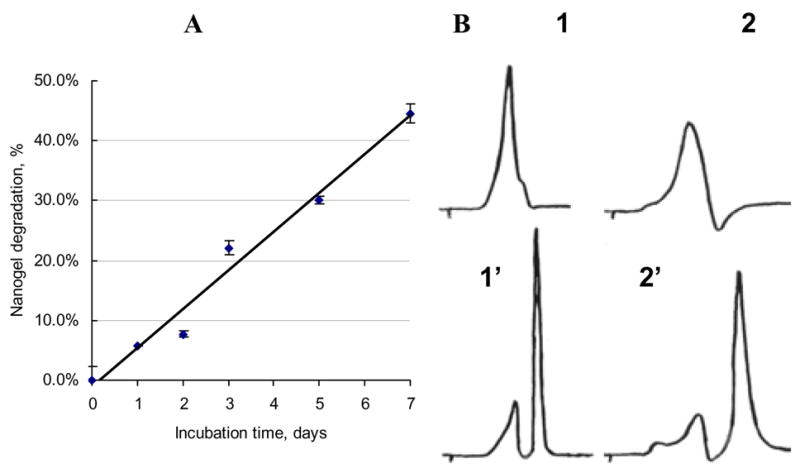
Degradation of nanogels: (A) Kinetics of in vitro degradation of NG(PEG) (PBS, 37°C) based on integrated GPC results; (B) GPC-profiles of NG(PEGss)(1) and NG(P85ss)(2) before and after (1′–2′) 1h-treatment with DTT.
To measure cellular accumulation of nanogel carriers, MCF-7 cells were treated with RITC-labeled nanogels. The obtained data demonstrated that the accumulation efficacy directly depended on lipophilic properties of polymers presented in nanogel structure. Hydrophilic/lipophilic balance of Pluronics® was decreased in the following order: P123<P85<F127. This well correlates with the observed 4-fold increase of cellular accumulation for NG(P123) over NG(PEG) compared to other nanogels (Figure 3).
Figure 3.
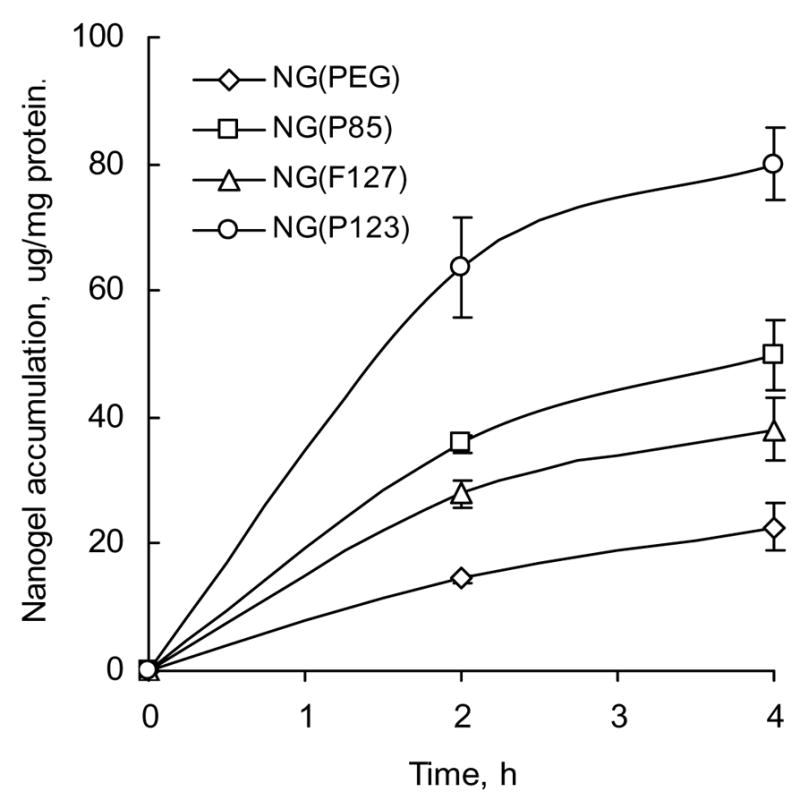
Accumulation of rhodamine-labeled nanogels (0.01 mg/ml) in MCF-7 cells (in triplicate ± SEM).
Tritium-labeled nanogels NG(PEG) conjugated with EP or BP at the density ca.40 EP or 90 BP peptide molecules per particle have been synthesized. Biodistribution of modified and non-modified nanogels between tumor and other organs was then tested following intravenous injection in mice carrying the CL66-induced murine mammary carcinoma. Our results demonstrated significant 3-fold increase in accumulation of EP-NG in tumor following modification of nanogel with EGFR-specific peptide [9]. Brain-specific BP-NG showed a 4-fold increase in brain accumulation versus non-modified nanogel, however, a certain level of brain accumulation was also observed for EP-NG. By another parameter, brain to plasma ratio, however, the BP-NG demonstrated 2-fold increased value compared to NG and EP-NG, which is indicative of a selective BP-NG affinity to the brain (Figure 4).
Figure 4.
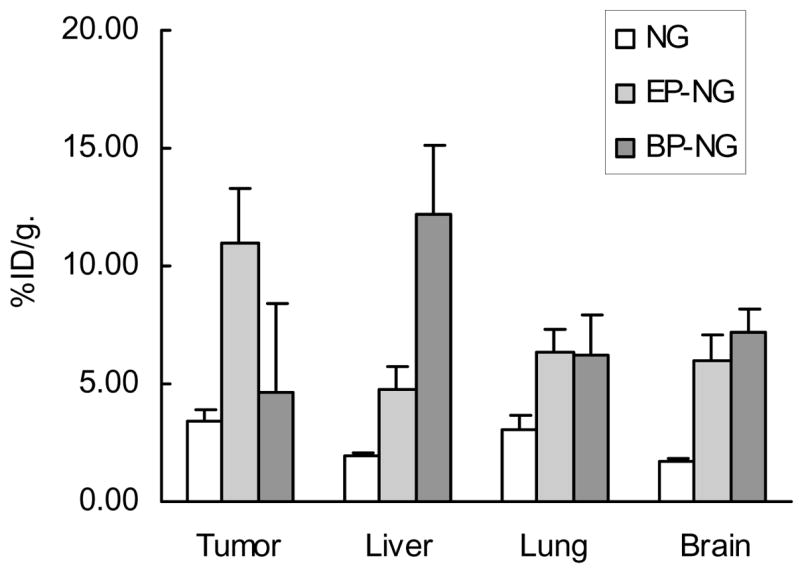
Biodistribution of tritium-labeled nanogels (specific activity 19.5 ± 0.5 uCi/mg) following injection of 2 uCi in the tail vein of mice with L66-induced murine mammary tumor (n = 5, average ± SEM).
Conclusions
Nanosized polymer carriers (nanogels) composed of biodegradable PEI backbone cross-linked by amphiphilic Pluronic® block copolymers have beenproduced using convenient micellar synthesis without usage of organic solvents. These nanogels demonstrated significantly lower cytotoxicity and increased cellular accumulation. It was shown that a multiple modification of nanogels with short EGFR- or brain-specific peptides could positively change biodistribution of intravenously injected carriers to the tumor site or targeted organs. Drug formulations of low- and high molecular nucleotide therapeutics based on the engineered nanogels will be developed for targeted delivery in vivo.
Acknowledgments
The financial support of this research was provided from NIH grants CA102791 and NS050660 for S.V.V. Technical assistance of H.-Y.Han was greatly appreciated.
References
- 1.Vinogradov SV, Bronich TK, Kabanov AV. Adv Drug Deliv Rev. 2002;54:135–147. doi: 10.1016/s0169-409x(01)00245-9. [DOI] [PubMed] [Google Scholar]
- 2.Vinogradov S, Batrakova E, Kabanov AV. Polym Prepr. 2000;41(2):1641–1642. [Google Scholar]
- 3.Vinogradov SV, Kabanov AV. Polymer Preprints (ACS) 2004;45(2):378–9. [Google Scholar]
- 4.Vinogradov SV, Batrakova EV, Kabanov AV. Bioconjug Chem. 2004;15:50–60. doi: 10.1021/bc034164r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinogradov SV, Zeman AD, Batrakova EV, Kabanov AV. J Contr Release. 2005;107:143–157. doi: 10.1016/j.jconrel.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vinogradov SV, Kohli E, Zeman AD. Mol Pharm. 2005;2:449–461. doi: 10.1021/mp0500364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vinogradov SV, Kohli E, Zeman AD. Pharm Res. 2006;23 doi: 10.1007/s11095-006-9788-5. (accepted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gosselin MA, Guo W, Lee RJ. Bioconjug Chem. 2001;12:989–994. doi: 10.1021/bc0100455. [DOI] [PubMed] [Google Scholar]
- 9.Lutsenko SV, Feldman NB, Severin SE. J Drug Target. 2002;10:567–571. doi: 10.1080/1061186021000038058. [DOI] [PubMed] [Google Scholar]